
- Academic Editor
Cardiovascular dysfunction induced by sepsis is one of the most common phenotypes of cardiovascular diseases (CVDs), which is closely related to the high mortality of sepsis and is an urgent health problem to be solved worldwide. Unfortunately, the exact pathogenesis and pathophysiology of sepsis-induced cardiovascular dysfunction are not clear. As a research hotspot in recent years, competing endogenous RNA (ceRNA) networks are involved in the modulation of the pathophysiological progression of many diseases, including sepsis-related CVDs. Both long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs) can specifically bind to microRNAs (miRNAs) as ceRNAs to target messenger RNAs (mRNAs), forming a ceRNA network composed of lncRNA/circRNA-miRNA-mRNA. This review demonstrates the potential regulatory mechanism of the ceRNA networks in sepsis-induced cardiovascular toxicity, hoping to provide novel therapeutic strategies and monitoring targets for sepsis-related CVDs.
Sepsis is a syndrome of th systemic inflammatory response caused by infection and, ultimately, multiorgan dysfunction [1], which endangers millions of patients worldwide each year and has high mortality rates ranging from one-in-six to one-in-three [2]. The cardiovascular system has been considered as the most frequently affected organ system during sepsis and plays a crucial role in the pathophysiology of septic organ dysfunction. Cardiac depression caused by sepsis is a common phenotype in septic cardiomyopathy and suggests a poor clinical prognosis. Septic cardiomyopathy is characterized by reversible systolic and diastolic dysfunction of the heart throughout the cardiac cycle under septic conditions, which involves complex responses to pathogens, excessive inflammation, oxidative response, metabolic energy impairment, endoplasmic reticulum (ER) stress, myocardial apoptosis and structural changes [3], as shown in Fig. 1. In addition, vascular dysfunction has been recognized as the other common phenotype in sepsis, and is associated with glycocalyx damage, endothelial injury, and vascular dystonia, leading to vascular paralysis, microcirculation disturbances, and septic shock [4]. Septic shock is usually characterized by fluid resuscitation-refractory hypotension and hyperlactatemia. With the development of training, monitoring and treatment in intensive care units, the hospital mortality of septic shock has dropped from 80% to 30%, but this condition is still life-threatening [5].
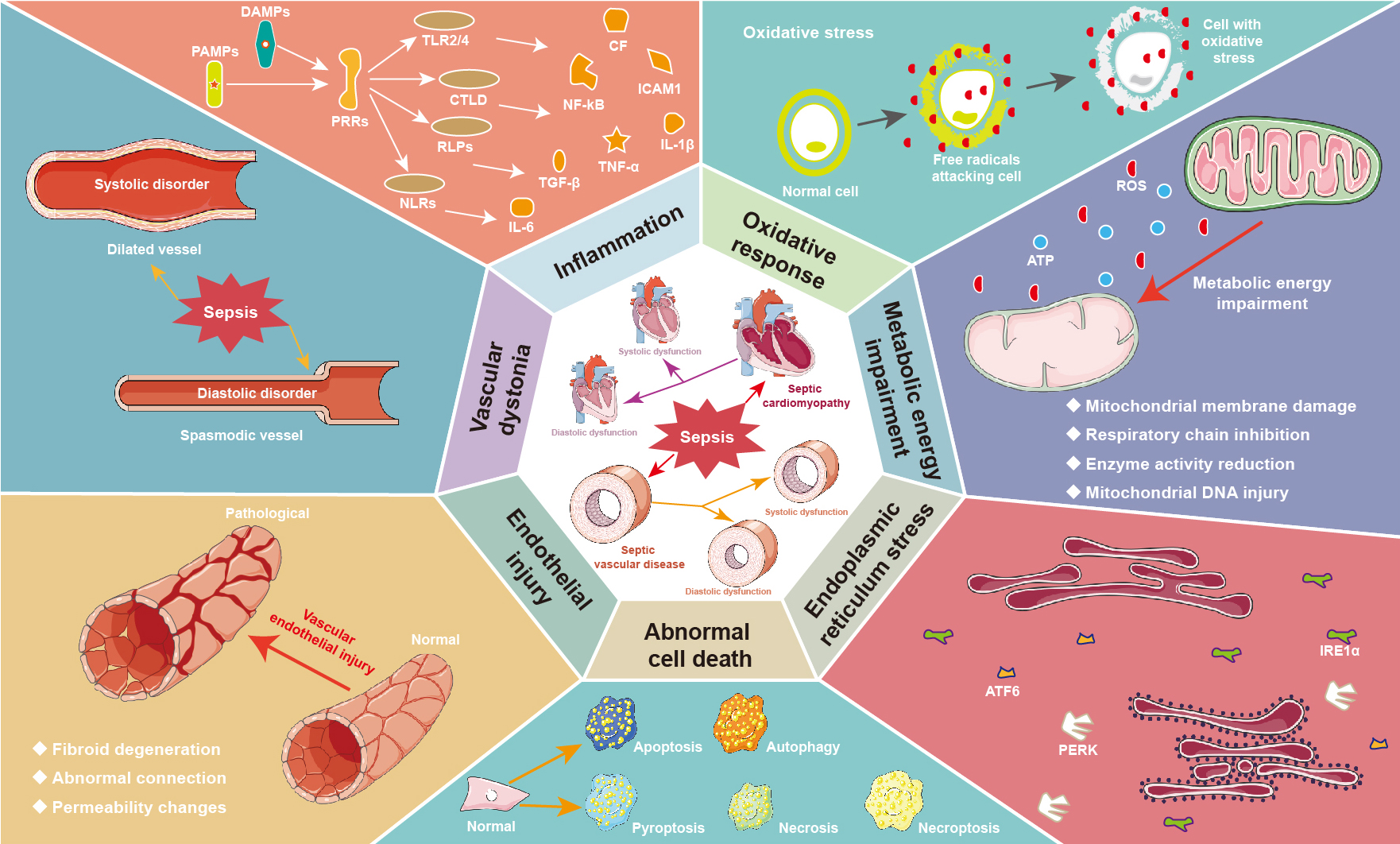 Fig. 1.
Fig. 1.Pathophysiological mechanism of septic cardiovascular diseases.
Septic toxicity often causes systolic and diastolic dysfunction of the heart and
vessels. The main mechanisms of septic cardiomyopathy include excessive
inflammation, oxidative response, metabolic energy impairment, endoplasmic
reticulum stress, and abnormal cell death. In addition, sepsis induced vascular
dysfunction is associated with glycocalyx damage, endothelial injury, and
vascular dystonia, leading to vascular paralysis and microcirculation
disturbances. ATF6, activating transcription factor 6; ATP, adenosine 5’-triphosphate; CF, cell factor; CTLD, C-type lectin-like domain; DAMPs, danger-associated molecular patterns; ICAM1, intercellular cell adhesion molecule-1; IL, interleukin; IRE1
Sepsis-induced cardiovascular diseases (SCVDs, a classic form of CVD) have been confirmed to be the major reason for the increased mortality in patients with sepsis and septic shock, which are considered serious healthcare problems [6]. Despite significant advances in anti-infection and organ supportive therapy, sepsis-associated death remains high. Even survivors who suffer from severe sepsis are often left with long-term sequelae and higher recurrence rates, resulting in a huge social, economic and public health burden [7]. A great number of studies on septic cardiovascular dysfunction have been carried out in recent decades, but the exact pathophysiology and pathogenesis are still unclear. Sepsis lacks ideal biomarkers and has no specific treatment beyond infection control and symptomatic support [8]. Early diagnosis and bundled treatment of sepsis within the first few hours can improve long-term outcomes in SCVDs [9]. Therefore, biomarkers for early detection and precision therapy are urgently needed to improve the survival rate and living quality of patients with SCVDs. Elucidating the molecular mechanism of cardiovascular dysfunction induced by sepsis can provide novel monitoring and therapeutic targets for SCVDs.
Over the past decade, numerous competitive endogenous RNA (ceRNA) species have
been discovered in eukaryotic genomes, which exhibit complex expression and
regulatory mechanisms [10]. Although previous study suggested that noncoding RNAs
(ncRNAs) cannot encode proteins, they participate in the regulation of many
pathophysiological processes (such as SCVDs) via ceRNA networks [11].
NcRNAs include long non-coding RNAs (lncRNAs, lncRs), circular RNAs (circRNAs,
circRs) and microRNAs (miRNAs, miRs). LncRNAs regulate transcription by splicing
and degrading RNA, while posttranscriptional regulation is controlled by decoy
and sponge proteins, as well as nuclear compartmentalization and epigenetic
modification [12]. Different from linear RNA, circRNA consists of a covalently
closed loop, which has neither a poly-A tail nor 5
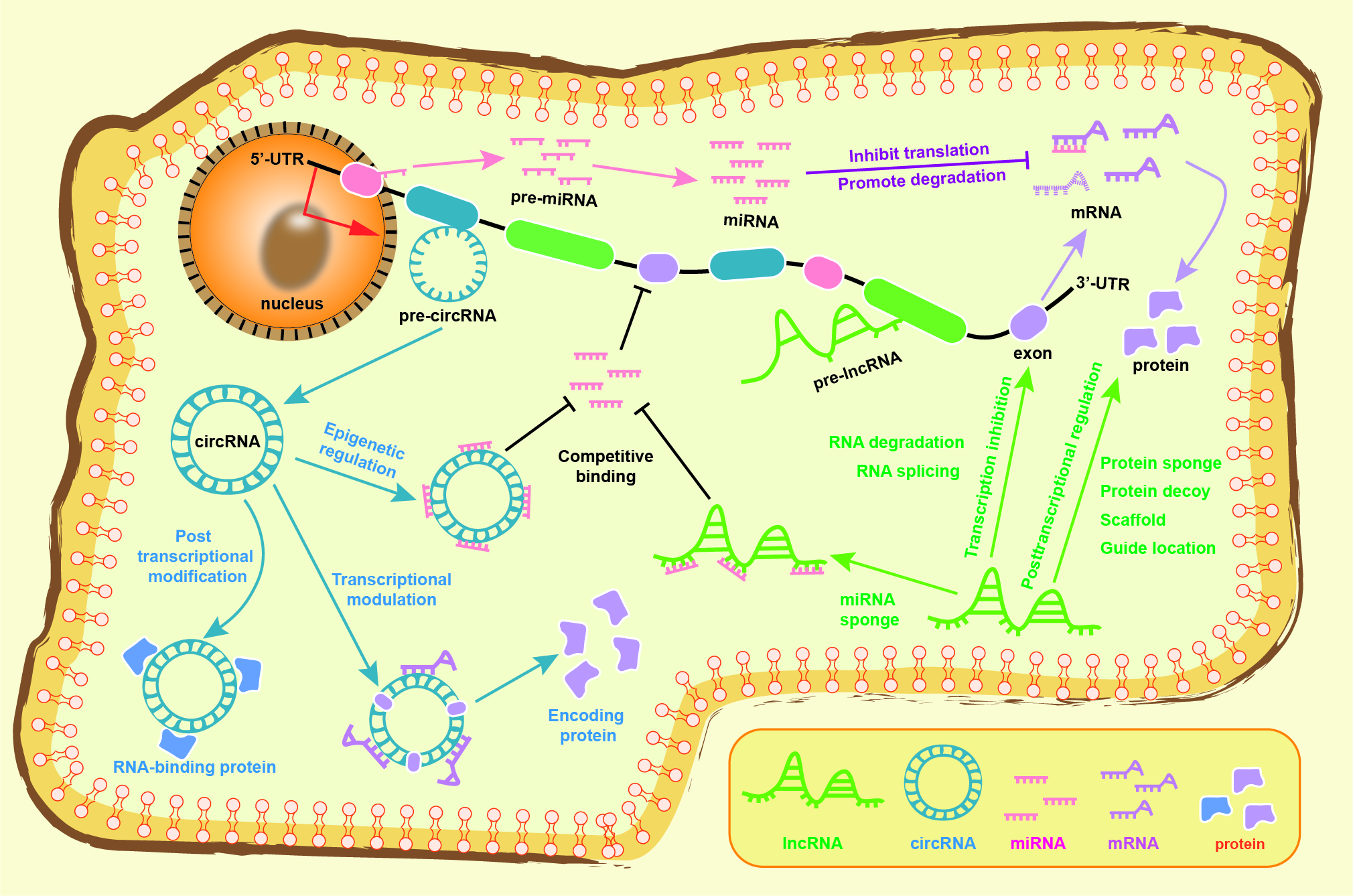 Fig. 2.
Fig. 2.The regulatory mechanism of the competitive endogenous RNA networks in transcription, translation, and epigenetics. LncRNA regulates transcription by splicing and degrading RNA, while posttranscriptional regulation is controlled by decoy and sponge proteins, as well as epigenetic modification. CircRNA regulates transcription and translation by binding to miRNA and interacting with RNA-binding protein, particularly in certain circRNA can code protein. Both lncRNA and circRNA can serve as competitive endogenous RNA that bind with miRNA to regulate the translation of targeted mRNA, thereby inhibiting mRNA translation and promoting mRNA degradation. See text for details. circRNA, circular RNA; lncRNA, long non-coding RNA; miRNA, microRNA; mRNA, messenger RNA; UTR, untranslated regions.
Increasing evidence has shown that ncRNAs are specifically expressed in some tissues and developmental stages and can be used as biomarkers for disease diagnosis and prognosis and as potential therapeutic targets [16]. The lncRNA/circRNA-miRNA-mRNA axis acts as a sophisticated interactive network that regulates cardiovascular structure and function, providing a promising breakthrough for improving cardiovascular dysfunction. This review will provide insight into sepsis induced cardiovascular toxicity and reveal directions for further research, contributing to the diagnosis, treatment, and monitoring of SCVDs.
LncRNAs are a typical class of ceRNAs and are defined as transcripts with a length of more than 200 bp without obvious protein coding functions. It has been demonstrated that lncRNAs participate in multitudinous cellular functions and pathological processes by regulating epigenetic, transcriptional, and posttranscriptional levels, and are potential therapeutic targets for septic CVDs. Lipopolysaccharide (LPS) is a glycolipid heteropolymer located on the outer wall of Gram-negative bacteria, and is often used to simulate sepsis toxicity in animal and cell experiments. Zhang et al. [17] focused on key lncRNAs and mRNAs in septic cardiomyopathy by using an LPS-induced rat model. A total of 74 differentially expressed lncRNAs (41 downregulated and 33 upregulated, of which 39 were novel lncRNAs) and 4011 differentially expressed mRNAs (2093 downregulated and 1918 upregulated) were identified by whole genomic RNA sequencing. Subsequent analysis showed that inhibiting the upregulation of lncRNA PVT1 (plasmacytoma variant translocation 1) significantly inhibited the expression of Myd88 and Bcl-2 and promoted the expression of c-Myc and Bax, thereby alleviating myocardial depression in response to LPS. Another study targeted septic vascular dysfunction by using an LPS-stimulated human umbilical vein endothelial cell (HUVEC) model [18]. A total of 30,584 differentially expressed lncRNAs (1068 downregulated and 871 upregulated) were screened by the Arraystar Human lncRNA Expression Microarray; among them, CTC-459I6.1 and AL132709.5 were the most downregulated and upregulated lncRNAs, respectively.
In addition, a systematic lncRNA survey was performed to assess the effects of
LPS stimulation on human monocytes [19]. A total of 221 differentially expressed
lncRNAs (39 downregulated and 182 upregulated) were screened in LPS-treated
granulocytes, and these differentially expressed lncRNAs were associated with the
human innate immune response. Notably, the LPS-induced differentially expressed
lncRNAs were enriched in NF-
| lncRNA | miRNA | Validation method | mRNA | Model | Mechanism | Ref. |
| ANRIL↑ | miR-125a↓† | Bioinformatics prediction | N.M. | Patients with sepsis | Acts as a diagnostic biomarker for the severity and prognosis of sepsis | [21] |
| CAIF↓ | miR-16↓ | RNA pull-down | CCL2, CXCL1↑† | Sepsis‐induced chronic heart failure patients and LPS-treated AC16 cells | Inhibits CMs apoptosis and inflammation by regulating miR-16 demethylation | [22] |
| CHRF↑ | miR-221↓ | RNA pull-down | NF- |
LPS-injured H9C2 cells | Promotes LPS-induced H9C2 cells injury | [23] |
| CRNDE↓ | miR-29a↑ | DLR, RNA pull-down | SIRT1↓ | LPS-treated Wistar rats and H9C2 cells | Inhibits CMs apoptosis, ROS content, and caspase-3 activity | [24] |
| CYTOR↓ | miR-24↑ | DLR, RNA pull-down | XIAP↓ | LPS-treated SD rats and H9C2 cells | Promotes CMs viability, inhibits CMs apoptosis and apoptosis-related protein release | [25] |
| FGD5‑AS1↓ | miR‑133a‑3p↑ | DLR, RNA pull-down | AQP1↓ | LPS-treated HL-1 cells | Reduces inflammatory cytokines (IL-1 |
[26] |
| GAS5↑ | miR‑26a↓ | DLR, RNA pull-down | HMGB1/NF- |
PA-stimulated H9C2 cells | Aggravates cardiac inflammatory damages | [27] |
| GAS5↑ | miR-214↓ | RNA pull-down | N.M. | Patients with sepsis, LPS-stimulated AC16 cells | Inhibits the apoptosis of CMs in sepsis | [28] |
| GAS5↑ | miR‑449↓ | DLR, RNA pull-down | HMGB1/NF- |
CLP-induced C57BL/6 mice | Promotes myocardial depression, injury, and inflammation responses in septic mice | [29] |
| H19↓ | miR-874↑ | DLR, RIP, RNA pull-down | AQP1↓ | LPS-treated UL-1 cells and BALB/c mice | Restores LPS dysregulated inflammatory responses and myocardial dysfunction | [30] |
| H19↓ | miR-93-5p↑ | DLR, RNA pull-down | SORBS2↓ | LPS-treated H9C2 cells | Reverses CMs growth inhibition and mitochondrial damage | [31] |
| HOTAIR↑ | miR-1-3p↓ | DLR, RNA pull-down | IL-6, TNF- |
LPS-treated H9C2 cells | Inhibits LPS-induced cardiomyocyte CMs proliferation, and induces cell apoptosis and inflammation. | [32] |
| KCNQ1OT1↓ | miR-192-5p↑ | DLR, RIP, RNA pull-down | XIAP↓ | LPS-treated SD rats and H9C2 cells | Facilitates the viability and impedes the apoptosis of CMs | [33] |
| LINC00472↓ | miR-335-3p↑ | DLR, RIP, RNA pull-down | MAOA↓ | LPS-treated C57BL/6 mice and AC-16 cells | Suppresses LPS-induced cardiomyocyte dysfunction activated by Yin Yang-1 | [34] |
| LUCAT1↑ | miR-642a↓ | DLR, RNA pull-down | ROCK1↑ | LPS-stimulated H9C2 cells | Decreases cell viability and migration, increases cell apoptosis | [35] |
| MALAT1↑ | miR-26a↓ | DLR, RNA pull-down | HMGB1, TLR4, NF- |
SFA-treated AC16 cells | Upregulates SFA-induced myocardial inflammatory injury | [36] |
| MALAT1↑ | miR-26a-5p↓ | DLR, RNA pull-down | RCAN2↑ | LPS-treated H9C2 cells and SD rats | Deteriorates LPS-induced inflammation and apoptosis | [37] |
| MALAT1↑ | miR-150-5p↓ | DLR, RIP, RNA pull-down | NF- |
LPS-treated H9C2 cells | Increases sepsis-induced cardiac inflammation | [38] |
| MAPKAPK5-AS1↑ | miR-124-3p↓ | DLR, RNA pull-down | E2F3↑ | LPS-treated H9C2 cells and SD rats | Deteriorates LPS-induced inflammation and apoptosis | [39] |
| MIAT↑ | miR-330-5p↓ | DLR, RNA pull-down | TRAF6/NF- |
LPS-treated BALB/c mice and HL-1 cells | Promotes inflammation response and oxidative stress | [40] |
| MIRT2↓ | miR-101↑ | DLR, RNA pull-down | PI3K/AKT↓ | CLP-induced SD rats | Inhibits myocardial inflammatory response and improves cardiac structure and function | [41] |
| NEAT1↑ | miR-144-3p↓ | DLR, RIP | NF- |
LPS-treated HL-1 cells | Suppresses CMs viability, promotes apoptosis and inflammatory response | [42] |
| PTENP1↑ | miR-106b-5p↓ | DLR, RNA pull-down | IL-6, TNF- |
CLP-induced C57BL/6 mice, LPS-treated H9C2 cells | Enhances cardiac myoblast viability and attenuates inflammation via Matrine treatment | [43] |
| RMRP↓ | miR-1-5p↑ | DLR, RNA pull-down | HSPA4↓ | LPS-treated C57B6/L mice and primary CMs | Attenuates LPS-induced CMs apoptosis and mitochondrial injury | [44] |
| SNHG1↓ | miR-181a-5p↑ | DLR, RNA pull-down | XIAP↓ | LPS-stimulated H9C2 cells | Facilitates CMs viability and represses inflammation and oxidative stress | [45] |
| TTN-AS1↓ | miR-29a↑ | DLR, RNA pull-down | E2F2↓ | LPS-treated SD rats and H9C2 cells | Attenuates mitochondrial ROS activity, and enhances mitochondrial membrane potential | [46] |
| XIST↑ | miR-150-5p↓ | DLR, RIP, RNA pull-down | c-Fos↑ | CLP-induced SD rat, LPS-treated H9C2 cells | Aggravates cardiac dysfunction, increases CMs apoptosis and pyroptosis | [47] |
| XIST↑ | miR-7a-5p↓ | RNA pull-down | PGC-1 |
LPS-stimulated mouse CMs | Reduces cell apoptosis and increases cell proliferation | [48] |
| ZFAS1↓ | miR-138-5p↑ | DLR, RNA pull-down | SESN2↓ | Patients with sepsis-induced myocardial injury, LPS-treated SD rats and H9C2 cells | Ameliorates sepsis induced CMs pyroptosis, myocardial injury and inflammatory response | [49] |
| ZFAS1↓ | miR-34b-5p↑ | DLR, RNA pull-down | SIRT1↓ | LPS-treated SD rats and H9C2 cells | Alleviates inflammatory response and cell apoptosis | [50] |
| ZFAS1↑ | miR-590-3p↓ | DLR, RIP, RNA pull-down | NLRP3↑ | CLP-induced C57BL/6 mice, LPS-treated primary CMs | Aggravates autophagy and pyroptosis of CMs activated by SP1 | [51] |
Note.
| lncRNA | miRNA | Validation method | mRNA | Model | Mechanism | Ref. |
| HULC↑ | miR-204-5p↓ | DLR, RIP, RNA pull-down | TRPM7↑ | LPS-stimulated HUVECs | Deteriorates cell apoptosis, inflammation, and oxidative stress | [52] |
| HOTAIR↑ | miR-211↓ | DLR, RNA pull-down | IL-6R↑ | CLP-induced C57BL/6 mice, LPS-treated monocytes | Aggravates the progression of sepsis, inhibits cellular proliferation, and promotes monocyte apoptosis | [53] |
| LUADT1↓ | miR-195↑ | DLR, RNA pull-down | PIM-1↓ | Sepsis patients, LPS-exposed HCAECs | Reduces LPS-induced cardiac endothelial cell apoptosis | [54] |
| MALAT1↓ | Hsa-miR-346↑ | DLR, RNA pull-down | SMAD3↓ | Patients with sepsis, LPS-treated RAW264.7 mouse macrophages | LPS decreases MALAT1 and SMAD3 levels, and increases has-miR-346 level | [55] |
| MALAT1↑ | miR-23a↓ | DLR, RNA pull-down | MCEMP1↑ | CLP-induced C57BL/6 mice, LPS-treated monocytes | Enhances the inflammation in septic mice and inhibits monocyte apoptosis | [56] |
| MALAT1↑ | miR-146↓ | DLR, RNA pull-down | NF- |
LPS-exposed HMEC-1 cells | Promotes LPS-induced inflammatory injury in microvascular endothelial cells | [57] |
| MALAT1↑ | miR-150↓ | DLR, RIP, RNA pull-down | NF- |
LPS-challenged HUVECs | Exacerbates endoplasmic reticulum stress and inflammatory response | [58] |
| PVT1↑ | miR-29a↓ | DLR, RNA pull-down | HMGB1↑ | LPS-treated C57BL/6 mice and primary macrophages | Enhances macrophage M1 polarization and sepsis-induced myocardial injury | [59] |
| SNHG15↑ | miR-362-3p↓ | RNA pull-down | TNF- |
LPS-exposed HUVECs | Aggravates LPS-induced vascular endothelial cell apoptosis, inflammatory factor expression and oxidative stress response | [60] |
| SNHG16↓ | miR-15a/16↑ | DLR, RNA pull-down | TLR4↓ | LPS-stimulated RAW264.7 cells | SNHG16 and TLR4 were downregulated, while miR-15a and miR-16 were upregulated | [61] |
Note.
CircRNAs (
The circRNA TLK1 (circTLK1, circ_009932) has recently been shown to be significantly upregulated in cecal ligation puncture (CLP)-induced septic rat hearts [65]. CircTLK1 acts as a ceRNA competitor of miR-17-5p. The overexpression of circTLK1 is related to downregulated miR-17-5p and increased levels of PARP1 and HMGB1, which consequently leads to mitochondrial dysfunction and DNA oxidative damage in LPS-treated human cardiomyocytes. Similarly, circRNA PTK2 was highly expressed in the hearts of CLP mice, and in LPS-exposed human umbilical vein endothelial cells (HUVECs) and human vascular smooth muscle cells (HUVAMCs), enhanced expression of circRNA-0044073 promotes the proliferation of vascular endothelial and smooth muscle cells, thereby alleviating atherosclerosis [66]. CircRNA-0044073 activates the JAK/STAT signaling pathway by sponging miR-107, as evidenced by RNA-pulldown and dual-luciferase reporter assays. Interestingly, exosomes derived from mesenchymal stem cells alleviate cardiotoxicity damage by upregulating the circRTN4/miR-497-5p/MG53 axis, and circRTN4 acts as a functional medium to suppress oxidative stress in CLP rats and LPS-treated H9C2 cells [67]. The circRNA-mediated ceRNA networks regulate cardiovascular toxicity in sepsis, as shown in Table 3 (Ref. [65, 66, 67, 68]) and Fig. 3.
| circRNA | miRNA | Validation method | mRNA | Model | Mechanism | Ref. |
| circRNA-0044073↑ | miR-107↓ | DLR, RNA pull-down | JAK/STAT↑ | LPS-stimulated HUVSMCs and HUVECs | Promotes the proliferation and invasion of HUVSMCs and HUVECs | [66] |
| circTLK1↑ | miR-17-5p↓ | DLR, RIP, RNA pull-down | PARP1/HMGB1↑ | CLP-induced SD rats, LPS-treated human CMs | Aggravates mitochondrial dysfunction, DNA oxidative damage, and CMs apoptosis | [65] |
| circRNA-PTK2↑ | miR-29b-3p↓ | DLR, RNA pull-down | BAK1↑ | CLP-induced C57BL/6 mice | Promotes inflammatory response and myocardial damage | [68] |
| circRTN4↓ | miR-497-5p↑ | DLR, RIP, RNA pull-down | MG53↓ | CLP-induced wistar rat, LPS-treated H9C2 and AC16 cells | Mesenchymal stem cells-derived exosomes prevent sepsis-induced myocardial injury | [67] |
Note.
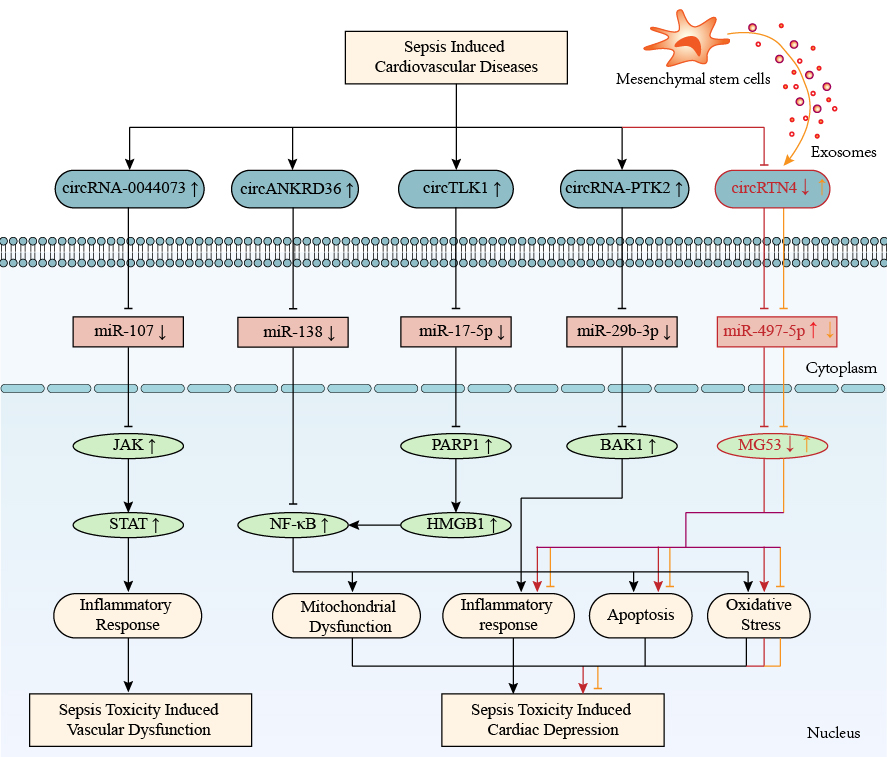 Fig. 3.
Fig. 3.CircRNA-mediated competitive endogenous RNA networks in
sepsis-induced cardiovascular diseases. CircRNAs 0044073, ANKRD36, TLK1, PTK2,
and RTN4 are involved in sepsis-induced cardiovascular toxicity by regulating
inflammation, mitochondrial dysfunction, oxidative stress, and apoptosis. The
up-arrow indicates upregulation and the down-arrow indicates downregulation. The
arrow-line represents promotion and the T-line represents inhibition. ANKRD36, ankyrin repeat domain 36; BAK1, BCL2-antagonist/killer 1; HMGB1, high mobility group protein 1; JAK, janus kinase; MG53, tripartite motif/TRIM72; PARP1, poly ADP-ribose polymerase 1; PTK2, protein tyrosine kinase 2; RTN4, reticulon 4; STAT, signal transducer and activator of transcription; TLK1, tousled-like kinase 1; circRNA, circular RNA; NF-
MiRNAs are highly conserved and small ncRNAs with a length of approximately 21
nucleotides that can control many developmental and cellular processes in
eukaryotes [69]. RNA polymerases II and III transcribe pre-miRNAs to form
precursors, which then undergo complex slicing and splicing events to synthesize
mature miRNAs. MiRNAs regulate biological functions through the silencing complex
(RISC) induced by RNA, which activates the complex to target miRNA-specified
mRNAs [70]. Recently, Liu et al. [71] identified 19 different expression
miRNAs (5 downregulated, 14 upregulated) and 323 different expression mRNAs (11
downregulated, 312 upregulated) in RAW264.7 cells under RT conditions, and 713
miRNA-mRNA networks were enrolled in the T-cell receptor, MAPK, and JAK-STAT
signaling pathways. TargetScan prediction and validation experiments revealed
that miR-155-3p could bind to GAB2 and reduce TNF-
Extensive knowledge suggests that miRNA activation or repression plays a vital
role in the regulation of septic CVDs by interacting with lncRNAs, circRNAs and
their target mRNAs. MiR-145 [73] and miR-29a [74] were downregulated in
LPS-stimulated H9C2 cells, while geniposide (an iridoid glycoside of
Gardenia jasminoides Ellis) and gracillin (a steroidal saponin
of Dioscorea quinqueloba) impeded apoptosis and inflammation by
upregulating miR-145 and miR-29a levels, and blocking the MEK/ERK and
NF-
The typical clinical phenotypes of sepsis are systemic inflammatory response
syndrome (SIRS), which results in fever, tachypnea, tachycardia, and peripheral
leukocytosis and is a dysregulated host response to infection that leads to a
hyperinflammatory cytokine storm and even immunodepression [78]. Sepsis consists
of two stages, acute immune activation and chronic immune depression. During the
initial activation phase of sepsis, necrotic tissue and microorganisms produce
damage-associated molecular patterns (DAMPs) and pathogen-associated molecular
patterns (PAMPs), which then activate pattern recognition receptors (PRRs, such
as TLRs) on the cytomembrane, triggering a series of intracellular signal events
and leading to a cascade of release of inflammatory factors (such as
NF-
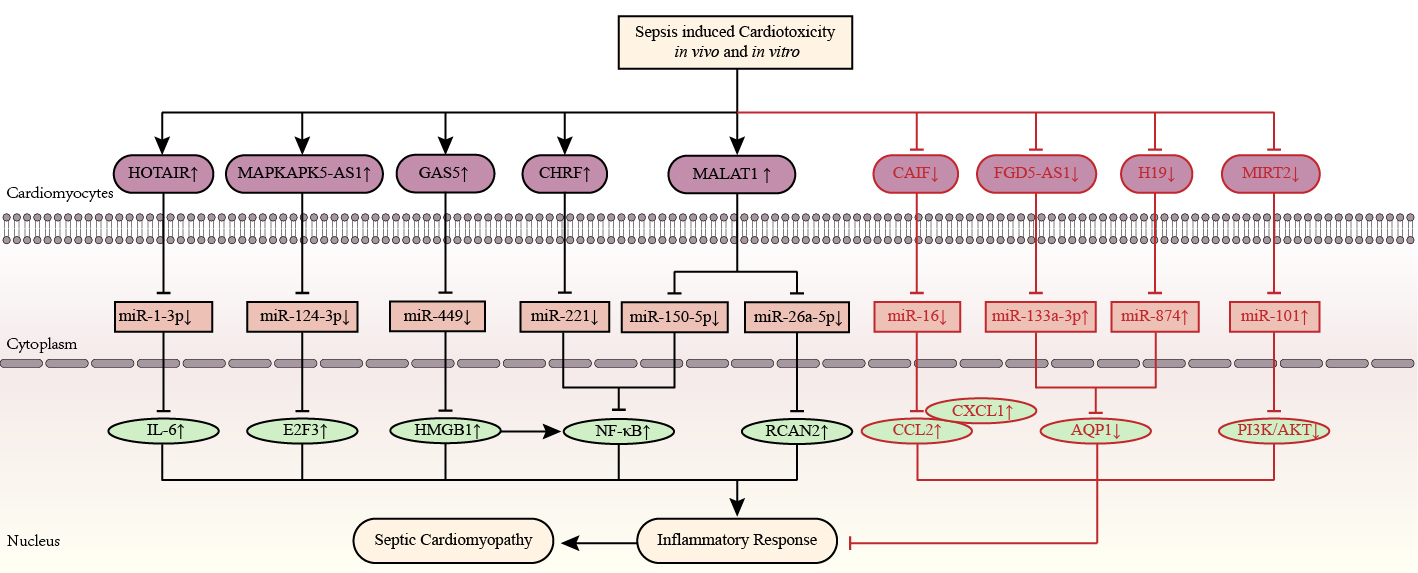 Fig. 4.
Fig. 4.Inflammation-related competitive endogenous RNA networks in
sepsis toxicity-induced cardiomyopathy. LncRNAs HOTAIR, MAPKAPK5-AS1, GAS5,
CHRF, and MALAT1 are upregulated, while lncRNAs CAIF, FGD5‑AS1, H19, and MIRT2
are downregulated in septic cardiomyopathy. The up-arrow indicates upregulation
and the down-arrow indicates downregulation. The arrow-line represents promotion
and the T-line represents inhibition. AQP1, aquaporin protein 1; CAIF, cardiac autophagy inhibitory factor; CCL2, chemokine ligand 2; CHRF, cyocardial hypertrophy related factors; CXCL1, chemokine (CXC motif) ligand 1; E2F3, E2F transcription factor 3; FGD5‑AS1, FGD5 antisense RNA 1; GAS5, growth arrest specific 5; H19, H19 imprinted maternally expressed transcript; HMGB1, high mobility group protein 1; HOTAIR, HOX transcript antisense RNA; IL, interleukin; MALAT1, metastasis associated lung adenocarcinoma transcript 1; MAPKAPK5-AS1, MAPKAPK5 antisense RNA 1; MIRT2, myocardial infarction-related transcription factors 2; NF-
The lncRNA ribonucleic acid nuclear paraspeckle assembly transcript 1 (NEAT1)
was significantly increased in LPS-treated C57 mice and HL-1 mouse cardiomyocytes
[42, 79]. Silencing NEAT1 inhibited inflammation-mediated cardiomyocyte
apoptosis, and this protective effect was similar to that of miR-144-3p
overexpression [42]. Both in vivo and in vitro experiments
indicated that NEAT1 was an upstream regulator of NF-
Abnormal circulating ncRNAs can be used as biomarkers to evaluate the risk,
severity, and prognosis of sepsis. Gui et al. [21] found that the plasma
lncRNA antisense ncRNA in the INK4 locus (ANRIL) was significantly elevated in
sepsis patients (aged 56.6
Along with inflammation, oxidative stress-mediated injury participates in detrimental pathways activated during sepsis-related organ dysfunction, ultimately causing multiple organ failure and death. Sepsis accelerates the excessive production of reactive oxygen species (ROS) and the disruption of antioxidant systems, disequilibrating redox homeostasis to a prooxidative state. Once the pathogen-caused prooxidant state is established, the subsequent cascade release of ROS exacerbates further self-damage to injured cells, independent of the original pathogen itself. Excessive ROS levels directly cause cardiomyocyte apoptosis, mitochondrial damage and cardiac insufficiency in the septic myocardium, simultaneously, it leds to glycocalyx degradation, increased permeability and impaired vasoreactivity in the septic vascular endothelium [80]. Accumulating knowledge implicates that ceRNA networks are involved in regulating the pathogenic process of sepsis-related CVDs, as shown in Fig. 5.
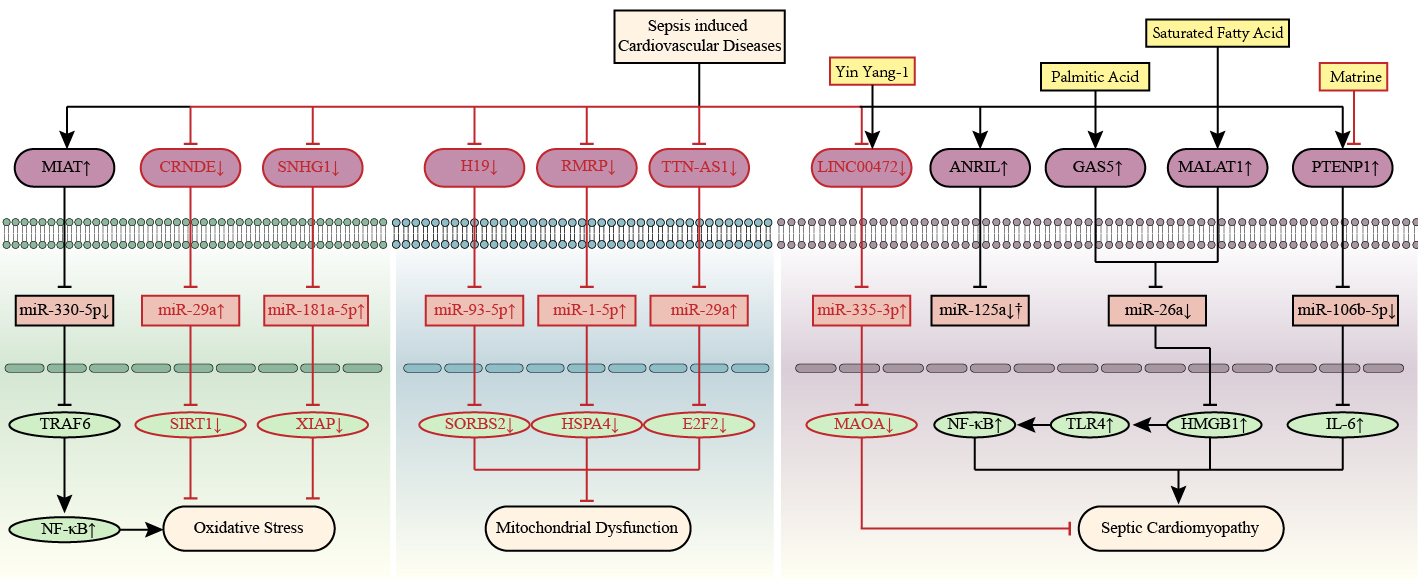 Fig. 5.
Fig. 5.LncRNA-mediated competitive endogenous RNA networks in septic
cardiomyopathy. LncRNAs CRNDE, MIAT, and SNHG1 are associated with septic
cardiomyopathy by regulating oxidative stress. LncRNAs H19, RMRP, and TTN-AS1 are
associated with septic cardiomyopathy by regulating mitochondrial injury. Yin
Yang-1 and matrine alleviate septic cardiomyopathy by upregulating LINC00472 and
downregulating PTENP1, respectively. Palmitic acid and saturated fatty acid
aggravate septic cardiomyopathy by upregulating GAS5 and MALAT1, respectively.
The up-arrow indicates upregulation and the down-arrow indicates downregulation.
The arrow-line represents promotion and the T-line represents inhibition. ANRIL, antisense ncRNA in the INK4 locus; CRNDE, colorectal neoplasia differentially expressed; E2F2, E2F transcription factor 2; GAS5, growth arrest specific 5; H19, H19 imprinted maternally expressed transcript; HMGB1, high mobility group protein 1; HSPA4, heat shock 70 kDa protein 4; IL-6, interleukin-6; MALAT1, metastasis associated lung adenocarcinoma transcript 1; MAOA, monoamine oxidase A; MIAT, myocardial infarction associated transcript; NF-
LncRNA myocardial infarction associated transcript (MIAT) is overexpressed in
LPS-treated BALB/c mice and HL-1 cells [40]. Transfection of MIAT siRNA
antagonized LPS-induced oxidative responses in HL-1 cells, whereas miR-330-5p
mimics partially reversed these effects, as evaluated by ROS and malondialdehyde
(MDA) assays, mitochondrial membrane potential (MMP), and the glutathione
reduction/oxidation (GSH/GSSG) ratio. MIAT regulates miR-330-5p directly as an
endogenous sponge and activates the TRAF6/NF-
The vascular endothelium acts as an important biological barrier of the circulatory system that controls systemic fluid regulation and plays key roles in hemodynamics, circulatory immunity, and tissue metabolism [81]. Endothelial dysfunction occurs in the early stage of sepsis toxic injury, which triggers the circulatory system and causes insufficient blood supply to vital organs, followed by the collapse of the immune system leading to systemic inflammation [82]. Sepsis-induced endothelial dysfunction increases the risk of CVDs, but the specific molecular mechanism is not clear. Singh et al. [18] examined the expression of lncRNAs and mRNAs in HUVECs stimulated with LPS, and 30,584 lncRNAs (1068 downregulated and 871 upregulated) and 26,106 mRNAs (536 downregulated and 733 upregulated) were significantly differentially expressed; among them, CTC-459I6.1 was the most downregulated, and AL132709.5 was the most upregulated lncRNA, which are associated with sepsis-induced endothelial dysfunction.
A variety of lncRNAs have been shown to participate in septic endothelial
dysfunction, such as MALAT1 (metastasis-associated lung adenocarcinoma transcript
1), LUADT1 (lung adenocarcinoma transcript 1), and HULC (highly upregulated in
liver cancer), as shown in Fig. 6. MALAT1 regulates miR-146 to inhibit the
activation of NF-
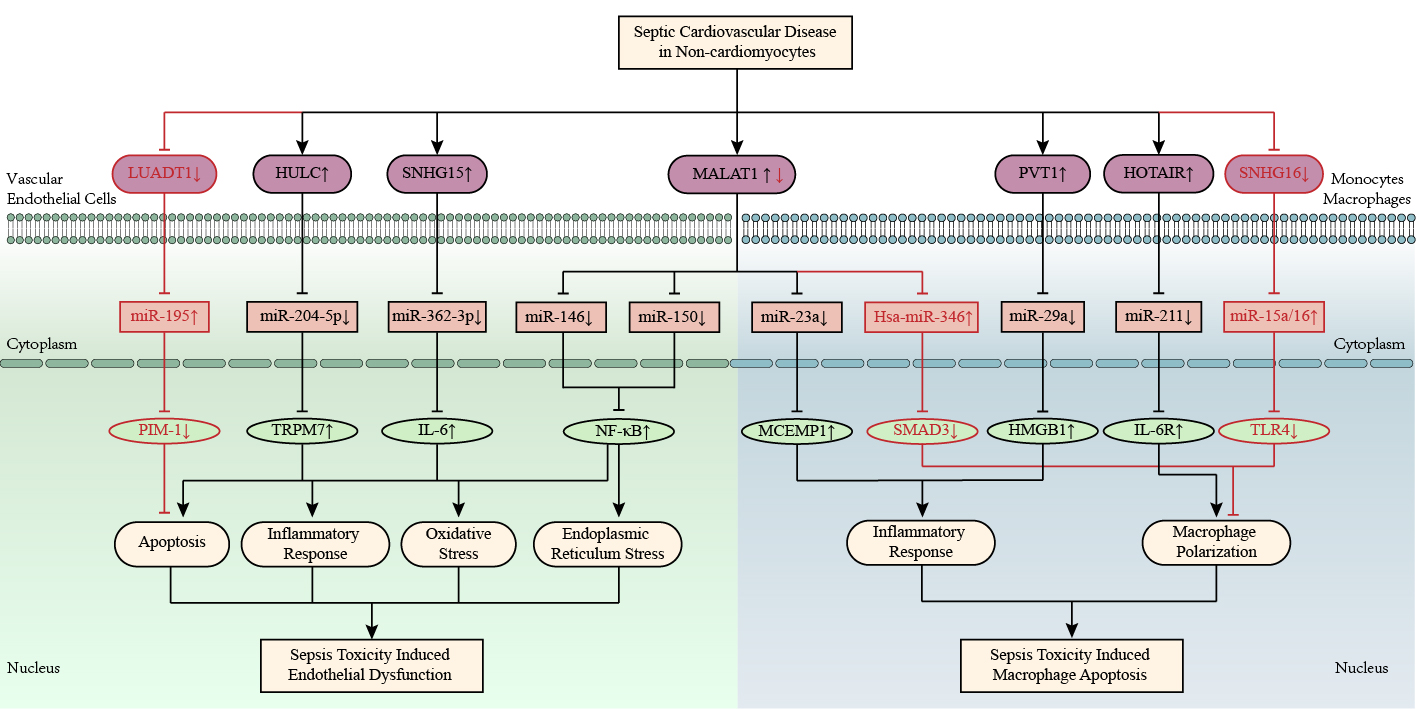 Fig. 6.
Fig. 6.LncRNA-mediated ceRNA networks in vascular endothelial cells and
mononuclear macrophages under sepsis environment. LncRNAs LUADT1, HULC, and
SNHG15 are involved in sepsis endothelial dysfunction. LncRNAs PVT1, HOTAR, and
SNHG16 are involved in sepsis-induced macrophage polarization. In general, lncRNA
MALAT1 aggravates sepsis-induced endothelial dysfunction and macrophage
polarization, but MALAT1 inhibits LPS-induced RAW264.7 macrophage apoptosis by
regulating has-miR-346/SMAD3 axis. The up-arrow indicates upregulation and the
down-arrow indicates downregulation. The arrow-line represents promotion and the
T-line represents inhibition. HMGB1, high mobility group protein 1; HOTAIR, hox transcript antisense RNA; HULC, highly upregulated in liver cancer; IL-6, interleukin-6; LUADT1, lung adenocarcinoma transcript 1; MALAT1, metastasis associated lung adenocarcinoma transcript 1; MCEMP1, mast cell expressed membrane protein 1; NF-
Macrophages are involved in the regulation of innate and acquired immunity and
can be transformed into classically (M1) and alternatively (M2) activated
macrophages under different pathophysiological conditions. M1/M2 polarization of
macrophages is positively correlated with the severity of sepsis, M1-type
macrophages release proinflammatory factors to participate in the occurrence and
maintenance of sepsis, and M2-type macrophages release anti-inflammatory factors
to participate in the resolution of sepsis [83]. Downregulation of lncRNA PVT1
suppresses M1-type macrophage polarization [59]. PVT1 inhibited miR-29a
expression and upregulated HMGB1 expression, subsequently aggravating
sepsis-caused myocardial injury in LPS-treated C57BL/6 mice and primary
macrophages. A recent study reported that MALAT1 was downregulated in patients
with sepsis and LPS-induced RAW264.7 murine macrophages, while hsa-miR-346 was
upregulated [55]. MALAT1 promoted the expression of SMAD3 by downregulating
hsa-miR-346 levels. The viability of RAW264.7 cells was inhibited by MALAT1 and
promoted by hsa-miR-346. Moreover, knockout of NEAT1 significantly decreased the
levels of IL-1
In contrast, plasma lncRNA SNHG16 (small nucleolar RNA host gene 16) expression
was significantly elevated in atherosclerosis patients [85], but significantly
decreased in neonatal sepsis patients [61]. SNHG16 was significantly elevated in
THP-1 cells in composite medium (containing ox-LDL, IL-1
Mitochondria are not only the core organelles that produce ROS, but also the energy metabolism factories that produce adenosine triphosphate (ATP). Sepsis promotes the production of ROS and inhibits the production of ATP in myocardial mitochondria, resulting in mitochondrial biogenesis (apoptosis and mitophagy), oxidative stress, calcium overload, energy imbalance, and cardiac dysfunction. In recent years, the epigenetic regulatory mechanisms in sepsis-related CVDs, including ncRNA regulation, chromatin remodeling, DNA methylation, and histone modifications, have attracted great attention from the life science community [86]. Evidence-based studies indicate that ceRNA networks play crucial roles in biological activities, especially in the regulation of mitochondrial function in septic CVDs, as shown in Fig. 5.
Recently, Shi and colleagues [87] identified 1275 dysregulated lncRNAs and 2769 dysregulated mRNAs in septic mouse hearts, among which 11 differentially expressed mitochondria-related mRNAs were highly correlated with 14 lncRNAs. According to recent reports, the lncRNAs H19, RMRP (RNA component of mitochondrial RNA processing endoribonuclease), and TTN-AS1 (TTN antisense RNA 1) were downregulated in cardiomyocytes treated with LPS, whereas miR-93-5p, miR-1-5p, and miR-29a were upregulated [31, 44, 46]. Overexpression of H19 alleviated mitochondrial damage and reversed cardiomyocyte growth inhibition and apoptosis via the miR-93-5p/SORBS2 axis [31]. RMRP increased the MMP in LPS-induced primary cardiomyocytes from male C57BL/6 mice by regulating the miR-1-5p/HSP70 axis, thereby preventing mitochondrial dysfunction and cardiomyocyte apoptosis [44]. TTN-AS1 inhibited the sepsis-induced reduction of the MMP in H9C2 cells by regulating the miR-29a/E2F2 pathway and suppressed inflammatory cytokine release and ROS activity [46]. Thus, H19/miR-93-5p/SORBS2, RMRP/miR-1-5p/HSP70, and TTN-AS1/miR-29a/E2F2 perform pivotal roles in the modulation of LPS-induced mitochondrial dysfunction and related metabolic disorders.
In response to misfolded proteins in the endoplasmic reticulum (ER) and
dysregulation of calcium homeostasis, ER stress (ERS) acts as a protective stress
response by reducing intracellular unfolded proteins to prevent their aggregation
[88]. Activating transcription factor 6 (ATF6), pancreatic endoplasmic reticulum
kinase (PERK), and inositol-requiring enzyme 1
The lncRNA discrimination antagonizing ncRNA (DANCR) is recognized as a
protective factor for myocardial infarction, and a recent study revealed its
mechanism in ERS-induced myocardial injury [90]. The upregulation of DANCR
promotes autophagy and inhibits apoptosis to protect cardiomyocytes against
tunicamycin-caused ERS injury. Mechanistically, DANCR enhances autophagy and ERS
to maintain cellular homeostasis, leading to a reduction in apoptosis by
adsorbing miR-6324. In addition, lncRNA MALAT1 affects ERS in CLP-induced septic
mice and LPS-challenged HUVECs via the miR-150/NF-
Apoptosis refers to autonomous programmed cell death, which is strictly controlled by genes to maintain cellular homeostasis. Sepsis promotes an uncontrolled inflammatory cascade and immunocyte apoptosis that leads to immune paralysis. Targeted regulation of apoptosis can improve the survival of patients with sepsis [91]. Excessive apoptosis of immune cells induces immunosuppression, and apoptotic cardiovascular cells have the potential to exacerbate secondary heart failure and microvascular dysfunction during sepsis. Although apoptosis is a crucial event in the pathology of sepsis-induced CVDs, the underlying mechanisms are not fully understood. Increasing knowledge indicates that ncRNAs are involved in the regulation of apoptosis in sepsis-related CVDs, as shown in Fig. 7.
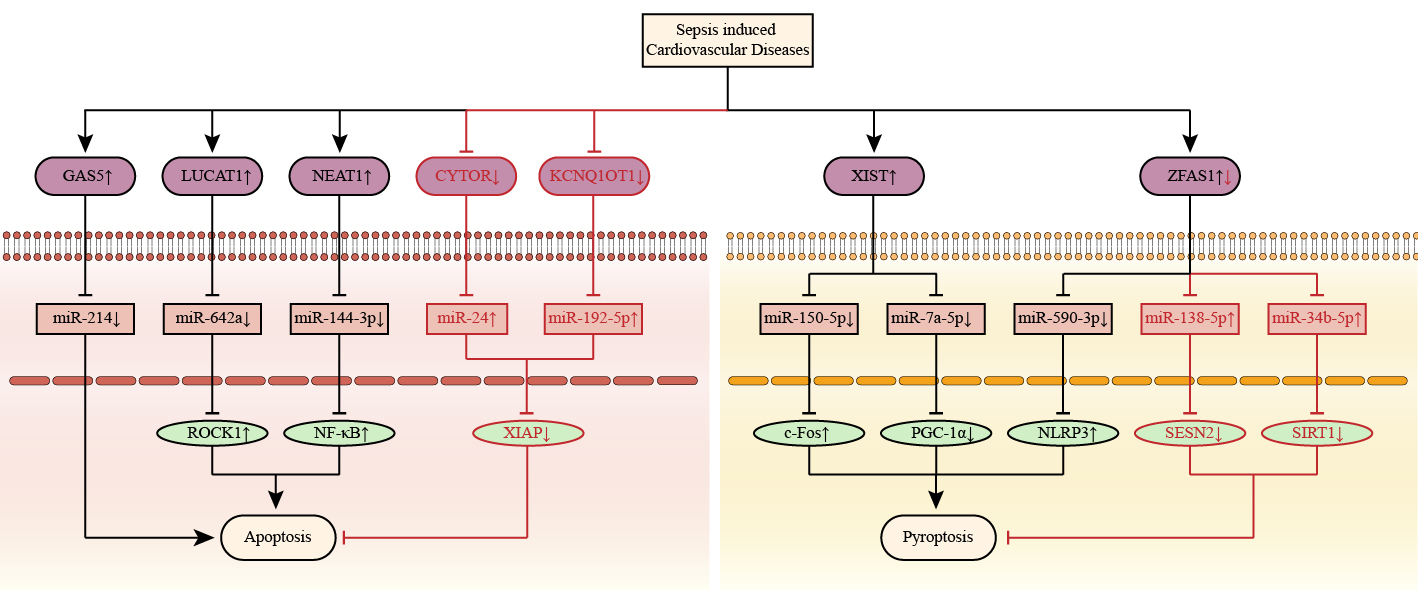 Fig. 7.
Fig. 7.Apoptosis and pyroptosis associated competitive endogenous RNA
networks in septic cardiomyopathy. LncRNAs CYTOR, GAS5, KCNQ1OT1, LUCAT1, and
NEAT1 are associated with septic cardiomyopathy by regulating apoptosis. LncRNAs
XIST and ZFAS1 are associated with septic cardiomyopathy by regulating
pyroptosis. The up-arrow indicates upregulation and the down-arrow indicates
downregulation. The arrow-line represents promotion and the T-line represents
inhibition. c-Fos, A nuclear phosphoprotein; CYTOR, cytoskeleton regulator RNA; GAS5, growth arrest specific 5; KCNQ1OT1, KCNQ1 opposite strand/antisense transcript 1; LUCAT1, lung cancer-related transcript 1; NEAT1, nuclear paraspeckle assembly transcript 1; NF-
In LPS-stimulated H9C2 cells and septic patients, the level of miR-642a was significantly decreased [35]. Silencing of the lncRNA lung cancer-related transcript 1 (LUCAT1) attenuated LPS-induced cardiomyocyte apoptosis, and LUCAT1 could regulate the secretion of ROCK1 by interacting with miR-642a. LUCAT1 could function as a sponge for miR-642a to modulate the expression of ROCK1 in LPS-exposed H9C2 cells. Beyond that, the plasma levels of the lncRNA cardiac autophagy inhibitory factor (CAIF) and miR-16 were decreased in patients with chronic heart failure (CHF) caused by sepsis [22]. The overexpression of CAIF or miR-16 repressed LPS-caused cardiomyocyte apoptosis by enhancing Bcl-2 levels and reducing Bax levels, and the expression of IL-6, CXCL1, and CCL2 was downregulated. CAIF upregulation inhibited cardiomyocyte inflammation and apoptosis by demethylating miR-16 in sepsis-associated CHF. In addition, the lncRNA hox transcript antisense RNA (HOTAIR) was observed to be upregulated in LPS-treated H9C2 cells and monocytes, which targets miR-1-3p and miR-211 to regulate IL-6 and IL-6R, respectively [32, 53]. These results showed that the HOTAIR-miR-1-3p/ miR-211-IL-6 pathway was involved in sepsis-related cardiovascular toxicity.
Interestingly, some transcription factors and herbal extracts, such as Yin Yang 1 (YY1) [34] and matrine [43], can inhibit sepsis-induced cardiovascular toxicity and cardiomyocyte apoptosis by regulating lncRNA-miRNA-mRNA networks. YY1 promoted the expression of lncRNA LINC00472 in LPS-exposed AC-16 cardiomyocytes [34]. Knockdown of LINC00472 reversed the inflammatory response and cardiomyocyte apoptosis caused by LPS and YY1 treatment, whereas the downregulation of miR-335-3p and upregulation of MAOA partly abrogated the protective effects mediated by LINC00472 knockdown. Thus, YY1 contributed to SCVD progression by activating the LINC00472/miR-335-3p/MAOA pathway. Besides, matrine inhibited the expression of the lncRNA PTENP1 (PTEN pseudogene-1) and promoted the expression of miR-106b-5p in CLP-induced mice and LPS-treated H9C2 cells [43]. PTENP1 upregulation or miR-106b-5p downregulation reversed the protective effects of matrine, and miR-106b-5p overexpression abolished the protective effects of PTENP1. It seemed that matrine-mediated PTENP1 deactivation protected cardiomyocytes from sepsis-induced apoptosis by targeting miR-106b-5p.
Pyroptosis has been identified as a specialized programmed cell death caused by
an uncontrolled cascade of inflammatory factors and is one of the important
mechanisms involved in septic CVDs. In contrast to apoptosis-induced nuclear
destruction, a typical feature of pyroptosis is that the nucleus usually remains
intact. Inflammasomes (such as NLRP3, also known as NACHT, LRR, and PYD
domain-containing protein) and LPS can activate certain proteins in the caspase
family (caspase-1, -4, -5, and -11) to proteolytically cleave N-terminal of
Gasdermin D (GSDMD) [78]. IL-18 and IL-1
Acting as a circulating biomarker for sepsis, high plasma levels of the lncRNA zinc finger antisense 1 (ZFAS1) are negatively associated with disease risk, inflammatory levels, and long-term mortality [92]. ZFAS1 has been reported to be involved in sepsis-induced multiple organ dysfunction, including acute injury of the heart, lung, and kidneys. In LPS-induced SD rats and H9C2 cardiomyocytes, the overexpression of ZFAS1 ameliorated sepsis-induced cardiomyocyte pyroptosis and myocardial damage by targeting the miR-138-5p/SESN2 and miR-34b-5p/SIRT axes [49, 50]. Both miR-138-5p and miR-34b-5p are ceRNAs of ZFAS1, while miR-138-5p and miR-34b-5p can negatively regulate SESN2 and SIRT, respectively, indicating that the ZFAS1/miR-138-5p/SESN2 and ZFAS1/miR-34b-5p/SIRT axes play critical roles in sepsis-mediated pyroptosis. Furthermore, SP1 (a zinc finger transcription factor) upregulated ZFAS1 expression in LPS-induced mouse neonatal cardiomyocytes and inhibited cardiomyocyte pyroptosis by regulating Notch signaling [93]. In contrast, another study showed completely opposite results for SP1- and ZFAS1-mediated regulation of septic cardiac dysfunction [51]. ZFAS1 was highly expressed in the septic myocardiopathy model in vivo and in vitro. SP1-activated ZFAS1 exacerbated cardiomyocyte pyroptosis by regulating miR-590-3p/AMPK/mTOR signaling. Knockdown of ZFAS1 alleviated LPS-induced pyroptosis. In conclusion, ZFAS1 acts as a double-edged sword in the regulation of pyroptosis-caused septic CVDs through the ceRNA-miRNA-mRNA axis.
Accumulating evidence has shown that ceRNA networks play vital roles in the pathophysiological progression of septic cardiomyopathy and vascular paralysis, as shown in Fig. 8. As mentioned previously, lncRNAs and circRNAs regulate miRNAs by sponging or decoying miRNAs in sophisticated ceRNA networks. MiRNA activation or inhibition leads to the degradation or renaturation of target mRNAs, thereby modulating the transcriptional and translational modification of downstream genes. Based on existing knowledge, lncRNAs MALAT1, GAS5, ZFAS1, and XIST and the circRNAs TLK1 and ANKRD36, are related to the pathogenesis of septic cardiomyopathy by regulating inflammation, oxidative response, endothelial dysfunction, macrophage polarization, apoptosis and pyroptosis, along with metabolic energy impairment and ERS. Moreover, the lncRNAs LUADT1, HULC, and circRNA-0044073 are involved in septic vascular paralysis by modulating endothelial cell apoptosis and the inflammatory cytokine cascade.
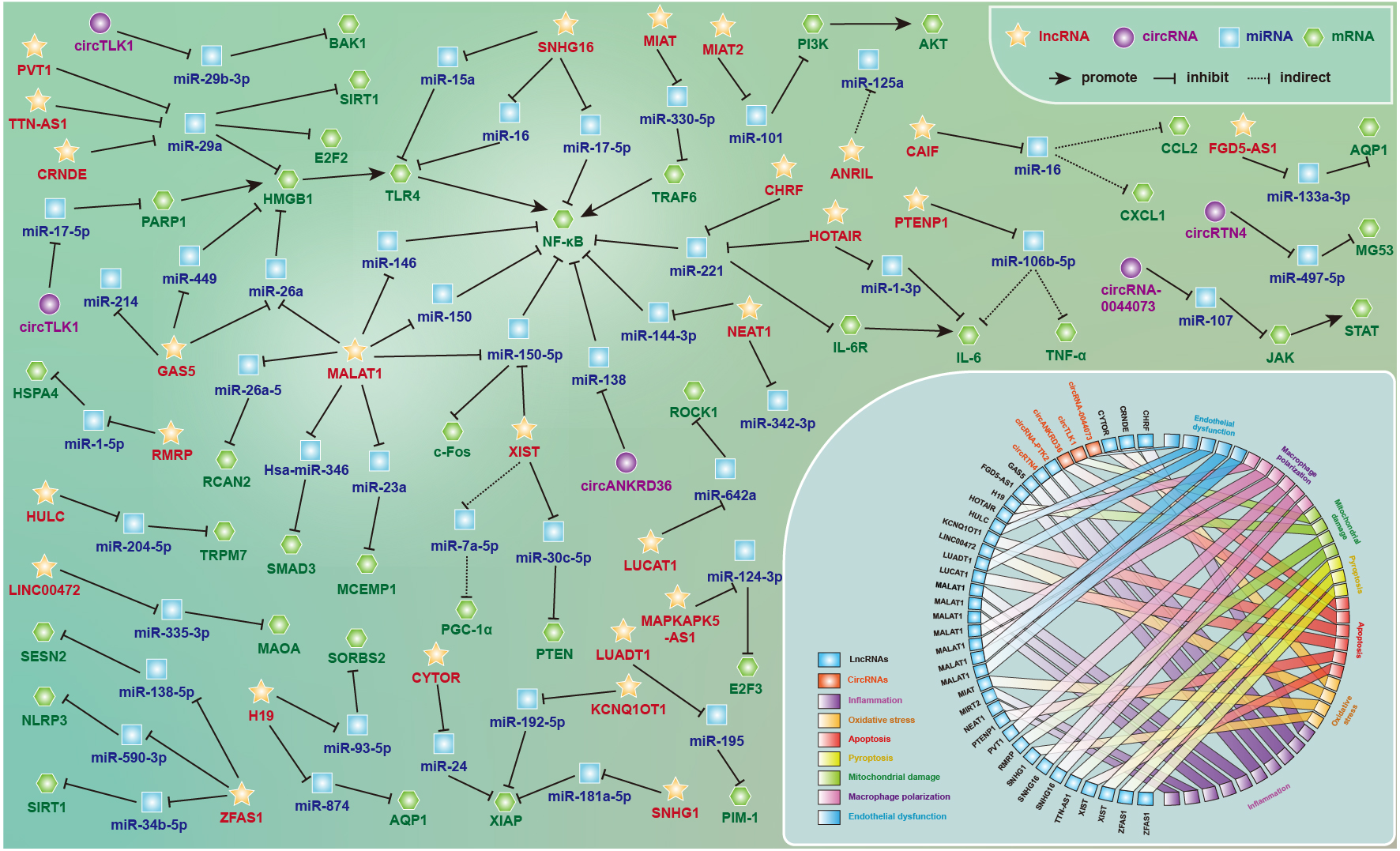 Fig. 8.
Fig. 8.The lncRNA- and circRNA-associated competitive endogenous RNA networks in septic cardiovascular dysfunction. The regulatory mechanisms and potential functions of ceRNA networks in septic cardiomyopathy and vascular paralysis by regulating inflammation, oxidative response, endothelial dysfunction, macrophage polarization, apoptosis and pyroptosis, along with metabolic energy impairment and endoplasmic reticulum stress. See text for details.
It is worth noting that MALAT1, GAS5 and NF-
Understanding the ceRNA networks contributes to further understanding of the molecular mechanisms of SCVDs and is expected to seek breakthroughs in the ncRNA-dependent treatment of septic CVDs. Based on the mechanisms discovered, it may be possible to focus on positively or negatively regulating key lncRNAs or circRNAs to prevent the pathological progression of SCVDs at the translational and posttranscriptional levels. Simultaneously, it might also be a good idea to restore or block certain miRNA functions to modulate target mRNAs for subsequent biological effects. On the other hand, previous studies partially explained the molecular mechanisms underlying the biological effects of SCVDs, suggesting that we can prevent cardiovascular dysfunction in sepsis by suppressing inflammation and oxidative stress, restoring endothelial and mitochondrial function, and inhibiting apoptosis and pyroptosis [8, 72, 80, 81, 86, 88].
Despite the great potential of the ceRNA network as a therapeutic target and diagnostic biomarker for SCVDs, there are numerous limitations affecting its clinical applications. In the first place, a great quantity of animal and cell experiments have demonstrated that ceRNA axes play important regulatory roles in SCVD models in vivo and in vitro, but there is a lack of further confirmation in large-sample clinical trials, especially in multicenter prospective studies. In addition, the clinical characteristics of sepsis are complicated and involve multiple systems and organs. Using only a certain ncRNA for early diagnosis and prognostic monitoring cannot fully reflect the severity and outcome of SCVDs, and a comprehensive evaluation should be combined with disease progression and treatment feedback. Moreover, the ceRNA network is not a simple one-to-one linear axis but a crisscross and interrelated map with multiple targets and pathways. The same lncRNA or circRNA can act on different miRNAs, and different miRNAs can regulate the same target mRNA, thereby mediating different biological effects. Consequently, a satisfactory therapeutic effect cannot be obtained through specific ncRNA-targeted therapy, and multiorgan support and early bundled treatment are needed. Although there are still limitations in the transformation from bench to bedside, it is undeniable that revealing the ceRNA network is beneficial for deciphering the pathogenic mechanism of SCVDs and providing directions for further clinical diagnosis and treatment.
AQP1, Aquaporin protein 1; ATP, Adenosine triphosphate; ATF6, Activating
transcription factor 6; CeRNAs, Competitive endogenous RNAs; CircRNAs, Circular
RNAs; CVDs, Cardiovascular diseases; CHF, Chronic heart failure; CHRF, Cardiac
hypertrophy related factor; CRNDE, Colorectal neoplasia differentially expressed;
DAMPs, Damage-associated molecular patterns; DANCR, Discrimination antagonizing
ncRNA; DLR, Dual-luciferase reporter gene assay; ER, Endoplasmic reticulum;
GSH/GSSG, Glutathione reduction/oxidation; HCAECs, Human primary coronary artery
endothelial cells; HMECs, Human microvascular endothelial cells; HULC, Highly
upregulated in liver cancer; HUVAMCs, Human vascular smooth muscle cells; HUVECs,
Human umbilical vein endothelial cells; IRE1
WX, SYF, YHZ, XQL and JG designed the research study and wrote the manuscript. JG and YHZ drafted the table. WX, SYF and XQL drew the figures. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We would like to thank peer reviewers and associated editors for their precious opinions and suggestions
This research was funded by Natural Science Foundation of Sichuan, China (grant number 2022NSFSC1404 and 2023NSFSC0694), Medical Scientific Project of Sichuan, China (grant number S21026), Medical Youth Innovation and Scientific Project of Sichuan, China (grant number Q21056), Ziyang Scientific Project, China (grant numbers ZYKJJSC20-YYJC-2022-09 and -2022-10), Ziyang Social Science Project, China (grant numbers ZY2021S0030/31 and ZY2022S0055/56). We wish to thank all our colleagues in the Sichuan University and University of Electronic Science and Technology.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.