



Frontiers in Bioscience-Landmark (FBL) is published by IMR Press from Volume 26 Issue 5 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Applied Biology, Kyoto Institute of Technology, Kyoto, Japan
Abstract
Traumatic brain injury (TBI) causes substantial mortality and disability, but effective treatments are unavailable. An external force causes primary injury, which is followed by secondary injury that triggers chronic neurodegenerative diseases. Therefore, understanding the mechanisms underlying post-TBI secondary injury might provide insights into neurodegenerative diseases. The secondary injury is known to share some physiological features with neurodegenerative diseases. So far, many TBI models in mammals exist, but models in other species are required from the viewpoint of lifespan and animal welfare. In Drosophila, closed and open TBI models are available. Both models have focused on TBI-induced changes in innate immunity. Aging strongly induces innate immunity responses, and neuroinflammation plays an important role in both mammalian models of TBI and humans with TBI. Although Drosophila models do not mimic all phenomena involved in post-TBI secondary injury in mammals, further experiments with Drosophila models and other animal models could elucidate the mechanisms involved in post-TBI secondary brain injury, which would in turn elucidate neurodegenerative processes.
Keywords
- Drosophila
- TBI
- Neurodegenerative disease
- Inflammation
- Review
Traumatic brain injury (TBI) induces the destruction or deterioration of brain cells. TBI is caused by external forces, such as a blow or gunshot to the head. There are two main types of TBI; open TBI and closed TBI. Open TBI (also called penetrating TBI) is an injury caused by physical penetration of an object into the brain parenchyma. Closed TBI is caused by a forceful impact from outside the skull, with brain injury subsequently occurring within the skull. TBI is one of the leading causes of death and disability across all age groups worldwide, leading to suffering of patients and huge costs to society. For instance, every year in the United States, approximately 1.4 million people sustain a TBI, of which 50,000 die and another 235,000 are hospitalized and survive the injury, with a direct cost estimated at 45 billion dollars per year (1, 2). It is known that a significant prognostic factor for mortality from TBI is being middle-aged or older (3). In Japan, it has been reported that the frequency of occurrence of TBI decreased in young people and increased in people of older ages from 1998 to 2009 (4). This was also observed in other countries (5, 6). According to the World Health Organization, TBI will be the third leading cause of death and disability by 2020 worldwide. However, no effective clinical treatment has so far been identified, possibly because potential TBI therapies are often screened for neuroprotective properties in models involving young animals.
Brain injury is classified, based on its severity, into mild, moderate, or severe using the Glasgow Coma Scale and head component of the Abbreviated Injury Score (7). Moderate-to-severe TBI seems to be associated with a risk of future dementia in young and middle-aged adults (8). A recent study indicated the existence of a time- and dose-dependent risk of developing dementia more than 30 years after TBI (9). Therefore, TBI is not only a problem itself but also appears to contribute to neurodegenerative disease. Furthermore, some molecular mechanisms associated with TBI share some physiological features with neurodegenerative diseases (10-14). Therefore, elucidating the mechanisms associated with TBI could contribute to neurodegenerative pathophysiology. In this review, I will focus on TBI models in Drosophila and briefly compare the pathophysiologic features of these models with the pathophysiologic features of mammalian TBI models. Furthermore, I will provide an example of the effective use of a Drosophila TBI model.
In general, symptoms of neurodegenerative diseases become expressed as people age, so when the TBI model is used as a model of neurodegenerative disease, age is a primary factor to be considered. However, this is difficult to analyze because mammals have a long lifespan. Moreover, animal experiment protocols are categorized by the Scientists Center for Animal Welfare as “D” when a vertebrate model of TBI is used. Therefore, other models using invertebrates are needed. Here, I will discuss models of TBI in Drosophila.
Two types of TBI models have been developed in Drosophila, closed TBI and penetrating TBI (Table 1). The high-impact trauma (HIT) device was developed to induce the closed TBI model (15). The HIT device consists of a fly vial connected to a metal spring that is clamped at one end to a wooden board. The vial is beaten against a polystyrene pad by lifting it up and releasing it. At that moment, flies in the vial suffer brain injury. Using this method, the level of TBI can be manipulated by the number of strikes to the vial. The second method for closed TBI uses the Omni Bead Ruptor-24 Homogenizer (16). Like in the seizure model in which flies receive a mechanical shock by brief vortexing (17), flies are placed into empty 2-mL screwcap tubes that are secured individually into the Omni Bead Ruptor-24 system where they receive shaking shocks. By modulating the intensity of the power (shaking speed in meters/second), the duration of the shock, and the number of shocks, the level of TBI can be determined. The closed TBI models in aged flies show an increased mortality rate than those of young. The model of penetrating TBI has been established by piercing the fly’s brain using a thin needle in our study (18). In this study, a medical 35G needle with a diameter of 0.15 mm was used. The tip of the needle was cut sharply, and the length of the cut face was 0.5 mm (Figure 1A). Using this cut face as a guide, the right side of the fly’s head was stabbed by the needle from the right eye (Figure 1B). Like penetrating TBI in humans, this model shows a greater mortality rate in aged flies. As it is described in section 4.2., innate immunity is strongly activated in TBI model flies. It has been thought that intestinal barrier dysfunction associated with altered immune signaling is linked to death in aging flies (19), which is thought to contribute to mortality in TBI flies. Interestingly, although the closed TBI models expressed disruption of the intestinal barrier in a dye permeability assay (Smurf assay) 24 h after TBI (16, 20), called the Smurf phenotype, the penetrating TBI model did not (18). Even in the aged fly (30 days after eclosion), single stabbing to the head, the thorax, or the abdomen did not show the Smurf phenotype (Figure 2). A recent study reported that the POU/Oct gene nubbin (nub), which encodes the transcription factor isoform nub-PB, is sufficient to drive gene expression related to innate immunity, and enterocyte-specific nub-RB overexpression significantly shortens median longevity. However, the nub-RB–overexpressing flies do not display the Smurf phenotype (21). Therefore, the difference among these models implies that in addition to the brain injury, a systemic injury has occurred in closed TBI models that induces the intestinal barrier disruption.
 Figure 1
Figure 1Penetrating TBI in Drosophila.A. Tip of the 35G medical needle (React System, Japan) and Drosophila. The diameter of the needle is 0.15 mm, and the length of the cutting face of the needle is 0.5 mm (inset). B. Induction of the penetrating TBI fly model using the needle. The brain damage is induced by stabbing the cut face of the tip of the needle into the fly from the right eye. TBI: traumatic brain injury
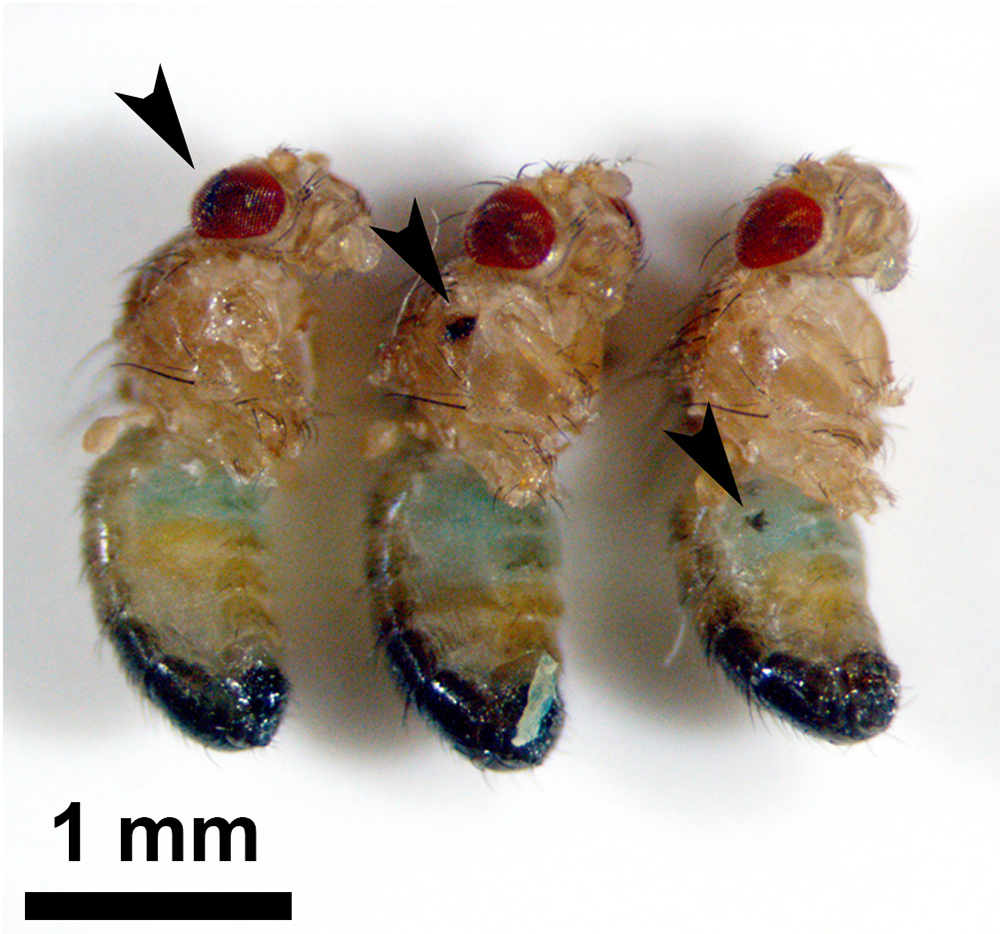 Figure 2
Figure 2Smurf assay in stabbed flies. The fly’s head (penetrating TBI, left), thorax (middle), and abdomen (right) were stabbed using a 35G thin needle. No fly exhibited the Smurf phenotype at one day after injury. Arrowheads indicate the stab wounds. TBI: traumatic brain injury
| Type of TBI | Method | Level of TBI | Smurf | References |
|---|---|---|---|---|
| Closed | Hit using HIT device | Moderate-to-severe TBI | Yes | (15) (20) |
| Closed | Shaken using an Omni Bead Ruptor-24 Homogenizer | Mild-to-severe TBI | Yes (severe model) | (16) |
| Open | Stabbed using a 0.15-mm diameter thin needle | Moderate-to-severe TBI | No | (18) |
TBI is divided into two stages: primary brain injury and secondary brain injury. The exogenous physical impact to the brain causes primary injuries that include direct trauma to the brain parenchyma, cerebral contusion, blood-brain barrier (BBB) disruption, hemorrhage, and hematoma. These primary injuries induce a cascade of molecular events that cause secondary brain injury (22-24) (Figure 3). The secondary injuries have a complicated molecular pathogenesis that includes an entire series of steps or stages of cellular, chemical, tissue, and blood vessel changes in the brain. These changes produce further cell death and lead to neurodegeneration in the hours to days following the initial damage (Figure 3). Negative effects from secondary brain injury continue throughout the life-course and may cause neurodegenerative diseases in later life stages in humans. Thus, it is very important to understand the molecular mechanisms of secondary brain injury to treat TBI and neurodegenerative diseases. Here, I will discuss the neuroinflammation and excitotoxicity involved, both of which are important initial phenomena in secondary brain injury from TBI.
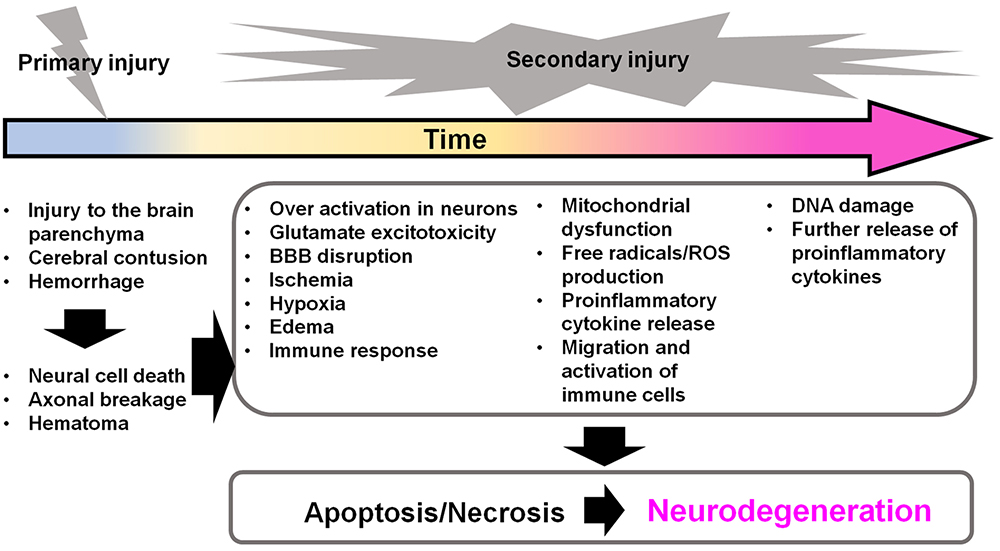 Figure 3
Figure 3Secondary brain injuries in TBI induce further neurodegeneration. After the primary injury, secondary injuries are induced immediately and persist for a long time. The secondary injuries contribute to further cellular apoptosis and necrosis in the brain, which can proceed to neurodegeneration. BBB: blood brain barrier; ROS: reactive oxygen species; TBI: traumatic brain injury
Neuroinflammation is thought to be the key reason for secondary injury in TBI. Even in the absence of microbial infection, the innate immune system is stimulated by danger-associated molecular patterns (DAMPs) that are released from damaged or dying cells (25). Numerous nuclear, cytosolic, and mitochondrial molecules have been known to act as DAMPs. Like many pathogens, pattern recognition receptors (PRRs; e.g., Toll-like receptors, which are homologs of the Toll receptor first identified in Drosophila) recognize DAMPs and activate the nuclear factor-kappa B protein (25). PRRs are expressed in most cells in the central nervous system (CNS), including microglia, astrocytes, oligodendrocytes, and neurons, and stimulate innate immunity in the CNS. Among the CNS cells, microglia express PRRs most abundantly. Microglia are innate immune cells of myeloid origin, like macrophages, in the CNS (26). Microglia are activated like macrophages and can be classified into neurotoxic M1 and neuroprotective M2 phenotypes. DAMPs convert resting microglia to M1 microglia, which transcribe the nuclear factor-kappa B gene at higher rates. Nuclear factor-kappa B then activates its target genes, which in turn produce proinflammatory cytokines (27, 28). Brains exposed to exogenous immune cells and abnormal levels of cytokines due to breakdown of the BBB after TBI show severe immune responses. Because cytokine receptors are widely expressed in CNS cells (29), a chain reaction of neuroinflammation is induced in the brain after TBI.
In Drosophila models of TBI, activation of antimicrobial peptide (AMP) gene expression has been reported (15, 16, 18). AMPs have toxic effects on invading bacteria via cell membrane disruption, and thus contribute to innate immunity in the fly (30). In the penetrating TBI model, compared with young flies, aged flies show increased mortality, increased apoptotic cell numbers in the brain, and age-dependent hyperexpression of AMP genes at the same time (18). The expression of AMP genes is mainly regulated by the Toll and immune deficiency (IMD) pathways through nuclear factor-kappa B activation. The IMD pathway related nuclear factor-kappa B gene, Relish, rather than the Toll pathway, contributes to the hyperactivation of innate immunity in the aged penetrating TBI model (18, 31). It has been reported that overexpression of AMP genes in the brain causes neurodegeneration (31), and overexpression of AMP genes in the whole body or the fat body induces a significantly shortened lifespan (32, 33). Therefore, I suppose that hyperactivation of innate immunity is one of the factors leading to the neurodegeneration and the increased mortality in aged TBI flies. Mutations in the ATM (ataxia-telangiectasia mutated) gene, which encodes a serine/threonine protein kinase and regulates the DNA damage network (34), is a cause of progressive degeneration of Purkinje and granule neurons in the cerebellum, called ataxia-telangiectasia in humans (35). Interestingly, ATM mutant flies, which show neuronal and glial cell death in the adult brain and a reduction in mobility and longevity, have elevated expression of AMP genes (36). Moreover, in the Drosophila model of retinal degeneration, norpA mutants, the locus was found to encode an eye-specific phospholipase C that is essential for photoresponses, and Relish was found to regulate neuronal cell death (37). These studies indicate that hyperactivation of innate immunity in TBI is a common feature throughout species and proper control of the immune system could be an important target for the treatment of TBI and neurodegenerative deceases.
Glutamate is the principal excitatory neurotransmitter in the brain and a ligand of three families of ionotropic receptors (iGluRs), named after their preferred agonists, N-methyl-d-aspartate (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionicacid (AMPA), and kainate. These receptors pass cations, such as Na+, K+, or Ca2+, through their channels upon binding with glutamate (38). Glutamate excitotoxity was originally identified in an experiment in which the parenteral administration of sodium l-glutamate damaged the inner layers of the mouse retina (39). In TBI, overactivation of glutamate receptors results from cell damage that induces glutamate excitotoxity in the brain. Excitotoxic glutamate increases the influx of extracellular Ca2+ into the cell, and intracellular Ca2+ elevation activates nitric oxide synthesis, the generation of free radicals, and programmed cell death (40). Glutamate transport into glial cells is the primary mechanism for the inactivation of synaptically released glutamate, disruption of which induces neurodegeneration (41). Two of the five mammalian glutamate transporter types, glutamate aspartate transporter (GLAST)/excitatory amino acid transporter (EAAT)-1 and glutamate transporter 1 (GLT-1)/EAAT2, are expressed on astrocytes (42) and contribute to the rapid removal of glutamate from the extracellular space.
By sequence analysis of the Drosophila genome, 14 iGluRs that are similar to vertebrate AMPA, kainate, and NMDA receptors were identified (43). Nine of these iGluRs are expressed in the brain (44, 45). In Drosophila, a single glutamate transporter (dEAAT1), which is an ortholog of the mammalian GLAST, is expressed in a glial subset at all developmental stages (46-48), and knockdown of dEAAT1 induced a shortened lifespan and brain neuropil degeneration (48). This neurodegeneration was rescued by the expression of a human GLT-1/EAAT2 gene (48). Huntington’s disease is a late-onset neurodegenerative disorder, and its model in Drosophila, which is made by expressing poly-Q peptides, displays reduced protein and gene expression of dEAAT1 in head extracts (49). These findings indicate that glutamate excitotoxity is a common mechanism involved in neurodegeneration across species. However, it must be noted that the effects of excitotoxity in Drosophila TBI models have not been determined.
An example of the Drosophila TBI model’s utility as a tool for screening potential neuroprotective agents can be found in the literature surrounding the tetracycline-like antibiotic minocycline. Because minocycline has known neuroprotective effects in rodents and flies (50, 51), my colleagues and I (18) examined its effects in a Drosophila model of penetrating TBI. As noted in section 4.2., TBI upregulates the expression of innate immunity–related genes, especially AMP genes, so we examined how administering minocycline at a 0.05-mM concentration affected AMP gene expression in young and aged TBI-exposed flies. We found that minocycline administration reduced AMP gene transcription in both young and aged TBI-exposed flies. Minocycline administration also increased the lifespans of young TBI-exposed flies. These findings suggest that Drosophila TBI models can be used to screen neuroprotective agents. It was interesting that minocycline administration exerted fewer effects in aged TBI-exposed flies, and I speculate that strongly activated immune reactions in aged TBI flies might have diminished minocycline’s efficacy. This has important implications for past investigations that have usually screened neuroprotective drugs in young model animals.
With regard to neuroinflammation, microglia have an essential role in mammals. However, microglia have not been found in flies. Furthermore, in mammals, macrophages migrate into the brain due to disruption of the BBB following TBI and play important roles in phagocytosis and cytokine secretion. Through these actions, macrophages play a role similar to that of microglia. However, plasmatocytes, the fly’s macrophages, do not appear to migrate into the brain in the penetrating TBI model.
In mammals, TBI can follow intracranial hemorrhage that damages the brain. Heme is the main component of hemoglobin, which is primarily released from hemolyzed red blood cells following TBI. Extracellular heme leads to superoxide anion and hydrogen peroxide release (52), which has been known to promote tau phosphorylation and beta-amyloid deposition (53). Released heme is taken up into cells and degraded by heme oxygenase proteins into Fe2+, carbon monoxide, and one isomer of biliverdin, which rapidly reduces to free bilirubin. Free iron causes further chain reactions of free radical–induced damage (52). However, Drosophila have an ortholog of hemoglobin that is not exported into the hemolymph (54). This means that it will be difficult to use Drosophila to understand hemorrhage in secondary injury in TBI.
Although many things differ between mammals and Drosophila, by focusing on the detailed pathways involved in secondary injury, part of the mechanism will be uncovered. For example, one of the downstream injuries caused by TBI hemorrhage is induced by an increase in reactive oxygen species (ROS). So far, methods involving the use of mutations or RNA interference–based gene knockdown to increase ROS levels have been used in studies of destructive agents in the context of nervous system aging, disease, and degeneration. However, abnormalities in these models in developmental stages have been observed. Moreover, it has been reported that ROS signaling is necessary for neuronal development (55, 56). Therefore, the methods using mutants or simple gene knockdown to increase ROS are difficult to use as an adequate model for adult TBI. One of the solutions for this is a current technique to manipulate protein expression levels. The plant-derived auxin-inducible degradation system enables the disruption of a specific protein in a spatiotemporal manner by combination with the GAL4 system (57). This system works well in the brain (58). This system allows the flies to develop normally and can therefore be used to model various disorders with sudden onsets in adulthood, such as TBI in an adult who experiences sudden physical trauma.
This work was supported by the Leading Initiative for Excellent Young Researchers (LEADER) initiated by MEXT, Japan, and JSPS KAKENHI (grant no. 18K06485).