


1 Department of Advanced Medical and Surgical Sciences, University of Campania “Luigi Vanvitelli”, I-80138 Naples, Italy
2 Department of Precision Medicine, University of Campania “Luigi Vanvitelli”, I-80138 Naples, Italy
†These authors contributed equally.
Academic Editor: Maurizio Pieroni
Abstract
Cardiac hypertrophy develops in response to increased workload to reduce
ventricular wall stress and maintain function and efficiency. Pathological
hypertrophy can be adaptive at the beginning. However, if the stimulus persists,
it may progress to ventricular chamber dilatation, contractile dysfunction, and
heart failure, resulting in poorer outcome and increased social burden. The main
pathophysiological mechanisms of pathological hypertrophy are cell death,
fibrosis, mitochondrial dysfunction, dysregulation of Ca
Keywords
- cardiac hypertrophy
- heart failure
- pathophysiology
- diabetic cardiomyopathy
- metabolism
The main function of the heart is to sustain peripheral organ perfusion in both normal and stress conditions. To achieve this goal, cardiac tissue exhibits plasticity that allows the heart to respond to environmental demands [1]. Cardiac hypertrophy (CH) mainly develops in response to an increased preload and/or afterload to preserve cardiac output, and rarely due to genetic mutations or exposure to growth factors. In case of increased workload, heart hypertrophy determines an increased contractility, at least at the beginning, a decrease in left ventricular wall stress due to increased wall thickness (Laplace’s law), and changes in gene expression with a consequent modification in heart metabolism, contractility, and cardiomyocytes survival. CH can be divided into physiological and pathological. Both conditions share cardiomyocytes enlargement and are a response to cardiac stress [1].
Cardiac growth during ontogenetic development, instead, is characterized by both hyperplastic and hypertrophic growth and is not considered CH [2]. Physiological CH is characterized by a mild increase in cardiac mass (10–20%), mainly due to cardiomyocytes’ hypertrophy in response to body growth or exercise, with an adequate capillary network expansion to provide appropriate cardiomyocyte nourishment. In this setting, no structural or functional cardiac abnormalities can be detected, and physiological hypertrophy is generally not considered to be a risk factor for heart failure (HF) [2]. In fact, heart contractility is preserved or increased in absence of interstitial or replacement fibrosis or cell death. Moreover, physiological hypertrophy is fully reversible. However, it should also be recognized that in case physiological hypertrophy becomes quantitatively excessive or sustained for a prolonged period, it can evolve into maladaptive. Pathological hypertrophy, instead, can be adaptive at the beginning with concentric growth of the ventricle as compensatory response, and is reversable if the primary stimulus is reversed before the development of intrinsic disease. If the stimulus is not solved, physiological hypertrophy progresses to ventricular chamber dilatation with wall thinning through lengthening of individual cardiomyocytes, contractile dysfunction, and, finally, HF with both preserved and reduced ejection fraction, arrhythmias, and death [3]. Aim of this review is to provide an insight into the most updated main pathophysiological mechanism of CH and consequent HF development.
Cardiac disfunction due to hypertrophy develops when, beyond cell growth and
protein synthesis, following further processes are established: cell death,
fibrosis, mitochondrial dysfunction, dysregulation of Ca
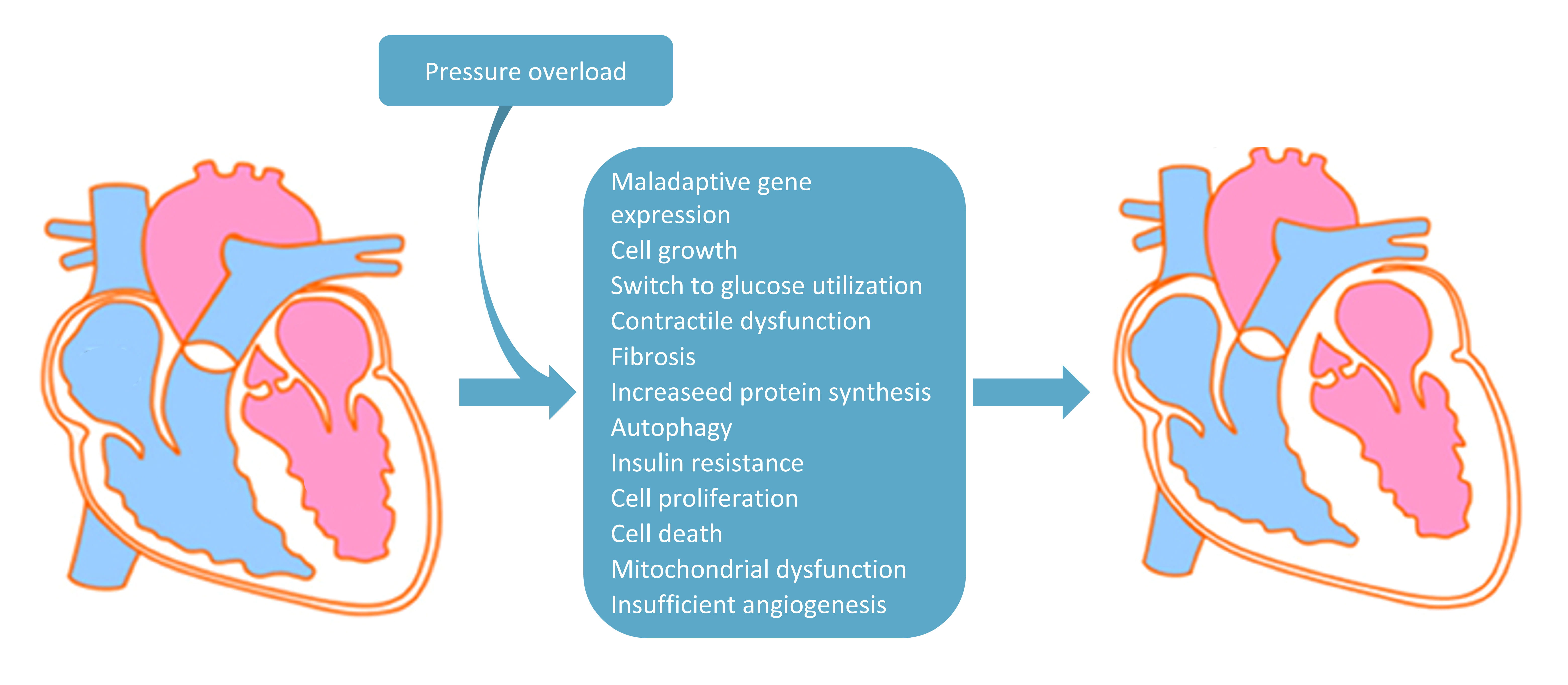 Fig. 1.
Fig. 1.Main pathophysiological changes due to pressure overload inducing cardiac hypertrophy. In the diagram are listed all the main pressure overload-induced pathophysiological mechanisms involved in the shift from non-hypertrophic (on the left) to hypertrophic heart (on the right).
Myocardial angiogenesis plays a key role in maintaining an adequate nutrient
supply, to avoid myocardial disfunction onset [4, 5]. Moreover, angiogenesis
stimulation during pressure-overload (PO) can prevent the switch from
compensatory CH to HF [6]. Furthermore, in patients with aortic stenosis and
preserved ejection fraction it was found a strong correlation between myocardial
blood vessel density and left ventricular mass index [7]. On the other hand, a
significant reduction in myocardial capillary density was associated with
advanced pathological remodelling and HF [8, 9]. Hypoxia-inducible factor
1
Angiotensin II and endothelin 1 are peptide hormones that bind to the G
protein-coupled angiotensin II receptor and endothelin 1 receptor, respectively,
resulting in the activation of G proteins such as G
Calcineurin is a Ca
Increased sympathetic nerve activity produces an increased concentration of
blood catecholamine, which in turn is related to a poor prognosis in patients
affected by HF [28]. Moreover, high adrenergic activity is associated with CH and
the basal plasma levels can also predict CH extent in patients affected by
hypertension, independently of systolic blood pressure and body mass index [29].
The role played by
Finally, catecholamines are also implied in another pathway which leads to CH, through G protein MAPK cascade activation. In fact, MAPK activate both MEK3/MEK6 and MEK4/MEK7, which in turn activate p38 kinases and JUN N-terminal kinases (JNKs), respectively, which ends with GATA4-mediated pathological hypertrophy gene transcription [42].
Increased protein synthesis or decreased degradation is necessary for
cardiomyocyte cell growth. In this setting a key role is played by mTOR (mammalian Target Of Rapamycin)
signalling cascade, which is involved in the modulation of growth factors
signalling and amino acids availability, of cell metabolism and growth. mTOR is a
serine/threonine-protein kinase that works as part of two distinct complexes,
mTORC1 and mTORC2 [43]. mTORC1 activity is increased in both physiological and
pathological hypertrophy and represent an essential adaptive mechanism during
acute PO [44]. However, mTORC1 sustained activation can be harmful as it may lead
to autophagy suppression with consequent protein quality control deterioration.
mTORC1 induce ribosomal protein production through a direct activation of
ribosomal protein S6 kinase
Insulin is an anabolic hormone involved in cardiac physiological hypertrophy. Growing evidence have suggested that insulin, insulin receptor activation and following Akt signalling are beneficial when maintained in physiological range [49]. In fact, insulin signal suppression leads to physiological hypertrophy inhibition, whereas overstimulation results in pathological CH and disturbs homeostasis induction. It seems that chronic hyperinsulinemia can promote these pathological changings through the activation of angiotensin II signalling, and intensive glycaemic lowering by insulin has also been associated with increased cardiovascular events and future risk of HF [49, 50]. In chronic PO murine model was reported an increase in cardiac insulin signalling with a mismatch development of cardiomyocytes size and vascularity, leading to myocardial hypoxia development, thus resulting in cardiomyocytes death and cardiac dysfunction. On the other hand, cardiomyocyte hypertrophy and cardiac dysfunction due to PO were significantly reduced in heart heterozygous insulin receptor deletion or Akt deletion [51]. Moreover, Akt-mTOR signalling was reported to be reduced in CH murine model, whereas it was found increased in physiological hypertrophy, making it easy to suggest its implication in the type of hypertrophy differentiation [52]. Insulin resistance prevalence is increased in patients affected by systolic dysfunction and is accompanied by an enhanced risk of HF development. It has been hypothesized a possible link between metabolic disfunction and HF, though the underlying mechanisms are still obscure [53, 54, 55, 56, 57].
Atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) are both
secreted by cardiomyocytes and can inhibit CH by activating cyclic GMP-dependent
protein kinase (PKG) signalling [58, 59]. Cardiac-specific deletion of the ANP
receptor 1 (NPR1) encoding gene can develop mild hypertrophy that worsen with PO
stimulus, which can in turn evolve into pathological hypertrophy and cardiac
remodelling [60]. PKG signalling activation, due to natriuretic peptide binding
with their receptor, leads to inhibition of calcineurin–NFAT, TRPCs, and
RHOA–RHO kinase pathways [61]. cGMP can be degraded by cGMP specific
3
Fibroblasts are one of the most prevalent cell types in cardiac tissue and
crosstalk with other heart’s cells, such as immune and endothelial cells, and
cardiomyocytes [64, 65, 66]. Their role is to contribute to homeostasis maintenance
and tissue remodelling in response to stress. Cardiac fibroblasts express
extracellular matrix (ECM) receptors coupling mechanical stimuli to functional
responses, with the aim of adapting the composition and stiffness of the ECM and
fibrotic response to mechanical stress [66, 67]. Their involvement in CH is not
well defined, and target genes depletion study resulted nonspecific. However,
fibroblasts manage to communicate with cardiomyocytes by secreting humoral
mediators such as growth factors, including fibroblast growth factors (FGFs),
transforming growth factor-
Rodent model studies have underlined the role of inflammation in HF, suggesting
its unfavourable impact on cardiac homeostasis [80, 81, 82, 83], also showing a
correlation with pro-inflammatory cytokines blood levels, such as tumor necrosis
factor (TNF)-
Endothelial cells modulate the cardiomyocyte growth through paracrine regulation
[99]. In response to PO, endothelial cells secrete IL-33, which binds to
membrane-bound ST2 (IL-1RL1) in cardiomyocytes [100]. IL-33 or IL-1RL1 deletion
is associated with increased PO hypertrophy, while recombinant IL-33 infusion
ameliorates it, together with fibrosis, due to NF-
TRPCs are a family of nonselective cation channels that can control pathological hypertrophy development mainly through calcineurin and NFAT signalling effectors [104]. TRPC3 and TRPC6 promote pathological hypertrophy development through calcineurin-dependent signalling, which is inhibited by their depletion in murine model [105, 106]. PKG phosphorylation of both TRPC3 and TRPC6, instead, is implied in channel conductance reduction, thus inhibiting TRPC-mediated hypertrophy [107]. TRPC3, TRPC4, and TRPC6, dominant negative gene variant overexpression was found to be protective against PO pathological hypertrophy development [27]. In myocardial infarction TRPC4 knockdown mouse model it was reported an improvement in pathological hypertrophy, cardiac performance, progression to HF, and increased survival when compared to wild type animals [108].
Stromal interaction molecule 1 (STIM1) is a Ca
Epigenetic modifications regulate chromatin structure, thus controlling gene
expression by filtering promoters and enhancers access to DNA. In particular,
histone acetylation on lysine residues promotes chromatin relaxation, with
enhanced transcriptional activation, whereas histone acetylation suppression is
implicated in chromatin condensation and gene expression inhibition. Therefore,
epigenetic modifications can induce pathological hypertrophy onset by modulating
genome architecture and stability, and gene expression. It was reported that
trimethylation of histone H3 at lysine 4, 9, or 27 and demethylation of H3 at
lysine 9 and 79 modulate a pro pathological hypertrophy gene expression [112].
Histone lysine-specific demethylase 4A (KDM4A) is increased during pathological
hypertrophy, leading to FHL1 enhanced expression that further promotes
hypertrophy and HF development [113]. Histone acetylation is modulated by histone
acetyltransferases (HATs) and histone deacetylases (HDACs). In mice, HATs
overactivation induces CH and left ventricular remodelling [114, 115]. HDACs,
instead, are divided into three classes: class I (HDAC1, 2, 3, and 8), class II
(HDAC4, 5, 6, 7, 9, and 10), and class III. Class I HDACs mediate cardiac
hypertrophic responses, with HDAC2 acting as an indirect down regulator of
Akt/GSK-3
MicroRNA (miRNA) is a class of small non-coding RNAs involved in translation
repression or transcriptional degradation of target mRNAs [119]. A single miRNA
may interact with multiple target genes, making it easy to suggest a possible
role in CH development, and HF, mostly due to MiRNAs targeting mRNAs encoding
Ca
Moreover, the long non-coding RNA (lncRNA) CH-associated epigenetic regulator (Chaer) is involved in pathological hypertrophy development, by interacting with Polycomb repressor complex 2 (PRC2) in response to hypertrophic stimuli [126]. This interaction leads to the inhibition of H3 lysine 27 methylation in the promoter regions of fetal genes related to CH, such as Acta1, Anf, and Myh7 in mice, thus developing pathological hypertrophy [42].
Most of the aforementioned signalling mechanisms contributing to pathological hypertrophy are initially activated as an adaptive response. However, sustained activation of these signalling mechanisms has a major role in inducing cardiac pathological hypertrophy and HF development. In fact, while short-term AKT activation promotes cardiomyocytes physiological growth, sustained activation of AKT has been associated with pathological hypertrophy and HF development [42]. Finally, the functional consequences of each stimulus and cardiomyocytes response depend on the balance between cardioprotective and detrimental effects.
The heart consumes a huge amount of ATP to perform its action. Impaired energy metabolism adaptation during hypertrophic response enhances pathological hypertrophy and cardiomyocyte death, thus preceding HF development [127, 128]. About 70–90% of the physiological heart ATP production derives from fatty acids (FA) oxidative phosphorylation, whilst 10–30% derives from glucose, lactate, and ketone bodies oxidation [129]. During pathological hypertrophy and HF development a metabolic impairment leads to a shift in cardiac energy production from FA to glycolysis, anaplerosis, and other forms of metabolism [130, 131, 132]. This metabolic reprogramming is associated with mitochondrial energy transduction and respiratory pathways downregulation, and it begins during hypertrophy early stages [133]. However, changes in FA ATP production are more consistent than glycolysis and of the other metabolic substrates, thus causing a progressive reduction of ATP synthesis, which in turn leads to energy deficiency and HF development [129, 134]. In murine model it was reported that Acacb deletion, which encodes in acetyl coenzyme A carboxylase 2, enhance FA oxidation, thus ameliorating both pathological hypertrophy and HF development by preserving the substrate utilization profile, making it easy to suggest that metabolic reprogramming might be a direct cause of pathological hypertrophy [134, 135].
In the heart, the nuclear receptors peroxisome proliferator activated
receptor-
Glucose is carried into cellular cytoplasm through glucose transporters (GLUTs). In particular, GLUT1 is most expressed in fetal heart, whereas GLUT4 in the adult one. However, in pathological hypertrophy, insulin-independent HepG2 glucose transporter GLUT1 levels are increased, whereas GLUT4 levels are reduced, together with an enhanced glucose uptake and glycolysis, but not glucose oxidation [141]. Consequently, glycolysis and glucose oxidation rates are mismatched, leading to glycolytic intermediates accumulation, such as glucose-6-phosphate, which regulates insulin-mediated and carbohydrate-mediated cell growth through mTORC1 activation [142, 143]. Moreover, increased accumulation of glycolytic intermediates improves both hexosamine biosynthetic pathway and pentose phosphate pathway, which in turn are involved in pathological hypertrophy development due to the biosynthesis of glycoproteins, protein O-GlcNacylation, and excessive accumulation of NADPH [141, 144, 145].
Finally, increased glucose reliance per se is not detrimental if the energetic demand is met in a healthy heart. However, in the long term, the metabolic remodelling coupled with increased glucose consumption could impair heart flexibility to use other substrates, thus promoting HF progression and onset [144].
It has been reported an upregulation of the fructose metabolism in pathological
hypertrophy. Hypoxia inducible factor-1
Ketone bodies represent a good alternative fuel source produced by liver
mitochondria from fatty acids, due to several stimuli, such as exercise, fasting,
ketogenic diet or untreated diabetes. In the heart, ketone bodies are metabolized
into acetyl-CoA, which in turn is used for energy production tricarboxylic acid
cycle and/or oxidative phosphorylation [147]. Sodium-glucose cotransporter 2
inhibitors (SGLT2i), a class of drugs recently approved for non-diabetic HF
patients, enhance ketone bodies production, in particular
Adenosine monophosphate-activated protein kinase (AMPK) regulates heart energy metabolism and is mainly activated by increased AMP and ATP depletion. AMPK main role is to enhance ATP production and reduce energy-consuming biosynthetic pathways by negatively regulating mTOR [153, 154]. ATP is the “molecular unit of currency” of intracellular energy transfer, and, when used during metabolic process, it is converted into AMP. During energy depletion, AMP levels are increased, and the binding and activation of AMPK is facilitated, through liver kinase B1, calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2), and TAK1, with glucose metabolism enhancement [155]. AMPK protein expression and activity is increased in human HF [156]. Several studies show that a pharmacological AMPK activation is associated with a reduced PO-induced hypertrophy [157, 158, 159], while mice with AMPK depleted activity experienced increased pathological hypertrophy and contractile dysfunction [160, 161, 162, 163]. However, results from rodent studies should be careful interpreted, also considering possible different regulatory mechanisms of AMPK cascade, also including isoform-specific function of AMPK across the species [156].
Sirtuins (SIRT1–SIRT7) are a family of NAD
Increased mitochondrial protein acetylation may contribute to impaired mitochondrial fuel oxidation and respiration, contributing to the vicious cycle of “energy starvation”, which in turn promotes HF development [152].
It has been suggested the heart role in other organs metabolism, mainly through cardiokines secretion. In particular, cardiac miRNA208a and RNA polymerase II transcription subunit 13 (MED13) targets liver and white adipose tissue inducing metabolic gene expression upregulation and mitochondria increased number [173, 174]. Obese and diabetic patients show an increased pro-inflammatory adipokines expression, promoting a low-grade inflammation which is involved in the metabolic dysfunction onset [175]. In these settings, the anti-inflammatory adipokines, such as adiponectin which have proven to suppress PO-induced hypertrophy through cardiomyocytes AMPK activation, are reduced [160, 176]. Leptin is a hormone associated with white fat tissue, satiety process, and increases energy expenditure. In the heart it has been associated with morphological and functional alterations, with increasing cardiac muscle size and decreasing cardiac output [177]. In fact, it has been reported that leptin activated downstream proteins leads to rho-associated protein kinase (ROCK) activation, which in turn induces pathologic hypertrophy through several pathways (ERK1/2, MAPK and AKT/mTOR) [178].
It is also thought that adipose tissue may modulate pathological hypertrophy
through exosomes secretion, though exosome research is still in its infancy [179, 180]. Finally, PPAR
Metabolic intermediates can accumulate in the heart, thus inducing
cardiomyopathy. In obese, insulin resistant or diabetic patients a new entity
called “diabetic cardiomyopathy”, also known as lipotoxic cardiomyopathy, has
been described [180, 181, 182]. The key role of glycaemic control on the pathogenesis
and outcome of coronary heart disease is well known [183, 184, 185]. Nevertheless,
diabetic cardiomyopathy (DCM) is characterised by both hypertrophy and gradual HF
with damaging cardiac remodelling, such as fibrosis and diastolic and systolic
dysfunction, which is not directly related to coronary artery disease [186]. The
main promoters of DCM are insulin resistance and subsequent hyperglycaemia,
associated with altered fatty acid metabolism, altered calcium homeostasis and
inflammation. Pro-inflammatory cytokines production (interleukin-6, tumour
necrosis factor-
Acylcarnitine production derives from long-chain fatty acids modification induced by acyl-CoA synthetases and by carnitine O-palmitoyltransferase 1, muscle isoform (CPT1M) in the cardiomyocytes’ outer mitochondrial membrane and is later carried into mitochondrial matrix through carnitine/acylcarnitine carrier protein, where it is metabolized in free carnitine and long-chain acyl-CoA. Heart acylcarnitine levels are increased in pathological hypertrophy setting, though it has been reported to be decreased 8 weeks after transverse aortic constriction and myocardial infarction [130, 208, 209]. In chronic HF patients, circulating long-chain acylcarnitine levels were independently associated with adverse clinical outcomes and, in end-stage disease, decreased after long-term mechanical circulatory support [210]. Nevertheless, the underlying mechanism leading to acylcarnitine level modification remains unknown.
Diacylglycerol, beyond being a lipid metabolite, is also a second messenger
which induces insulin resistance by indirectly suppressing IRS1 phosphorylation,
and inflammation through NF-
DCM could recognise two different phenotypes based on different molecular
adaptation and damage [215]. The first phenotype of DCM is related to increased
systemic inflammation and characterised by concentric hypertrophy with preserved
left ventricular diastolic and systolic function, increased myocardial stiffness
and high left ventricular telediastolic pressure [215]. This isoform has several
inflammatory/fibrosis biomarkers such as IL-1
CH mainly develops in response to an increased preload and/or afterload and can evolve to cardiac impaired contractility, with a worsening outcome and increased social burden. Despite current large knowledge, most of which based on murine models, the pathophysiology of CH, as well as diabetic cardiomyopathy development and progression is still far from being fully explained.
More in-depth knowledge of both etiologic and pathogenic mechanisms is an exciting challenge for target-specific treatments development and to prevent HF onset.
FCS, AC, EV, RG, TS designed the research study. AC, EV, RG, FCS performed the research. LR, GD, CS, MA provided help and advice on the figure; AC, TS, RM, RE, MA, CS, LR, GD, CS analyzed the literature data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflicts of interest statement. Raffaele Galiero, Celestino Sardu, and Ferdinando Carlo Sasso are serving as Editorial Board members/Guest Editors of this journal. We declare that Raffaele Galiero, Celestino Sardu, and Ferdinando Carlo Sasso had no involvement in the peer review of this article and have no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Maurizio Pieroni.