1. Introduction
In Canada, more than 100,000 patients with heart failure are diagnosed annually
and about 2.6 million adults aged 20 and over are living with this heart disease.
Since heart failure is one of the top reasons for hospitalization, the associated
healthcare costs have been estimated to reach 2.8 billion by 2030 in this
country [1, 2, 3, 4]. However, it should be pointed out that significant advances have
been made for the development of medical therapies, which are used for the
treatment of this disease. Several interventions have reduced morbidity,
mortality, and economic burden of this devastating disorder, and in fact a great
deal of effort is being made to further improve its pharmacotherapy [5, 6, 7, 8, 9, 10, 11].
Although extensive research is also being done to understand the pathogenesis of
heart failure, the exact mechanism for its progression remains unclear at present
[12, 13, 14, 15, 16, 17, 18]. Nonetheless, it is evident that heart failure is a complex problem,
which is associated with different disorders such as cardiac dysfunction, cardiac
arrhythmias, loss of adrenergic support, exercise intolerance and fluid
retention. Since a number of vasoactive hormones are elevated in heart
failure, several hormone receptor antagonists are now available for its therapy.
In this regard, guanine nucleotide protein coupled receptors (GPCRs) have been
identified as the most promising targets for drug discovery and a few of their
blockers have been shown to exert beneficial effects in heart failure [19, 20, 21, 22, 23, 24].
It is noteworthy that -adrenergic receptors (-AR) are the most
prominent class of GPCRs, which along with their modulators, are shown to play a
critical role in cardiac health and disease [25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38]. Since alterations in
-AR mechanisms are reported in heart failure, these targets have been
manipulated to achieve clinically relevant therapies [39, 40, 41, 42]. Furthermore,
attenuated responses of the heart to sympathetic stimulation have been observed
at different stages of heart failure [28, 43, 44, 45]. The activities of various
components of -AR system are unaltered, upregulated, or
downregulated in different types of heart failure [42, 46]. Since cardiac
hypertrophy is generally associated with development of heart failure [23, 47, 48, 49, 50, 51, 52], it is not clear whether upregulation or downregulation of
-AR mechanisms are involved in adaptive or maladaptive cardiac
hypertrophy [53, 54, 55, 56, 57, 58]. In this article, we have briefly reviewed the role of
-AR signaling activation in the regulation of cardiac function
upon stimulation of the sympathetic nervous system (SNS). Furthermore, the status
of this system in the development of cardiac hypertrophy and heart failure is
discussed. We have also reviewed the evidence regarding -AR signal
alterations in adaptive and maladaptive cardiac hypertrophy due to pressure
overload. In addition, some observations regarding changes in -AR
mechanisms in adaptive hypertrophy and heart failure due to volume overload are
described to evaluate the role of hypertrophic process in heart failure.
2. Role of -AR Signal Transduction in Cardiac Function
It is now well known that stimulation of -AR signal transduction
by activation of the SNS or exogenous catecholamines for a short duration
augments cardiac function and produces cardiac hypertrophy whereas its
stimulation for a prolonged period results in heart failure. Furthermore, several
-AR blockers have been reported to exert cardiodepressant action
under physiological conditions but improve cardiac function in heart failure
[27, 28, 29, 30, 31, 34, 35, 36, 37, 38, 39, 40, 41, 59, 60, 61, 62, 63]. The activation of -AR stimulates
adenylyl cyclase activity to form 3-5-cyclic adenosine monophosphate
(cyclic AMP) in the myocardium. The elevated level of cyclic AMP promotes protein
kinase A (PKA)-mediated phosphorylations of different Ca-handling proteins
in the sarcolemma and sarcoplasmic reticulum for increasing the intracellular
concentration of Ca ([Ca]) and producing positive inotropic
effect in the heart [39, 64, 65, 66, 67, 68, 69]. The increased activation of -AR
signal transduction is considered to provide circulatory support during early
stages of heart failure [70, 71, 72, 73] but prolonged stimulation triggers
-AR desensitization in the failing heart [42, 55, 69, 74, 75, 76, 77, 78, 79, 80].
Such changes due to elevated levels of circulatory catecholamines or prolonged
stimulation of -AR system are associated with worsening cardiac
outcome, cardiac dysfunction and sudden cardiac death [41, 63, 81, 82, 83, 84, 85, 86].
It is pointed out that the -AR family in healthy human heart comprises
subtypes that include 80% -AR, 20% -AR and about
3% -AR [22, 87, 88, 89]. -AR subtype displays
localization in the sarcolemma in the heart whereas -AR and
-AR subtypes are mainly confined to the T-tubular network [90, 91]. The density of -ARs is reduced by about 50% depending upon
the severity of heart failure, whereas the -AR density remains
unchanged. A substantial reduction in -AR receptor density in
heart failure has been shown to be due to downregulation of these receptors [44, 71, 72, 92, 93]. It should be mentioned that the activation of both
-AR and -AR subtypes occurs with different
potencies by catecholamines (norepinephrine and epinephrine) in general.
-ARs are coupled to G-proteins and
-ARs are coupled to both G-and
G-proteins. The acute activation of -AR through
G-proteins produces positive chronotropic and inotropic responses
as well as cardiac hypertrophy whereas the chronic stimulation of
-AR is associated with heart failure. The effects of both acute
and chronic stimulation of the SNS are illustrated in Fig. 1. It needs to be
emphasized that acute stimulation of -AR system results in
adaptive hypertrophy whereas prolonged -AR signaling accounts for
the development of maladaptive hypertrophy and subsequent heart failure [53, 94, 95, 96, 97]. Furthermore, overexpression of -AR in transgenic mice has
also been reported to exhibit depressed cardiac function, progressive
hypertrophy, and myocardial fibrosis [54, 98]. On the other hand,
G-protein mediated signaling via -AR is generally
believed to be cardioprotective due to its anti-apoptotic and anti-fibrotic
effects [99, 100].
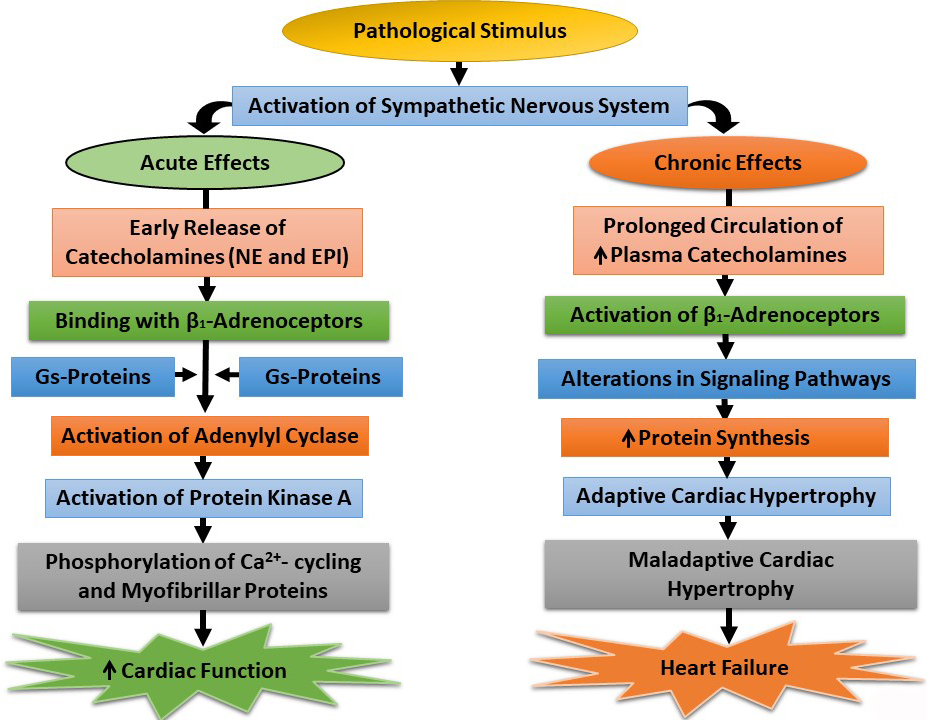 Fig. 1.
Fig. 1.
Acute and chronic effects of the sympathetic nervous system on
-adrenoceptor-mediated signal transduction components. NE,
norepinephrine; EPI, epinephrine; Gs-Proteins, stimulatory guanine nucleotide
proteins; , increased.
In certain types of heart failure such as that due to aortic stenosis, it has
been reported that -AR signaling may change to
-AR-like signaling, become more susceptible to ischemic injury and
contribute to the development of heart failure [101, 102]. It has been suggested
that such pathological manifestations of -AR overexpression are
mediated primarily by G- proteins rather than
G-proteins [102]. Thus, it has been indicated that
-AR signaling may be either protective or deleterious in the heart
depending on transducer coupling with G-proteins [103, 104, 105, 106, 107, 108]. It should also be
noted that both -AR and -AR subtypes are coupled to
-arrestins, which may induce cardioprotective signaling cascades in the
heart. Although the role of -AR in cardiac pathology is unclear,
some studies have suggested that -AR may be involved in the
development of heart disease [89, 109, 110, 111, 112]. The -AR expression in
the myocardium has been shown to be upregulated in heart failure [67, 113, 114].
In addition, -AR has been reported to signal through endothelial
nitric oxide synthase/nitric oxide/cyclic guanosine monophosphate
(eNOS/NO/cGMP) pathway for the attenuation of cardiac contractility [90]. While
extensive work needs to be carried out for establishing the exact role of both
-AR and -AR signaling systems in cardiac
hypertrophy and heart failure, there is overwhelming evidence that
-AR signal transduction is activated. In this regard, it is
noteworthy that blocking -AR signaling by several antagonists such
as carvedilol, metoprolol, atenolol, and bisoprolol has been shown
cardioprotection and other beneficial effects in heart failure [73, 108, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129].
3. Role of -AR Signal Transduction in Cardiac
Hypertrophy and Heart Failure
Several studies have indicated that a wide variety of both extrinsic and
intrinsic stimuli induce activation of different signal transduction pathways to
increase the muscle mass for the occurrence of cardiac hypertrophy. This process
is initiated by mechanical stress as well as different hormones, cytokines and
growth factors that are sensed by different receptors in the cell membrane of
cardiomyocytes. It is evident that cardiac hypertrophy at initial stages is an
adaptive process in which the heart does not show any structural abnormalities
and cardiac function is usually unaltered or augmented [25, 56, 130, 131, 132, 133, 134].
However, if the stimulus is not removed within a certain time period, there
occurs a transition of adaptive hypertrophy to maladaptive hypertrophy, which
exhibit a set of complexities, including cardiac remodeling, cardiac dysfunction,
metabolic alterations, electrophysiological defects and increased ventricular
wall stress. Progressive metabolic alterations in maladaptive hypertrophy are
considered to result in the progression of subcellular abnormalities for
Ca-handling, cardiac dysfunction and heart failure [51, 57, 58]. The loss
of inotropic mechanism in the hypertrophied heart has been reported to occur due
to changes in membrane receptors, protein kinase activities, and associated
signal transduction system as well as defects in subcellular organelles during
the progression of heart failure [23, 34, 49, 50, 52, 69, 135, 136, 137, 138, 139, 140, 141, 142, 143].
Involvement of -AR signaling in both adaptive and maladaptive
hypertrophy as well as in heart failure is now well established [30, 37, 42, 54, 140] and the SNS is considered to regulate the status of -AR
signal pathway during occurrence of these phenotypes. At early stages, activation
of the SNS and subsequent elevation in the levels of plasma norepinephrine and
epinephrine stimulate -AR and increase cardiac contractile force.
However, prolonged hyperactivity of the SNS and elevated plasma catecholamines
result in the derangement of one or more components of the -AR
signaling transduction system, including -AR, Gs-proteins,
adenylyl cyclase, -AR-Gs-protein coupling, and
Gs-protein-adenylyl cyclase interactions. It is pointed out that an increase
in Gs-protein or content results in augmenting cardiac function by
increasing the adenylyl cyclase activity whereas an increase in G-protein
activity or content is known to depress cardiac function by decreasing the
adenylyl cyclase activity. Furthermore, exposure of cardiomyocytes to high amount
of norepinephrine has been shown to cause a reduction in -AR
expression, adenylyl cyclase activity, and contractile activity. Thus, excessive
circulating levels of catecholamines can be seen to induce abnormalities in the
-AR signal transduction pathway and result in cardiac dysfunction
[133, 135, 144, 145, 146, 147, 148].
Depressed sensitivity of -AR to catecholamines as well as
reduction in -AR number are reported to occur in heart failure
[149]. Furthermore, overexpression of -AR in the heart in
transgenic mice was found to develop hypertrophy at young age followed by
progressive heart failure in later life [54, 98, 150, 151, 152]. Chronic stimulation
of -AR by agonists such as isoproterenol has also been observed to
induce cardiac hypertrophy [53] due to activation of PKA by elevated levels of
cyclic AMP. Another study has indicated that -AR signaling
stimulates hypertrophy in a PKA-independent manner via the activation of cyclic
AMP binding protein, Epac [153]. However, other investigators have shown that
mice overexpressing PKA are protected against isoproterenol-induced cardiac
hypertrophy [154]. It is also pointed that the level of Gi-proteins is elevated
in heart failure and this reduces cyclic AMP content for overall depression in
-AR-mediated signaling [68]. Since PKA signaling microdomains
regulate Ca-handling, it has been suggested that some PKA catalytic
subunit may cause maladaptive hypertrophy and result in heart failure [48]. It
should also be mentioned that PKA may directly enhance the stimulation of
calcium-calmodulin kinase (CaMKII) or calcineurin/nuclear factor of activated T cells (NFAT) signaling [155].
Furthermore, the activation of PKA has also been suggested to inhibit cardiac
hypertrophy via some signaling protein changes such as histone deacetylases (HDAC)5 phosphorylation or
HDAC4 proteolysis [156]. While most of these observations support the view that
-AR stimulation results in cardiac hypertrophy and progression to
heart failure [53, 93, 94, 118, 125, 157], the specific mechanisms remain unclear
because of the complex nature of -AR signaling transduction
pathway. It is also likely that changes in -AR signaling may
depend on the stage and type of hypertrophy and heart failure.
4. Dependence of Changes in -AR Signal Transduction on
Type and Stage of Pathological Stimulus
Since hypertrophy and heart failure are known to occur in response to several
pathological stimuli, it was considered of great interest to determine if
alterations in -AR signal pathway occur in different types of
cardiac diseases. It may be noted that pressure overload in cardiovascular
diseases such as hypertension, aortic stenosis, and aortic valve stenosis is
associated with an increase in the ventricular wall thickness (concentric cardiac
hypertrophy). On the other hand, volume overload in pathological conditions such
as anemia, heart block, regurgitant mitral or aortic valves, as well as atrial or
ventricular septal defects, and different congenital diseases, is associated with
dilatation of the left ventricle chamber (eccentric cardiac hypertrophy) [61, 158, 159]. Varying degrees of changes in -AR signaling system due
to both pressure overload [160, 161, 162, 163, 164] and volume overload [165, 166, 167, 168, 169] have been
observed at the end-stage heart failure. Alterations in -AR signal
transduction have also been reported to occur in other types of heart diseases
[170, 171, 172] and heart failure due to chronic myocardial infarction [173, 174, 175].
Downregulation of -AR has been shown to occur in patients with
left heart valvular disease as well as chronic mitral regurgitation [166, 176].
Depressions in myocardial -AR density, adenylyl cyclase activity,
and response to isoproterenol were observed after inducing volume overload [177].
A reduction in the adenylyl cyclase response to norepinephrine has been reported
due to volume overload [167]. Furthermore, upregulation of -AR
mechanisms was seen in the hypertrophic stage whereas these changes were
depressed in heart failure [178]. Alterations in -AR signaling
system, sensitivity of the myocardium to -AR stimulation, as well
as changes in the subcellular distribution of regulatory proteins namely
G-protein-coupled receptor kinase (GRK) isoforms and -arrestins were
observed at different stages of heart failure due to volume overload [165, 168].
Other studies have also shown increased -AR expression and GRK
activity as well as depressed activities of different components of
-AR signaling pathways in heart failure [169, 179, 180, 181]. Such
variable alterations in -AR signal transduction system in the
hypertrophied and failing hearts due to volume overload appear to be related to
the stage of heart disease.
Varying degrees of changes in -AR, adenylyl cyclase and
Gs-protein have also been identified in cardiac hypertrophy under several
conditions associated with pressure overload [160]. Modification of cardiac
adenylyl cyclase activities and changes in Gs-protein function have been
observed in hypertension [172, 182]. Pressure overload induced heart failure in
guinea pigs was accompanied by an increase in -AR density [183]whereas depressions in the density of -AR as well as
isoproterenol-induced increase in cardiac contraction and stimulation of adenylyl
cyclase activity were observed in dogs with heart failure due to pressure
overload [161, 184]. Overexpression of cyclic AMP-hydrolyzing protein
phosphodiesterase 4B (PDE4B), a key negative regulator of cardiac
-AR stimulation, was shown to blunt the -AR
signaling whereas its deficiency resulted in abnormal Ca-handling in
pressure overload induced cardiac hypertrophy [185]. Furthermore, overexpression
of a dominant negative mutant of Gs-proteins decreased
-AR responsiveness and protected against isoproterenol-induced
cardiac hypertrophy in transgenic Gs-DN-mice [186]. These observations
showing variable changes in -AR signaling transduction system due
to pressure overload also support the view that alterations in -AR
signaling are dependent upon the stage of cardiac hypertrophy and heart failure.
5. Experimental Evidence for Alterations in -AR
Mechanisms in Cardiac Hypertrophy
Since heart failure is commonly associated with cardiac hypertrophy, we have
evaluated the existing information to determine if alterations in
-AR mediated activities in the failing hearts are a consequence of
the hypertrophic process. In this regard, we monitored changes in
-AR signal transduction in pressure overload induced cardiac
hypertrophy which was induced upon occluding the abdominal aorta in rats for 4
and 24 weeks [34, 42, 172, 187, 188]. The results in Fig. 2 (Ref. [42]) indicate
that increased heart weight/body weight ratio (an index of cardiac hypertrophy)
at 4 weeks of pressure overload was accompanied by increased left ventricle
developed pressure (LVDP), left ventricle end-diastolic pressure (LVEDP) as well
as rates of both rise and decline of ventricular pressures ( dP/dt) without
any changes in the lung or liver weight to body weight ratios. On the other hand,
hypertrophy induced by pressure overload for 24 weeks was associated with
increased LVEDP and depressions in both LVDP and dP/dt parameters without
any changes in lung or liver weight to body weight ratios (Fig. 2). These
observations suggest that pressure overload for 4 weeks induces adaptive
hypertrophy whereas that for 24 weeks induces maladaptive hypertrophy without any
changes in lung or liver congestion (well-known indices of heart failure).
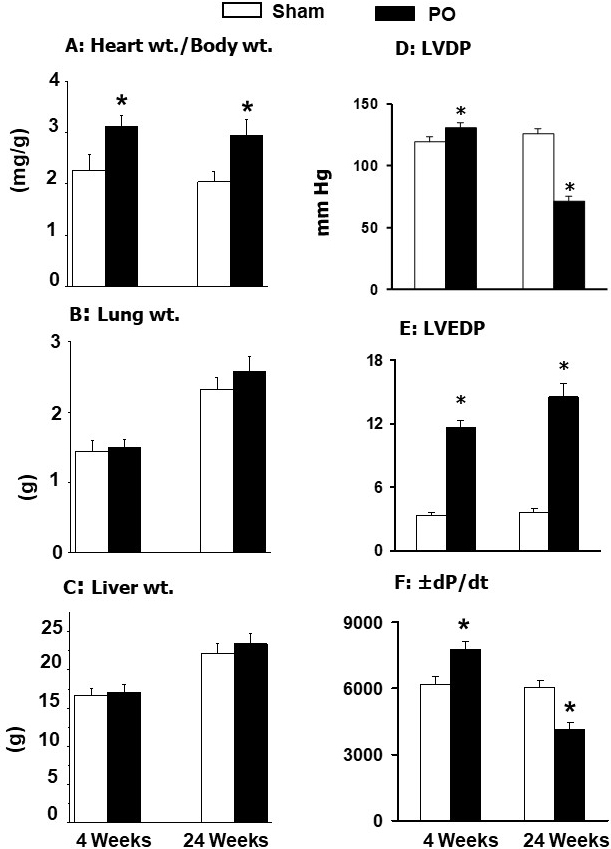 Fig. 2.
Fig. 2.
General characteristics and ventricular function in rats at 4
and 24 weeks due to pressure overload (PO) after occluding the abdominal aorta.
Data are based on the results described in our paper —Journal of Applied
Physiology. 2007; 102: 978–984 [42]. LVDP, left ventricle developed pressure;
LVEDP, left ventricle end diastolic pressure; dP/dt, rates of rise and
decline of ventricle pressures. *p 0.05 versus respective sham.
Fig. 3 (Ref. [42]) shows that increased cardiac function (as reflected by
increase in LVDP) and intracellular Ca-concentration ([Ca])
in cardiomyocytes by isoproterenol were not affected in adaptive hypertrophy due
to pressure overload at 4 weeks. In contrast, both isoproterenol-induced increase
in LVDP in the heart and [Ca] in cardiomyocytes were depressed in
maladaptive hypertrophy due to pressure overload at 24 weeks. Furthermore, the
results in Fig. 4 (Ref. [42]) show that pressure overload induced adaptive
hypertrophy for 4 weeks did not show any changes in -AR density
(B value); without any changes in dissociation constant (K value)
or isoproterenol-induced increase in adenylyl cyclase activity. In contrast,
pressure overload reduced maladaptive hypertrophy for 24 weeks showed depressions
in -AR density and isoproterenol-induced increase the adenylyl
cyclase activity (without any changes in K value) (Fig. 4). These data have
been interpreted to reflect that adaptive cardiac hypertrophy due to pressure
overload did not show any changes in -AR signal transduction
mechanisms whereas maladaptive cardiac hypertrophy due to pressure overload was
associated with some defects in the -AR signaling.
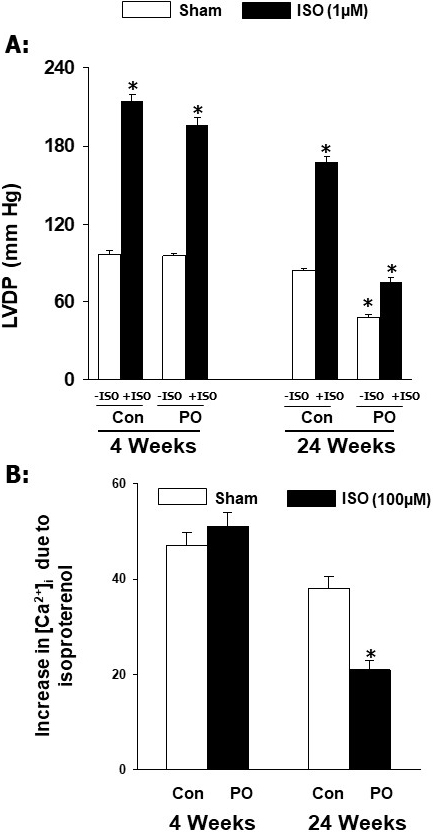 Fig. 3.
Fig. 3.
Effects of isoproterenol (ISO) on ventricular developed pressure
and [Ca] in cardiomyocytes at 4 and 24 weeks due to pressure
overload (PO) in rats. Data are based on the results described in our paper
—Journal of Applied Physiology. 2007; 102: 978–984 [42]. Con, control; LVDP,
left ventricle developed pressure. *p 0.05 versus respective sham.
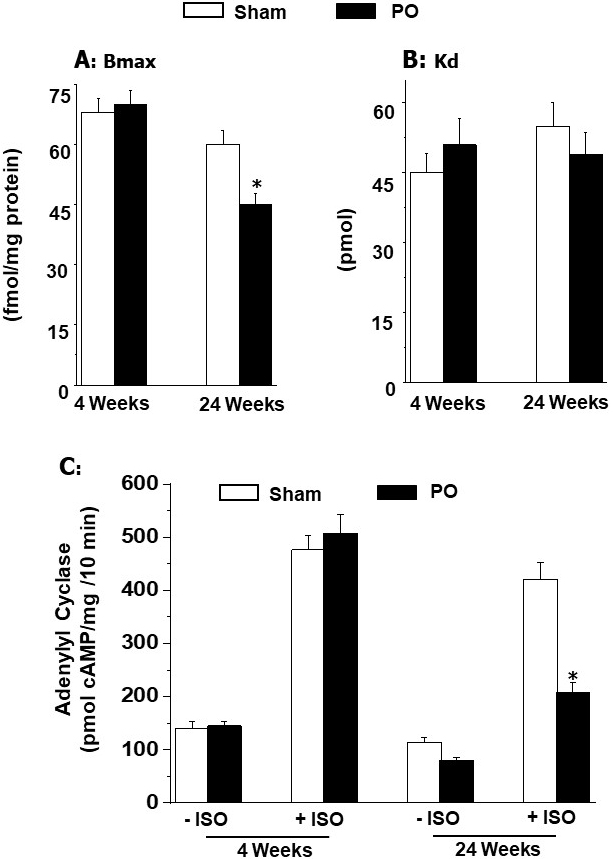 Fig. 4.
Fig. 4.
Ventricular B (maximal number of binding) and
K (dissociation constant) values for -adrenoceptors and
effect of isoproterenol (ISO) on adenylyl cyclase activity at 4 and 24 weeks due
to pressure overload (PO) in rats. Data are based on the results described in
our paper — Journal of Applied Physiology. 2007; 102: 978–984 [42]. *p 0.05 versus respective sham.
6. Experimental Evidence for Alterations in -AR
Mechanisms in Heart Failure
In order to show if changes in -AR signal transduction system in
heart failure are similar to those seen in adaptive cardiac hypertrophy, the data
from studies in which volume overload was induced by aorto-venous (AV) shunt in
rats at 4 and 24 weeks was evaluated [42, 84, 165, 168, 169, 189, 190]. The
results in Fig. 5 (Ref. [42]) show that increased heart weight to body weight
ratio was accompanied by increased LVEDP and lung weight to body weight
ratio without any changes in LVDP, dP/dt and liver weight to body weight
ratios upon inducing AV-shunt for a 4-week period. It is pointed out that since
no changes in cardiac function (as represented by LVDP and dP/dt
parameters) were evident upon inducing volume overload for 4 weeks, we believe
that cardiac hypertrophy at this stage is of adaptive type. Since lung weight to
body weight ratios was significantly increased at 4 weeks of inducing volume
overload, it can be argued that it may represent an early stage of heart failure.
However, this may not be the case as no changes in cardiac function were observed
at this stage. On the other hand, increases in heart weight to body weight ratio
and LVEDP upon inducing volume overload for 24 weeks were associated with
depressions of both LVDP and dP/dt as well as increases in both lung or
liver weight to body weight ratios, indicating the occurrence of heart failure.
These data are consistent with the view that adaptive cardiac hypertrophy and
heart failure due to volume overload become evident at 4 weeks and 24 weeks after
inducing AV-shunt, respectively.
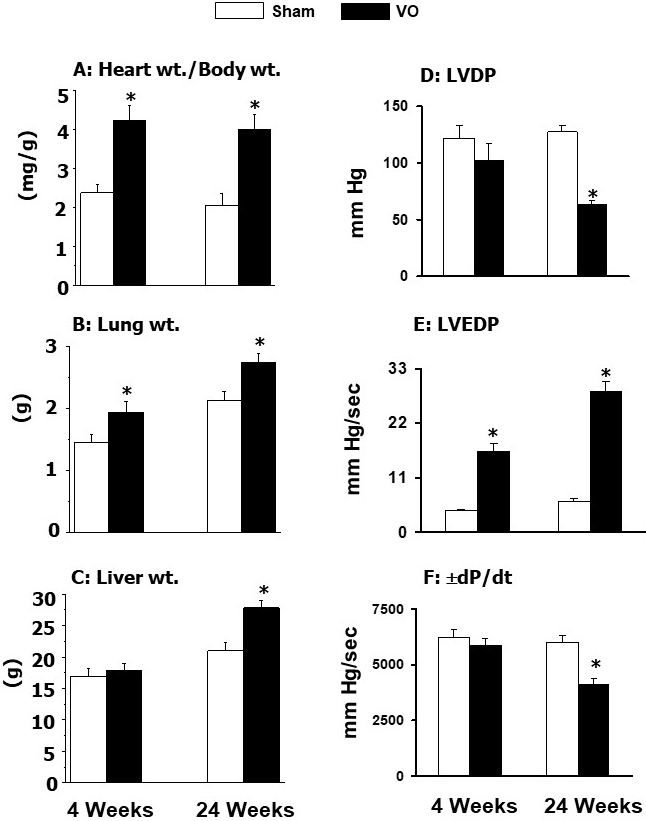 Fig. 5.
Fig. 5.
General characteristics and ventricular function in rats at 4
and 24 weeks due to volume overload (VO) after the aortocaval shunt. Data are
based on the results described in our paper — Journal of Applied Physiology.
2007; 102: 978–984 [42]. LVDP, left ventricle developed pressure; LVEDP, left
ventricle end diastolic pressure; dP/dt, rates of rise and decline of
ventricle pressure. *p 0.05 versus respective sham.
The results described in Fig. 6 (Ref. [42]) indicate that isoproterenol-induced
increases in LVDP in the heart and [Ca] in cardiomyocytes were
augmented by volume overload at 4 weeks of inducing AV-shunt whereas these
responses of the heart to isoproterenol showed marked depressions at 24 weeks
AV-shunt. Furthermore, -AR density as well as activation of
adenylyl cyclase by isoproterenol were markedly augmented by volume overload at 4
weeks after inducing AV-shunt whereas both -AR density and
isoproterenol-induced activation of adenylyl cyclase were attenuated at 24 weeks
after inducing AV-shunt. No changes in K values for -AR were
observed either at 4 weeks or 24 weeks after inducing AV-shunt (Fig. 7, Ref.
[42]). These data indicate that alterations in -AR signal
transduction pathways in the failing heart are not similar to those in adaptive
cardiac hypertrophy due to volume overload.
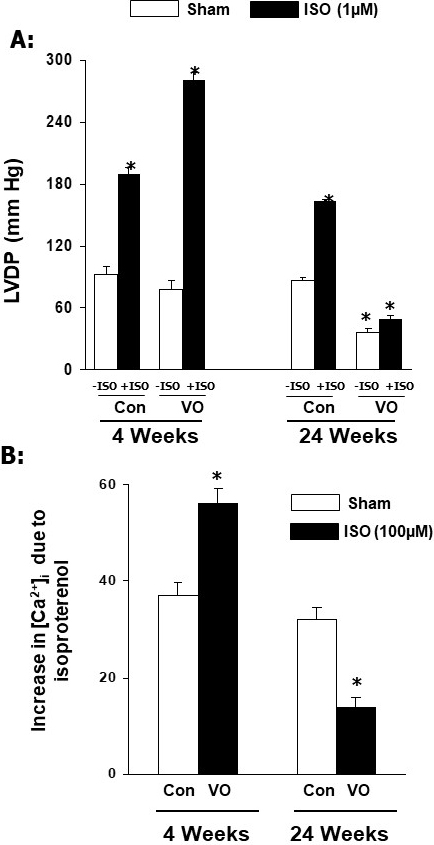 Fig. 6.
Fig. 6.
Effects of isoproterenol (ISO) on left ventricular developed
pressure (LVDP) in rats and [Ca] in cardiomyocytes at 4 and 24 weeks due
to volume overload in rats. Data are based on the results described in our paper
—Journal of Applied Physiology. 2007; 102: 978–984 [42]. LVDP, left ventricle
developed pressure; Con, control; VO, volume overload. *p 0.05 versus respective sham.
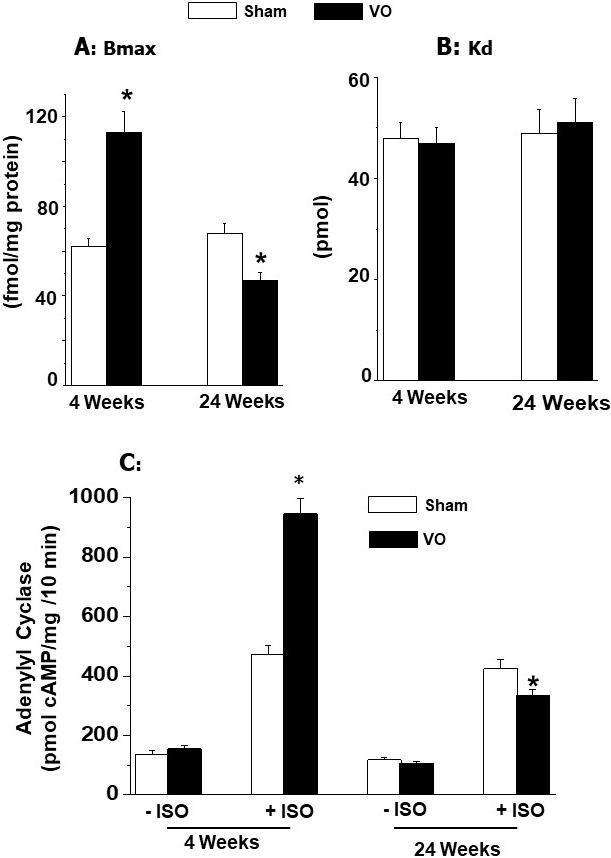 Fig. 7.
Fig. 7.
Ventricular Bmax (maximal number of binding) and Kd
(dissociation constant) values for -adrenoceptors and effect of
isoproterenol (ISO) on adenylyl cyclase activity at 4 and 24 weeks due to volume
overload (VO) in rats. Data are based on the results described in our paper
—Journal of Applied Physiology. 2007; 102:978–984 [42]. *p 0.05 versus respective sham.
7. Conclusions and Perspectives
Although heart failure is associated with cardiac dysfunction, there also occurs
a loss of adrenergic support, which is considered to maintain cardiac performance
in this syndrome. The depression of inotropic responses to stimulation of the SNS
or exogenously administrated catecholamines is considered to be a consequence of
a defect in the -AR signal transduction in heart failure. However,
the exact mechanisms for such an alteration are not fully understood. Since the
-AR signaling system is known to include -AR,
Gs-and Gi-proteins and adenylyl cyclase, it has been observed that alterations in
anyone of these components may result in reduced formation of cyclic AMP and
subsequent impaired PKA-mediated phosphorylation of subcellular proteins in the
failing heart. In view of the importance of -AR signaling and
PKA-induced phosphorylation of Ca- pump and Ca- release proteins in
the sarcoplasmic reticulum as well as troponin and other regulatory proteins in
myofilaments for regulating cardiac function, it is likely that augmentation and
depression of isoproterenol - induced responses of cardiac function in adaptive
cardiac hypertrophy and failing hearts are due to corresponding alterations in
PKA associated phosphorylations [13, 14, 32, 34, 63, 65], respectively. In fact,
various studies in heart failure have shown that the depressed -AR
signaling in failing hearts is due to desensitization of -AR [67, 74, 85] but these changes are considered to be dependent on the stage of heart
failure. Since catecholamines for a short period increase cardiac contractile
force whereas these responses are attenuated over a prolonged period, it appears
that downregulation of -AR signal transduction in heart failure
may be due to elevated levels of plasma catecholamines for a prolonged period. It
is also pointed out that oxidative stress plays an important role in the
pathogenesis of heart failure and it is likely that defects in
-AR signaling at the advanced stage of heart failure may be due to
the development of oxidative stress as a consequence of circulating
catecholamines and other vasoactive hormones such as angiotensin II [34, 80, 191]. Accordingly, it is suggested that therapy of heart failure with some
antioxidants may prove useful in preventing downregulation of -AR
mechanisms in the failing heart.
From the foregoing discussion, it is evident that not only changes in
-AR signal transduction are dependent upon the stage of heart
failure, marked differences in -AR signaling have also been
observed in adaptive and maladaptive cardiac hypertrophy. Particularly, it is
noteworthy that adaptive hypertrophy induced by pressure overload or volume
overload for a 4-week period was found to exhibit either unaltered or augmented
responses of heart function, [Ca] in cardiomyocytes and adenylyl
cyclase activity to isoproterenol as well as unaltered or increased
-AR density. On the other hand, all these responses or parameters
for -AR signal transduction mechanisms were depressed in
maladaptive hypertrophy at 24 weeks of inducing pressure overload as well as in
heart failure at 24 weeks of inducing volume overload. Such differences in
-AR signaling in adaptive and maladaptive cardiac hypertrophy as
well as heart failure can be explained on the basis of differences in the
development of progressive levels of oxidative stress as a consequence of
circulating catecholamines and other vasoactive hormones for a prolonged duration
[143, 191]. Furthermore, it is pointed out that, unlike the adaptive cardiac
hypertrophy, both maladaptive cardiac hypertrophy at 24 weeks due to pressure
overload and heart failure due to volume overload for 24 weeks were found to
exhibit a similar pattern of depressions in all parameters of -AR
signal transduction system. Thus, it appears that downregulation of the
-AR signaling in heart failure or maladaptive cardiac hypertrophy
may not be associated with the hypertrophic process per se. Although
occurrence of oxidative stress has been suggested to be involved in transition of
adaptive hypertrophy to maladaptive hypertrophy as well as progression to heart
failure [80, 143, 191], extensive research work needs to be carried out with
respect to establishing any relationship between oxidative stress and changes in
-AR signal transduction pathway during the development of heart
failure to make any meaningful conclusion.
Several investigators have reported a wide variety of changes in
-AR signal transduction in cardiac hypertrophy and heart failure
[25, 34, 35, 46, 54, 73, 149]; however, the exact mechanisms for such variable
alterations in this pathway have not been identified. It needs to be emphasized
that adaptive cardiac hypertrophy has been suggested to be a consequence of
changes in the redox status of myocardium due to formation of a small amount of
oxyradicals [137, 191]. On the other hand, excessive formation of oxyradicals for
the occurrence of oxidative stress is considered to be involved in the
development of maladaptive cardiac hypertrophy and subsequent heart failure [137, 191]. However, the participation of other mechanisms such as alterations in the
levels of proinflammatory cytokines and intracellular Ca - overload as
well as metabolic abnormalities [51, 60, 61, 136, 138, 155, 192] cannot be ruled
out for explaining the difference in the status of -AR signaling
in non failing and failing hypertrophied hearts. Since the activation of
baroreceptors in the heart is known to play a critical role in the regulation of
cardiac function and -AR mechanism [193], alterations in the
baroreflex mechanisms during the development of hypertension and heart failure
have been implicated in changing the intensity of adrenergic stimuli and
-AR signal transduction pathway [194, 195]. This view is also
supported by the observations that there occurs an increase in the sympathetic
activity and a decrease in the parasympathetic activity in patients with heart
failure [196]. In addition, newer approaches for activating the baroreflex system
or vagal stimulation have been shown to exert promising effects in correcting the
autonomic imbalance for improving cardiac performance in heart failure [197, 198]. Accordingly, progressive changes in the baroreflex system due to both
pressure and volume overload can also be seen to induce upregulation and
downregulation of -AR signaling during the development of cardiac
hypertrophy and heart failure. Thus, it appears that the pathophysiological and
molecular mechanisms in changing the status of -AR signal
transduction pathway in cardiac hypertrophy and heart failure are of complex
nature and require further studies for establishing the exact relationship among
diverse pathogenic factors for the induction of alterations in -AR
signaling.
Author Contributions
NSD developed the concept and outline for this project whereas SKB searched the
literature, prepared figures and wrote the first draft of this manuscript. AA,
KOM and CMLdeV participated in analysis and interpretation of data as well as in
editing and revising the manuscript. All authors have co ntributed sufficiently
in preparing, editorial changes and completing this manuscript and have approved
its submission for publication. All authors have read and approved the final
manuscript and have agreed to be accountable for all aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
We thank the St. Boniface Hospital Albrechtsen Research Centre for
infrastructural support. Thanks are also due to Ms. Khushman Kaur for her help in
editing this paper.
Funding
This research received no external funding.
Conflict of Interest
The authors declare no conflict of interest. Although the data presented in this
paper are based on earlier work from our laboratory, none of the figures in this
article show any similarity with those in our previous paper. Naranjan S. Dhalla is serving as one of the Editorial Board members of this journal. We declare that Naranjan S. Dhalla had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Zoltán Papp and Maurizio Pieroni.








 , Sukhwinder K. Bhullar 1, Adriana Adameova 2, Karina Oliveira Mota 3, Carla Maria Lins de Vasconcelos 3
, Sukhwinder K. Bhullar 1, Adriana Adameova 2, Karina Oliveira Mota 3, Carla Maria Lins de Vasconcelos 3