
Academic Editor: Demosthenes B Panagiotakos
Atherosclerosis ranks first among cardiovascular system diseases. It is the “disease of the century”, and more than 50% of people with circulatory pathology die of it. The clinical manifestation of atherosclerosis is observed at the middle and older ages, but it is known that the pathological process develops much earlier. There has been a clear trend in theoretical and practical cardiology in recent years to study the earliest atherogenic markers. Epidemiological, clinical, and morphological studies have proved the presence in children and adolescents of sexual, endogenous, exogenous, primary, and potentiating risk factors contributing to an early formation of a pathogenic foundation for atherosclerotic cardiovascular diseases. Disorders of lipid metabolism – dyslipidemias are attributed to the most significant risk factor for atherosclerotic cardiovascular diseases. The DLP prevalence in the pediatric population is extremely high. According to the results of conducted global studies, lipid metabolism disorders occur in more than 70% of children and adolescents. It causes the need for timely diagnostic, therapeutic and preventive measures. The need to extrapolate the risk factor concept to childhood age is justified by several reasons, the main of which include the broadest spread of atherosclerosis that has become a global pandemic, genetic determinism, and low variability of the lipid spectrum of blood serum: the levels of lipids and lipoproteins discovered in childhood are stable throughout life and have an independent prognostic value. That is why the most practical significance is inherent to the study of lipid and lipoprotein metabolism, starting in the early periods of lipid and lipoprotein ontogenesis. Since risk factors can be identified at the preclinical stage of the atherosclerotic process, dyslipidemia phenotyping will facilitate identifying children and adolescents at risk of developing cardiovascular pathologies in the future. The study objective is to examine the pathophysiological aspects of lipid and lipoprotein metabolism and examine DLP epidemiology – as the leading atherosclerotic cardiovascular disease risk factor in children and adolescents, DLP classification, modern approaches to DLP diagnosis and management.
Atherosclerotic cardiovascular diseases (ASCVD) rank first in the structure of morbidity and mortality in various countries of the world. According to WHO definition, atherosclerosis (AS) is a variable combination of changes in artery inner lining (intima), comprising aggregations of lipids, complex carbohydrates, fibrous tissue, blood components, calcium salts depositions, and the underlying changes of the medium lining (media) in the arterial wall. AS has been thoroughly examined: its etiology, pathogenesis, development forms, and stages, and quite a lot of scientific literature has been published about it. However, neither diet nor strict control over risk factors, drugs, or physical activity can decisively change the global AS spreading. Therefore, it remains the leading death cause of the most active and productive population.
The reasons catering for ASCVD are split into modifiable and non-modifiable risk factors (RF). Non-modifiable RFs include age, gender, and burdened heredity for cardiovascular diseases. Modifiable RFs comprise dyslipidemia (DLP), arterial hypertension (AH), diabetes mellitus, factors contributing to thrombosis, enhanced values of homocysteine, uric acid, inflammation markers, unhealthy lifestyle features (obesity, inactivity, consumption of excessive saturated fats and refined carbohydrates, harmful addictions) [1].
The concept of early onset of the atherosclerotic process refers to the period of intrauterine development. Epidemiological studies have shown coronary thickening of the intima already in fetuses and infants; their prevalence and progression significantly increase in children and adolescents. The studies revealed that the coronary changes range from focal lesions with moderate thickening of the intima to diffuse mild thickening with the presence of mixed or non-stenotic plaques. The thickening of the intima is considered the initial stage of atherogenesis and the pathomorphological basis for the accumulation of lipids with the subsequent formation of an atherosclerotic plaque. Studies have confirmed that the initiation of the atherosclerotic process begins in the womb and progresses in infancy and childhood [2, 3]. All this allows us to talk about atherosclerosis as a significant pediatric problem.
The main AS RFs in children and adolescents are [4, 5, 6, 7, 8]:
• strong family history or cases of premature manifestation of cardiovascular diseases in the family,
• arterial hypertension,
• comorbid conditions related to early AS development: DM, obesity, hypothyroidism, HIV infection, systemic lupus erythematosus, radically cured neoplasms, organ transplantation, kidney diseases, hypersympathicotonia, hypodynamia, stressful situations, and harmful habits.
However, despite numerous studies on AS markers in children and adolescents, this problem is far from being resolved. Moreover, the data obtained are contradictory, since the climatic and geographical conditions, nutrition features, social environment, and child’s somatotype greatly influence AS risk factors, in particular, the level of atherogenic and anti-atherogenic lipid and LP fractions in blood, blood pressure (BP) enhancement, and body weight.
For a long time, AS can develop latently. Then, only many years later, clinical disease manifestations occur in the form of stenocardia, cerebrovascular diseases, atherosclerosis of the peripheral arteries, myocardial infarction manifestations, ischemic stroke, and sudden death.
Lipoproteins are an extensive group of compounds that differ significantly in their chemical structure and functions (Table1) [9].
| Density | Diameter | TG (%) | Cholesterol esters (%) | Phospholipids (%) | Cholesterol (%) | Apolipoproteins | ||
| (g/mL) | (nm) | Main | Others | |||||
| Chylomicrons | 80–100 | 90–95 | 2–4 | 2–6 | 1 | ApoВ-48 | ApoА-I, A-II, A-IV, A-V | |
| VLDL | 0.95–1.006 | 30–80 | 50–65 | 8–14 | 12–16 | 4–7 | ApoВ-100 | ApoA-I, C-II, C-III, E, A-V |
| IDL | 1.006–1.019 | 25–30 | 25–40 | 20–35 | 16–24 | 7–11 | ApoВ-100 | ApoC-II, C-III, E |
| LDL | 1.019–1.063 | 20–25 | 4–6 | 34–35 | 22–26 | 6–15 | ApoВ-100 | - |
| HDL | 1.063–1.210 | 8–13 | 7 | 10–20 | 55 | 5 | ApoА-I | ApoA-II, C-III, E, M |
| Lp(а) | 1.006–1.125 | 25–30 | 4–8 | 35–46 | 17–24 | 6–9 | Apo (а) | ApoВ-100 |
| Note. VLDL, very low-density lipoproteins; IDL, intermediate-density lipoprotein; LDL, low-density lipoproteins; HDL, high-density lipoproteins; Lp(a), lipoproteins (a); TG, triglycerides. | ||||||||
No matter how much we may discuss causes and conditions of AS development and highlight the polyethological nature of AS as a disease, one thing remains unalterable: certain LP classes of are a primary substrate that, having penetrated the arterial wall, trigger the atherosclerotic plaque formation.
Blood lipids are: cholesterol (CH), triglycerides (TG), phospholipids (PL), free fatty acids (FFA).
Cholesterol is the main component of cell membranes. It is engaged in the synthesis of steroid hormones, sex hormones, and bile acid formation. The active cholesterol metabolites are its esters synthesized during free CH esterification with adding FFA radical and enzyme engagement of lecithin cholesterol acyltransferase. Hepatocytes secrete these metabolites into the blood flow as CH component of VLDL. CH is absorbed in the small intestine and participates in chylomicrons formation. Within the biochemistry process, 30% of the daily cholesterol demand is provided by food incoming from the body. Endogenous CH synthesis occurs in the smooth endoplasmic network of almost all vertebrate cells, and 70% of cholesterol is synthesized in hepatocytes from acetyl-CoA. At the first stage, acetyl-CoA is converted to 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) that under the influence of HMG-CoA reductase (3-hydroxy-3-methyl-glutaryl-CoA reductase, HMGR) is converted to mevalonate [10]. Mevalonate, bypassing intermediate metabolites (mevalonate-5-pyrophosphate, isopentenyl-5-pyrophosphate, geranyl-pyrophosphate, farnesyl-pyrophosphate, squalene, lanosterol), is converted to CH. The products of CH catabolism are neutral sterols and bile. Bile acids play an important role in the digestive process. They are absorbed in the small intestine back into the blood and come into the liver, representing an additional material for CH resynthesis [11]. Hypercholesterolemia leads to many negative effects, including vascular and endothelial shifts that are the most significant in atherogenesis. Against the background of endothelium-dependent vascular dilatation, platelet adhesion increases, the production of a plasminogen activator inhibitor is stimulated, the plasminogen activator is inhibited, the tissue procoagulation factor is induced, and, ultimately, the function of heparin-sulfate-proteoglycans is disrupted. These shifts inhibit the endothelium anticoagulation properties and initiate blood clot formation. In parallel, pro-inflammatory changes occur in the endothelium, resulting in monocytes’ accumulation (macrophages, T lymphocytes) [12].
Triglycerides (TG) are compound esters of glycerol and fatty acids. They are synthesized in many organs and tissues, but the most significant are the liver, intestinal wall, adipose tissue, and the lactating mammary gland. The body of an adult receives 60–80 grams of TG with food daily [10].
Chylomicrons mainly perform diet TG transport function. During absorption, TG cleavage products first enter the epithelial cell membranes in the small intestine villi, and then they are transferred in the cytoplasm to the TG resynthesis site — the smooth endoplasmic reticulum. Commonly, TG is deposited in adipose tissue (visceral and subcutaneous). Under stress, significant physical exertion, TG undergoes lipolysis with the formation of free fatty acids absorbed by muscle cells as a substrate for mitochondrial oxidation and ATP synthesis. After a meal, TG concentration in blood increases, but after 10–12 hours, it returns to its original level.
Hypertriglyceridemia is one of the main ASCVD predictors [13]. Hypertriglyceridemia blocks not only the endothelium-dependent relaxation factor by suppressing the nitric oxide synthesis and L-arginine, but also promotes hypersecretion of endothelin 1 — a potent vasoconstrictor which is ten times higher than the effects of norepinephrine and angiotensin II [14, 15, 16].
Phospholipids are an integral part of cell membranes and LP. Phospholipids include polar lipids: phosphatidylcholine and sphingomyelin. Recently, the enzyme lipoprotein-associated phospholipase A2 (Lp-FLA2) has been studied intensively [17]. It is secreted by white blood cells and manifests its effect in circulating LP composition. Lp-FLA2 hydrolyzes the phospholipids of the surface monolayer of oxidized LDL with the formation of endogenous inflammatory mediators-lysophosphatidylcholine and oxidized non-esterified fatty acids, which play a crucial role in atherogenesis [18].
Lipid carriers are transport macromolecules in the form of lipid-protein complexes-lipoproteins. Lipoproteins differ in size, density, and the amount of CH and TG comprised by them. The structural LP elements are specific apoproteins. The main functions of apoproteins are LP interaction with the receptor apparatus of hepatocytes and other tissue cells and lipolysis rate regulation. In LP center, there are hydrophobic molecules of CH and TG esters, outside LP there is free (non-esterified) cholesterol, phospholipids and apoproteins.
In the sequence of hydrated density increase and size reduction, the main plasma lipoproteins, are: chylomicrons, very low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), low-density lipoproteins (LDL), and high-density lipoproteins (HDP) in two subfractions (HDL 2 and HDL 3), and lipoprotein(a) (Lp(a)) [11].
The main chylomicron apoproteins are apoB48, apoE, and apoCII. The first two are ligands binding the chylomicron remnants to hepatocyte scavenger receptors. ApoCII is a cofactor that activates plasma lipoprotein lipase, an enzyme that hydrolyzes LP with a high TG content.
In VLDL and IDL, the main apoproteins are apoB100 and apoE that function as ligands while binding to the liver receptor apparatus. VLDL, like chylomicrons, are hydrolyzed by lipoprotein lipase, and they comprise the cofactor apoCII. In LDL, the main and only apoprotein is apoB100, thanks to which they are captured and removed from the bloodstream by LDL-hepatocyte receptors [11, 19].
LDL Cholesterol (LDL-C) and VLDL cholesterol (VLDL-C) are the most atherogenic LP classes. They are complex protein-lipid supramolecular complexes that transport cholesterol and polyunsaturated fatty acids to cells respectively. The receptor uptake of LDL-C by cells is the most important factor determining the cholesterol level in blood [20]. These LP fractions are the leading atherogenesis originators, they change the endothelium properties, promote blood cell adhesion and activation of monocyte/macrophage chemotaxis with subsequent proliferation of smooth muscle cells. The mechanism of cholesterol transportation into the cell as a component of LDL-C by receptor-mediated endocytosis is known due to the brilliant paper by American scientists M. Brown and J. Goldstein who were awarded the Nobel Prize in 1985 [21]. The receptor-mediated capture of LDL-C ensures maintaining of LDL-C levels in the blood within the limits of normolipidemia, preventing AS development.
The most important role in atherogenesis is played by peroxide-modified LDL-C that have strongly marked atherogenic properties. Peroxide-modified LDL-C is recognized by macrophages’ scavenger receptors, and it is quickly captured by them, resulting in accumulation of cholesterol esters and transformation of macrophages into foamy cells that contain products of oxidative degradation of fatty acids with high cytotoxicity, stimulating monocyte chemotaxis, delaying the migration of macrophages from the intima, stimulating the formation of leukotrienes. LP exhibit a “hormone-like effect” on cells. Pathophysiological effect of LDL-C is classified as damaging and having a causal relationship with the risk of ASCVD developing [22, 23]. All ApoB containing LP and their remnants are able to penetrate the endothelial barrier, especially under endothelial dysfunction. They initiate the deposition of lipids and formation of atherosclerotic plaques [24].
HDL Cholesterol (HDL-C) contains apoproteins ApoAI and AII. These apoproteins are ligands for binding HDL-C to peripheral tissue cells via specific ABCA1 cluster transporter receptors and scavenger B1 receptors in the liver. The main function of this LP class is the reverse CH transportation from peripheral tissues, the vascular wall, to hepatocytes. HDL cholesterol has a protective effect against AS, so they are called anti-atherogenic. HDL-C plays a key role in lipid metabolism and is an integrative indicator of the well-being of the lipid spectrum. HDL-C determines the amount of cholesterol in cells, stimulates utilization of TG-rich LP, and stabilizes LDL-C, protecting them from modification changes and atherogenic orientation [25, 26]. However, it should be noted that there is currently no evidence to support that an increase in HDL-C in plasma contributes to ASCVD risk reduction [19, 27] (Fig.1).
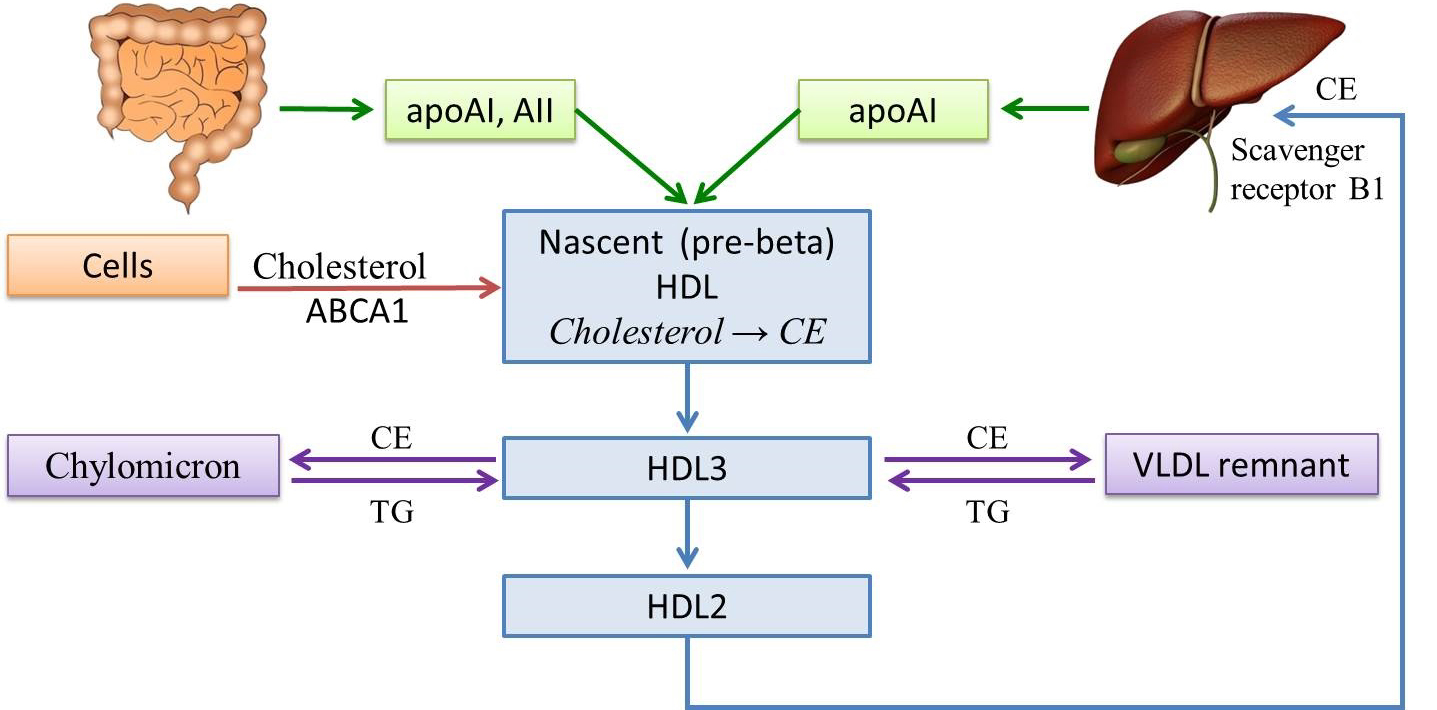 Fig. 1.
Fig. 1.High density lipoprotein metabolism. HDL, high density lipoprotein; VLDL, very low density lipoprotein; TG, triglycerides; AI, AII, apolipoproteins; CE, cholesteryl esters; ABCA1, ATP-binding cassette transporter A1.
Factors associated with low HDL-C: male gender, progestogens, obesity, hypertriglyceridemia, diabetes Type 2, high carbohydrate intake, alcohol, smoking.
Factors associated with high levels of HDL-C: female gender, estrogens, increased physical activity, weight loss, moderate alcohol consumption.
Lipoprotein(a) (Lp(a)) is a conjugate of LDL-C with a specific apoprotein (a) that is similar in its structure to plasminogen and can affect the process of fibrinolysis by suppressing it. Besides, Lp(a) has pro-inflammatory properties due to oxidized phospholipids transfer [28, 29]. High Lp(a) concentrations in plasma are associated with an increased ASCVD risk, but for most patients this risk factor is less significant than LDL-C [30, 31].
Individuals with excessively elevated Lp(a) levels
It is known that atherogenesis is based on endothelial dysfunction. The endothelium regulates vascular tone by balancing vasodilator and vasoconstrictor factors and controls the functional activity of smooth muscle cells [33, 34].
Vasodilator factors include:
(1) Nitrogen monoxide has anti-aggregation, anti-adhesive, anti-thrombogenic, anti-inflammatory, and neoangiogenesis-inhibiting properties.
(2) Endothelial hyperpolarization factor is identical to bradykinin, promotes hyperpolarization of smooth muscle cells with reduced sensitivity to vasoconstrictor factors and increased vasodilation through the induction of NO and prostacyclin synthesis.
(3) Prostacyclin I2 (PGI2) is a prostaglandin, acts as an antiplatelet and an anticoagulant factor.
(4) C-type natriuretic peptide and adrenomedullin are direct vasodilators due to increased cyclic adenosine monophosphate (cAMP) production due to adenylate cyclase activation [35].
Vasoconstrictor factors include:
(1) Endothelin-1 (ET-1) is a powerful vasoconstrictor, activator of mitogenesis, cell proliferation, and intimal fibrosis with subsequent increased vascular stiffness [36].
(2) Thromboxane A2 (THA2) has prothrombotic properties, stimulates platelet activation and aggregation. Platelet aggregation is achieved by mediating GPIIb/IIIa glycoprotein complex expression in the platelet cell membrane. Circulating fibrinogen binds these receptors on neighboring platelets, further strengthening the clot [12].
(3) Prostaglandin F2
(4) Superoxide-anion binds physiologically significant NO with vasodilation suppressing, synthesizes the highly toxic compound peroxynitrite (ONOO’) with subsequent cell damage [38, 39].
Under the risk factor impact, the ability of endothelial cells to secrete vasodilators decreases, while the synthesis of vasoconstrictors persists or increases. Such risk factors as DLP, hyperglycemia, hypertension and endotoxins negatively affect the glycocalyx structure. The combined impact of aggressive factors leads to endothelial dysfunction: disturbance of glycocalyx integrity increased endothelial permeability, and the formation of defects in the monolayer [40].
It is currently established that AS and thrombosis have a single mechanism in part of the affected endothelial cells that triggers and maintains pathological processes. The relationship of hemostatic parameters with atherogenesis, which are predictors of coronary incidents in patients with ASCVD, has been proven. First of all, these indicators include fibrinogen, blood viscosity, antithrombin III, inhibitor Type 1 of plasminogen activator, aggregation indicators of platelet, monocytes, red blood cells, and white blood cells. Atherogenic DLP, and incredibly high levels of LDL-C and TG, are the determining factor of pathological changes in the hemostatic system. The validity of these provisions has been confirmed by the reverse method, when following the normalization of the blood lipid spectrum with lipid-correcting drugs, the decrease was recorded in blood thrombogenic potential, as well as normalization of fibrinolytic activity and improvement of platelet functional state [41, 42, 43, 44, 45, 46, 47].
Homocysteine (HC) is a demethylated derivative of methionine. HC plasma level
increases under the deficiency of B
Currently, DLP prevalence varies significantly in the world, but there is a steady increase in these metabolic disorders. Of particular concern is the tendency to increase mortality from ASCVD between young people [50, 51].
In the analysis, DLP prevalence in the pediatric population in various countries of the world made: in Denmark – 6.4%, and in children and adolescents with obesity, DLP is four times more common – 28% [52]. The DLP prevalence in Chile makes 38%, in children and adolescents with obesity DLP prevalence makes 54% [53]. DLP structure in Iran is as follows: hypercholesterolemia occurs in 48%, hypertriglyceridemia – 50%, elevated hyper LDL-C – 20%, and decreased HDL-C – 88% [54]. In Thailand, hypercholesterolemia occurs in 1.2%, hypertriglyceridemia-10.6% [55]. In Turkey, in obese children and adolescents (2001–2013, Ankara), DLP was detected in 42.9% of cases. In DLP structure, hypercholesterolemia made 18.6%, hypertriglyceridemia – 21.7%, elevated LDL-C – 13.7%, and decreased HDL-C – 19.7%. With age and increase in body mass index, DLP prevalence also increased, while there were no significant differences in DLP frequency in girls and boys [56]. In Germany, a population sample from the LIFE Child Study was used to estimate the lipid spectrum. It was found that cholesterol and LDL-C concentrations reached their peak values in girls two years earlier than in boys. Hypercholesterolemia was detected in 7.8%, elevated LDL-C – 6.1%, and decreased HDL-C – 8.0%. Hypercholesterolemia in children aged 0 to 9 was found in 22.1%, and among children aged 10 to 16 this indicator is lower – 11.7% [57]. In the United States, from 1999 to 2012, there was a tendency to reduce DLP prevalence in children aged 8 to 17: hypercholesterolemia decreased from 10.6% to 7.8%; hypoalphacholesterolemia – from 17.9% to 12.8%; non-HDL-C – from 13.6% to 8.4% [58].
In the review by A.M. Perak et al. [59], the lipid spectrum analysis in 26047 children and adolescents aged 6 to 19 for 1999–2016 was made. (the average age of subjects was 12.4 years, 51% were females). The study showed a decrease in cholesterol level from 164 mg/dL (1999–2000) to 155 mg/dL (2015–2016) and an increase in HDL-C concentration from 52.5 mg/dL (2007–2008) to 55.0 mg/dL (2015–2016).
Studies conducted in Brazil have evidenced a high DLP prevalence between adolescents, ranging from 24.2% to 71.4%. A characteristic feature in DLP structure was a widespread hypoalphacholesterolemia occurrence. DLP was more common in adolescents with obesity and unhealthy lifestyle components [60, 61, 62, 63, 64, 65].
In the Russian Federation, in the European North area (Arkhangelsk, 2002, 779 children and adolescents aged 7 to 16 years were examined) – DLP was detected in 48.98%. The study showed a widespread low HDL-C levels, and it did not correlate with DLP presence: it was recorded in subjects with both normolipidemia – 54% and DLP – 51%.
The wide prevalence of HDL-C is regarded as an adaptive mechanism under high body’s demand for cholesterol and its increased consumption to ensure energy processes in the environment of the European North [66].
Dyslipidemia is a congenital or acquired (secondary) disorder in the body lipid transport system manifested as a qualitative and/or quantitative change in blood plasma lipids and LP regarded as important ASCVD risk factors. J. W. Hoffman was the first who discovered the correlation of lipid and LP metabolism disorders with ASCVD in 1950. Some time later, D. S. Fredrickson and co-authors (1967) developed these ideas into a DLP classification. The first and very successful DLP classification was approved by WHO experts and subsequently became widely used throughout the world (Table2) [19].
| Phenotype | Synonym | LP level enhancement | Liped level enhancement | Atherogenicity | Frequency (%) in DLP structure |
| I-а | Hereditary hyperchylomicronemia, lipoprotein lipase deficiency | Chylomicrons | TG | Not proven | |
| I-b | Hereditary apoprotein CII deficiency | ||||
| I-с | Hyperchylomicronemia in circulation of a lipoprotein lipase inhibitor | ||||
| II-а | Polygenic hereditary hypercholesterolemia | LDL | CH | + + + | 10% |
| II-b | Combined hyperlipidemia | LDL and VLDL | CH, TG | + + + | 40% |
| III | Hereditary dis- |
IDL | CH, TG | + + + | |
| IV | Hereditary hypertriglyceridemia | VLDL | TG | + | 45% |
| V | Mixed hyperlipidemia | Chylomicrons and VLDL | CH, TG | + | 5% (Pediatrics) |
This classification does not diagnose but establishes a DLP type, regardless of whether it is acquired or hereditary.
Hyperlipidemia by the nature of biochemical disorders, according to the classification by Fredrickson D. S., are split into types:
Type I – hyperchylomicronemia (induced by fats), characterized by severe hypertriglyceridemia and an increase in chylomicron concentration. In DLP structure, Type I makes less than 1%; it is usually caused by a hereditary defect in the gene encoding plasma lipoprotein lipase. DLP Type I is considered conditionally atherogenic. Prolonged circulation of chylomicron residuals in the blood may be accompanied by their accumulation in endothelium, provoking atherosclerotic plaque development. DLP Type I clinical markers are hepatosplenomegaly, pancreatitis attacks, and abdominal colic. It is manifested in children under 10 years of age.
Type IIa – hyper-
Type IIb – hyper-
Type III – dis-
Type IV – hyper-pre-
Type V – hyperchylomicronemia, or
hyper-pre-
Currently, it is important to differentiate between primary and secondary DLPs.
Primary DLP is characterized by stable changes in one or more lipid spectrum parameters in the absence of any other diseases. Primary DLP is characterized by specific disorders in the lipid and LP metabolism of a hereditary nature. In population studies of genetic relationships, more than 100 genes were identified that could affect the levels of cholesterol, TG, LDL-C, and HDL-C.
Familial hypercholesterolemia (FHCH) is one of the most common genetically determined diseases. Its cardinal signs are elevated blood levels of LDL-C, early development of ASCVD, as well as skin and tendon xanthomas [67, 68]. Heterozygous FHCH occurs in the general population with a frequency of 1:200–1:500, homozygous FHCH is a very rare pathology with a frequency of 1:160 000–1:320 000 [69]. The genetic basis of the disease is mutations of genes that encode the proteins of low-density lipoprotein receptors (LDLR/LDL), apoprotein B (apoB), proprotein converting subtilisin-kexin protein kinase type 9 (PCSK9). Homozygous FHCH has an extremely unfavorable prognosis due to an extremely early development of ASCVD [70]. So far, the genes are known with a proven FHCH association: LDLR, ApoB, PCSK9, and LDLRAP1 [71, 72, 73]. The phenotype determined by the LDLR, ApoB, and PCSK9 genes demonstrates autosomal and dominant transmission type, while the phenotype determined by mutations in LDLRAP1 gene has an autosomal recessive transfer.
In 20% of patients with FHCH, even with the use of modern molecular genetic research methods, no mutations are detected, but it does not exclude the diagnosis [74, 69].
Features of lipid metabolism under FHCH
As a result of LDL-C receptors’ deficiency, their connection with ApoB is disrupted. It leads to a decrease in LDL-C catabolism by the liver. Disorder of LDL cholesterol metabolism causes a sharp increase in the total cholesterol content in blood. The cholesterol enhances up to 600–1200 mg/dL (15–30 mmol/L) under homozygous FHCH, and up to 280–500 mg/dL (7–12.5 mmol/L) under heterozygous FHCH) [69, 72, 74, 75].
According to the classification by D. Frederikson, FHCH relates to hyperlipoproteinemia Type IIa. The level of HDL-C and apolipoproteins (Apo) A-I and A-II in homozygotes is reduced due to a combination of metabolic defects in the form of elevated catabolism and reduced apo A-I synthesis [72].
There are no clinical manifestations for heterozygous FHCH in children. This form, as a rule, is detected accidentally during a biochemical examination of child’s blood, against the background of other somatic or infectious diseases. In this case, an increased level of the total cholesterol and LDL cholesterol is revealed. Changes in these indicators in children can also be detected during a cascade screening examination [76, 77, 78].
Clinical manifestations in children with homozygous familial hypercholesterolemia, as a rule, occur as early as the first life decade. They are as follows:
Xanthomas (xanthomatous plaques, tuberous (tuberous) xanthomas, tendon xanthomas) are the earliest clinical manifestations and enable early diagnosis of the disease.
Ophthalmic changes are characterized by xanthelasmas. The most typical is the corneal lipidemic arch, i.e., a circular cornea opacity colored gray-white, 1 to 3 mm wide, with a smooth surface.
Cardiovascular system changes. The basis of cardiovascular lesion of atherosclerotic genesis is formed by a sharp increase in LDL-C level since the neonatal period. In patients with homozygous FHCH, the cardiovascular disease symptoms emerge before the age of 10. The process of atherosclerotic plaque formation is sharply accelerated. It results in stenosis of the vascular bed of the coronary, carotid, and renal arteries. The risk of early development of coronary heart disease is about 20 times higher than in the general population [69, 72, 74, 75].
In the histological study of the coronary arteries of children with homozygous FHCH, atherosclerotic changes correlate with final stages of the atherosclerotic process. There is a high frequency of sudden cardiac death against the background of acute myocardial infarction. The myocardial infarction development can occur as early as the first or second life decade [69, 75, 79, 80].
The aortic root, aortic valve, and elastic vessels are affected by the atherosclerotic process. Degenerative changes of the aortic valve flaps often occur. The cholesterol deposition on the aortic valve flaps can also lead to aortic insufficiency. The effort angina occurs due to both aortic stenosis and coronary atherosclerosis. In the case of renal vessel stenosis, the arterial hypertension is added [69, 81].
Family history data is very important for FHCH diagnosis. Early ASCVD initiation, myocardial infarction at a young age (under the age of 40), hypercholesterolemia and other clinical AS manifestations are very common.
Under heterozygous FHCH, the atherosclerotic process begins to manifest clinically after 17–20 years of life. Thus, in young men with heterozygous FHCH, the first cases of coronary heart disease are described since the age of 17, and in girls a little later, starting from the age of 25. According to examination of young patients aged 20 to 29 and diagnosed with heterozygous FHCH, the risk of fatal cardiovascular disease development increased 125-fold in women and 48-fold in men in the period before statin treatment, compared to those with normal CH levels [82].
Familial combined hyperlipidemia is currently defined as a common
metabolic disorder characterized by an elevated serum cholesterol or TG levels in
at least two members of the same family; variability in the hyperlipidemia type
both in patient (individual variability) and in his sick relatives (variability
within the family); an increased risk of early ASCVD [83]. In addition to the
clinical and biochemical criteria listed above, familial combined hyperlipidemia
is characterized by hyperapobetalipoproteinemia (apolipoprotein (apo) level) B
Primary hypertriglyceridemia is the main risk factor for ASCVD. It is caused by a deficiency of hepatic lipoprotein lipase (elevated TG, normal cholesterol). The main genes associated with elevated TG levels (as the main phenotype) are APOA5, APOC2, APOE, GPD1, GPIHBP1, HNF1A, LMF1, LPL, and SLC25A40. According to studies’ findings, carriers of APOA5 gene have a 3-fold higher risk of ASCVD development [86]. Clinically, primary hypertriglyceridemia is manifested by an abdominal pain from childhood (pancreatitis), hepatosplenomegaly, retinal vascular damage, peripheral neuropathy, and xanthomas. Weak or moderate hypertriglyceridemia can increase dramatically when exposed to provoking factors: uncompensated diabetes mellitus, hypothyroidism, alcohol abuse, and administration of combined oral contraceptives. In each case, TG level in plasma may exceed 10 g/L. Within exacerbation periods, patients develop mixed hyperlipidemia, i.e., the concentration of both VLDL and chylomicrons increases. High chylomicrons levels predispose to formation of eruptive xanthomas and pancreatitis development [87].
Sitosterolemia is a disease caused by an enhanced cholesterol absorption and accumulation of sterols, especially of plant origin. Clinical markers of the disease are the presence of xanthus, premature AS, hemolytic anemia, and liver pathology. The disease is associated with mutations in ABCG5 and ABCG8 genes.
Hypoalphalipoproteinemia is a disease related to a group of diseases with a low level of HDL-C and a high prevalence of early AS development. The genes associated with low HDL-C of monogenic etiology are ApoA1, ABCA1, LCAT, SAR1B, and ABCG1. ApoA1 gene is responsible for the phenotype of familial hypoalphalipoproteinemia with autosomal dominant inheritance. ABCA1 gene is associated with Tangier disease inherited on an autosomal recessive basis. Clinical markers of the disease are enlarged tonsils colored bright yellow-orange, splenomegaly, lymphodenopathy, muscle weakness in extremities, reflex weakening, loss of sensitivity. In the bone marrow punctate, “frothy” cells are found. LCAT gene mutation in Chromosome 16 is associated with “fish-eye” disease (partial insufficiency of lecithin cholesterol-acyltransferase – LCAT) is an autosomal recessive disease characterized by DLP and eye damage.
Thus, primary DLPs can be classified genotypically or phenotypically by biochemical analysis. In the genotypic classification, DLPs are split into monogenic (caused by mutations in a single gene) and polygenic (caused by associations of multiple mutations).
Congenital DLPs are disorders of lipid metabolism detected in a child on the first days of life; these disorders are not hereditary or familial but caused by adverse factors that affected the fetus in the intrauterine period (chronic fetal hypoxia, maternal diabetes, obesity, toxicosis, nephropathies [88, 89, 90].
Hypolipidemia in newborns. LP content in blood plasma in healthy newborns is significantly lower than in adults. Especially remarkable are chylomicrons’ absence and a significant decrease in VLDL-C value. The main blood class in newborns is HDL-C. In total, LDL-C and VLDL-C are 170 mg% in newborns, which is 5 times less than in maternal blood. The distribution of LP classes is as follows: HDL-C 49.7–56%; LDL-C 35.6–41%; VLDL-C 3–14.7%. LDL-C was found to contain higher TG levels (more than 50% of all TG in umbilical blood), protein, and cholesterol. The composition of HDL-C in newborns’ blood compared to maternal blood is characterized by a higher content of phospholipids, cholesterol, and lower esterified cholesterol and TG values. With age, HDL-C level decreases, and LDL-C level increases [91].
Secondary DLPs. Today, several diseases are known to result in secondary DLP development (Table3) [92]. There is a large number of pathological conditions associated with secondary hypercholesterolemia that require a differential diagnosis:
| Disease or condition | Changes in lipid levels | Changes in lipoprotein levels | |||
| Chylomicrons | High-density lipoprotein | Low-density lipoprotein | Very low-density lipoprotein | ||
| Diabetes mellitus | - | ||||
| Hypothyroidism | - | Normal or |
- | ||
| Chronic kidney failure | - | Normal or |
|||
| Nephrotic syndrome | - | ||||
| Cholestasis | - | - | |||
| Alcohol abuse | - | Normal or |
- | ||
(1) endocrine system pathologies (hypothyroidism, hypopituitarism, polycystic ovarian syndrome, lipodystrophy, porphyria);
(2) kidney pathologies (chronic kidney diseases, hemolytic-uremic syndrome, nephrotic syndrome);
(3) liver diseases (obstructive liver diseases, cholestasis, biliary cirrhosis, Alagille syndrome);
(4) systemic connective tissue diseases (juvenile rheumatoid arthritis, systemic lupus erythematosus);
(5) accumulation diseases (glycogenosis, Gaucher disease, Niemann-Pick disease, Tay-Sachs disease);
(6) kawasaki disease;
(7) anorexia nervosa, idiopathic hypercalcemia;
(8) klinefelter syndrome, Turner syndrome;
(9) medications (glucocorticoids, beta-blockers, antiretroviral drugs);
(10) exogenous causes (alcohol).
Secondary DLPs (mostly hyperlipidemia) is a frequent metabolic syndrome in children and adolescents, and it is confirmed under many diseases. It is no exaggeration to say that every disease, especially in young children, is accompanied to a greater or lesser extent by lipid metabolism disorders. Enhanced lipid peroxidation, functional and structural disorders of the phospholipid spectrum of cell membranes are regarded as the main pathochemical mechanism of many pathological processes in childhood. Secondary DLPs are more often combined, when changes in several lipid and LP fractions are detected, and isolated DLPs, as a rule, are found to be primary.
Today, there is no doubt that lipid metabolism disorders are at the heart of atherogenesis; it has been proven by the National Cholesterol Education Program (NCEP) designed to normalize and reduce the level of cholesterol in blood and resulted in progress in ASCVD prevention.
DLP is a molecular disease associated with lipid and LP metabolism disorders in human body. The more narrow is the term “hyperlipidemia” which reflects an increase in the amount of various classes of lipids and LP. It is of the greatest interest from a practical point of view, since it is the major ASCVD risk factor.
2001 NCEP ATP III recommendations are widely used as criteria for normolipidemia in clinical practice (Table4) [93].
| Category | Cholesterol total, mmol/L | LDL-C, mmol/L |
| Desirable | 4.40 ( |
2.85 ( |
| Borderline high | 5.15 (from 170 to 199 mg/dL) | 3.35 (from 110 to 129 mg/dL) |
| High | 5.20 ( |
3.40 ( |
| HDL – more than 1.0 mmol/L | ||
| TG up to 10 years – 1.1 mmol/L, over 10 years 1.69 mmol/L | ||
The classification proposed by Koletzko et al. [94] deems to be more up-to-date (see Table5).
| Indicators | Levels of serum lipids, lipoproteins, and apolipoproteins | |||
| Optimal | Borderline | High | Low | |
| below percentile 75 | percentile 75–95 | over percentile 95 | ||
| Cholesterol total | 170–199 mg/dL (5.15 mmol/L) | - | ||
| TG | - | |||
| Aged 0–9 | 75 mg/dL ( |
75–99 mg/dL (0.85–1.12 mmol/L) | ||
| Aged 10–19 | 90 mg/dL ( |
90–129 mg/dL (1.0–1.46 mmol/L) | ||
| LDL-C | 110–129 mg/dL (2.85–3.35 mmol/L) | - | ||
| Non-HDL-C | 120–144 mg/dL (3.2–3.6 mmol/L) | - | ||
| HDL-C | 35–45 mg/dL (0.9–1.2 mmol/L) | |||
| ApoВ | 90–109 mg/dL | - | ||
| ApoАI | 115–120 mg/dL | |||
| Lipoprotein (а) | - | - | ||
| Note. * Cholesterol: 1 mg/dL = 0.0259 mmol/L; triglycerides: 1 mg/dL = 0.01 mmol/L. 1 non-HDL = total cholesterol – HDL-C. High triglycerides levels are often revealed in infants at high fat content in food and in cases when blood test was performed not in the fasted state. | ||||
The content of LDL-C is determined by the calculation method according to W. T. Friedewald’s formula: LDL-C = Cholesterol total – (HDL-C + TG/2.2) (when determining the indicators in mmol/L);
LDL-C = Cholesterol – (HDL-C + TG/5) (when determining the indicators in mg/dL).
The level of non-HDL-related cholesterol (Cholesterol-HDL-C) is recommended to
be determined in individuals with TG level
Currently, the non-HDL-related cholesterol (non-HDL-C) indicator is recommended to be used as a target level in patients with diabetes mellitus and metabolic syndrome, since they are more likely to have hypertriglyceridemia and low HDL-C levels. The target value of non-HDL-C for the corresponding risk categories is calculated by adding 0.8 mmol/L to the target level of LDL-C.
Diagnosis of familial hypercholesterolemia is carried out on the basis of Dutch (Dutch Lipid Clinic Network), British (Simon-Broome Registry) or American (MedPed-US Make Early Diagnosis Prevent Early Death) criteria [72, 74, 76, 77, 78, 95].
According to the European Guidelines for the diagnosis of familial hypercholesterolemia in children, the British criteria (Broome familial hypercholesterolemia Registry) should be applied. In children under 10, the main criterion for familial hypercholesterolemia diagnosis is LDL cholesterol level over 4 mmol/L (160 mg/dL). If the hypercholesterolemia is diagnosed as a part of a cascade screening (when in a parent, mostly in a father, hypercholesterolemia and myocardial infarction were genetically verified at a young age), the diagnosis can be confirmed at LDL cholesterol level over 3.5 mmol/L (130 mg/dL). If the child is not burdened by early cardiovascular disease heredity, FHCH diagnosis should be assumed at higher LDL-C levels – over 4.9 mmol/L (190 mg/dL). At the same time, enhanced LDL cholesterol values shall be confirmed by two tests with an interval of 3 months [72, 76, 77].
Simon Broome Registry criteria
The Simon Broome Registry criteria [78] are used to diagnose heterozygous FHCH in adults, as well as in children and adolescents under the age of 16. The heterozygous FHCH is diagnosed if the patient meets the criteria for a “certain” or “probable” FHCH diagnosis.
A definite heterozygous FHCH is diagnosed if:
• Total CH
• Total CH
Plus one of the following:
• The presence of tendon xanthomas in the patient or a first-degree relative (parents, children, brothers, sisters), or in a second-degree relative (grandparents, uncles or aunts).
• A positive DNA diagnostic test confirming the presence of a pathogenic or probably pathogenic variant of the nucleotide sequence in LDLR, APOB, or PCSK9 genes.
A probable heterozygous FHCH is diagnosed if:
• Total CH
• Total CH
Plus one of the following:
• They have a history of myocardial infarction in a second-degree relative up to 50 years, in a first-degree relative up to 60 years.
• Total CH
• Total CH
Criteria for heterozygous FHCH diagnosis in children and adolescents
The Modified Criteria of Experts of the European Society for Atherosclerosis for
the Diagnosis of Heterozygous CFS in Children and Adolescents
• The level of LDL-C
• LDL-C levels
• The level of LDL-C
• Any LDL-C level when confirming the presence of a pathogenic or a probably pathogenic variant of the nucleotide sequence in LDLR, APOB, or PCSK9 genes detected in a parent diagnosed with heterozygous FHCH.
Criteria for homozygous FHCH diagnosis
Criteria proposed by experts of the European Society for Atherosclerosis for homozygous FHCH diagnosis in adults and children.
According to these criteria, the homozygous FHCH is diagnosed in children and adults when one of two conditions below is found [78]:
(1) The presence of two mutant alleles in LDLR, APOB, PCSK9 or LDLRAP1 genes.
(2) The LDL-C level over 13 mmol/L (500 mg/dL) without hypolipidemic therapy or LDL-C level over 8 mmol/L against the background of lipid-lowering therapy.
• Emergence of tendon or skin xanthomas before the age of 10.
• The LDL-C level without hypolipidemic therapy correlating with heterozygous FHCH in both parents.
During the diagnostic search, secondary forms of hypercholesterolemia shall be excluded. They can be caused by impaired thyroid function (hypothyroidism), nephrotic syndrome, chronic renal failure, liver diseases occurring with cholestasis, anorexia, and the use of medications (glucocorticoids, protease inhibitors) [72, 76, 77].
The LDL-C level over 13 mmol/L (500 mg/dL) indicates the presence of homozygous hypercholesterolemia, although lower values are possible due to genetic heterogeneity. In addition, the existence of a transition zone between heterozygous and homozygous hypercholesterolemia shall be taken into account, when the LDL-C level ranges from 8 to 13 mmol/L (300–500 mg/dL) [69, 77, 95].
Types of hypercholesterolemia screening
Various types of screening are currently used to detect HCH: cascade, targeted, and universal.
Cascade screening is currently used in most countries of the world. In cascade screening, a sequential measurement of the blood lipid spectrum is performed in the closest relatives of a person with diagnosed familial heterozygous hypercholesterolemia (index patient).
Cascade screening is the most appropriate way to diagnose familial heterozygous HCH, as it enables identifying patients at the preclinical stage. This type of screening is the most effective and economical [72, 76, 77, 78].
The sensitivity and specificity of cascade screening for the diagnosis of heterozygous hypercholesterolemia in the Russian population makes 93 and 82%, respectively [78]. If mutations that cause the disease are detected in patients with familial hypercholesterolemia, the effectiveness of cascade screening increases [72, 76, 78]. At the same time, 50% of first-degree relatives, including children, are diagnosed with familial heterozygous hypercholesterolemia. Subsequent genetic testing of first-degree relatives has 100% sensitivity and specificity, while when diagnosed only on the basis of clinical data, sensitivity and specificity range from 70 to 85% [96].
In children with clinical manifestations of hypercholesterolemia, whose parents suffer from this disease, mutations in the genes encoding lipid metabolism are detected with a frequency of up to 95% [97]. If both parents are ill, then in 25% of cases, homozygous hypercholesterolemia can be diagnosed. In the United States, European countries, and Russia, it is recommended that children be screened as part of cascade screening starting at the age of two [76, 78, 98]. In the UK, the beginning of cascade screening is at the age of 10 years [77]. If homozygous hypercholesterolemia is suspected, screening is performed as early as possible.
Targeted screening includes examination of children with a history of early cardiovascular diseases of atherosclerotic origin (in parents and/or second-degree relatives) [76, 78, 96]. Screening begins in children since the age of 2. If the LDL-C level is more than 4.0 mmol/L (160 mg/dL), the test should be repeated twice, to exclude secondary causes of hypercholesterolemia. In the case of death of parents from CHD, even with moderate hypercholesterolemia, a genetic test and determination of lipoprotein(a) are required.
Universal screening means examination of all children in the population. In European countries, it is performed in Slovenia in newborns and children aged 5 [97]. The German Society of Pediatrics and Adolescent Medicine recommends screening for high cholesterol in all children aged 5 [77, 99].
In the United States, universal screening of children aged 9–11 years is done in part because selective screening based on data on early cardiovascular disease-burdened heredity, was not effective enough to identify children with high LDL-C levels [99, 100].
The age of 9–11 years is considered optimal, since hormonal imbalances in the puberty period in children, starting from the age of 12, can reduce LDL-C levels, thereby leading to false negative results. Universal screening can also be performed in adolescents aged 17–21 [101, 102, 103]. Screening may be performed as part of a scheduled visit to a doctor. If the detected LDL-C level is more than 5 mmol/L (190 mg/dL), the analysis is repeated twice, and secondary cases are excluded. If familial hypercholesterolemia is confirmed, a cascade screening of the closest relatives is performed.
Genetic screening is not used in normal clinical practice, but can be used as needed. Revealing the specific mutation often increases the patient’s adherence to therapy. Cascade DNA screening is indicated if a mutation is identified in the family. Children with suspected heterozygous hypercholesterolemia can be screened since the age of 5 [72, 76]. Girls and boys are screened at the same age [72, 76, 77].
If a high level of total cholesterol is detected in a child during an examination against the background intercurrent disease or cascade screening, the level of LDL-C shall be determined. If LDL-C concentration is higher than 4 mmol/L, a repeated lipid level should be found and the causes of secondary hypercholesterolemia forms should be excluded. With a steady increase in LDL-C concentration (more than 4 mmol/L), DNA testing is recommended to determine mutations in the main genes that cause hypercholesterolemia: LDLR, APOB, PCSK9.
Mutation revealing confirms the familial hypercholesterolemia diagnosis. Children with LDL-C levels over 3.5 mmol/L are prescribed medication. Children with a lower LDL-C level are taken for dispensary observation with subsequent control at the age of 8, 10 years, then annually.
In the absence of mutations, further tactics depend on the LDL-C level:
• At the level of over 5 mmol/L (190 mg/dL), the presence of hypercholesterolemia is likely, the drug treatment is indicated;
• At the level of over 4.0 mmol/L (160 mg/dL) in combination with a family history of early cardiovascular diseases, familial hypercholesterolemia is also likely - the drug treatment is indicated;
• At the level of below 3.5 mmol/L, the diagnosis of familial hypercholesterolemia is unlikely, and dynamic monitoring continues.
The second strategy option is based on detecting high cholesterol in child’s parents. In such a situation, the reason for hypercholesterolemia detecting shall be detailed (accidental detection during a routine examination without clinical symptoms, combination with symptoms of early coronary heart disease, examination as part of a cascade screening). Further on, a DNA examination of the parents is done.
In case their mutation is confirmed, a DNA test in children is done:
• If the child also bears this mutation, the diagnosis of familial hypercholesterolemia is verified, and the therapy is prescribed;
• If the mutation is not revealed in the child, monitoring is started, lipoprotein control is carried out at the age of 5 and 8 years, and the diagnosis of familial hypercholesterolemia is unlikely.
If the mutation is not detected in parents, then the further tactics are determined by LDL-C level in the child:
• If the LDL-C level is over 5.0 mmol/L, and the secondary causes of hypercholesterolemia are excluded, the familial hypercholesterolemia diagnosis in the child is likely; and the drug treatment is indicated;
• If the LDL-C level is over 4.0 mmol/L, and there is a heredity of early cardiovascular diseases (a disease in parents), the familial hypercholesterolemia diagnosis is likely; the drug treatment is indicated;
• If the LDL-C level is within the normal range, the familial hypercholesterolemia diagnosis is discarded.
• Non-drug therapy: dietary cure, physical inactivity prevention, overweight and obesity prevention activities, refusal of bad habits.
• Hypolipidemic therapy.
• Extracorporeal treatment methods.
Dietary cure is the lifestyle and diet that AS prevention is based on. For children under two years of age, there are no strict recommendations for limiting the use of fat and cholesterol, since growth and development require a lot of energy resources. After two years, children begin to develop their own diet. By the age of five, their diet should contain no more than 30% and no less than 20% of calories derived from fat. Proper nutrition can eliminate several risk factors simultaneously: hyperlipidemia, hypertension, and obesity.
The main caloric content of the diet comes in the first half of the day: 1st breakfast – 25%, 2nd breakfast – 15%, lunch – 30–35%, afternoon tea – 10%, dinner – 15% of the daily caloric content. For children with DLP and normal body weight, a 4-times meal is recommended, for children with obesity – 5–6 meals a day, as with rare meals, glucose tolerance decreases, and hypercholesterolemia and hypertriglyceridemia are formed.
It is recommended to reduce the total fat intake to 20–30% of the diet caloric content. The diet should include no more than 8–10% saturated fat. The consumption of monounsaturated fats should be more than 10%, and polyunsaturated fatty acids – from 7 to 10% of the total diet caloric content. The consumption of cholesterol should be reduced to 75 mg per each 1000 kcal [104].
A hypolipidemic effect can be achieved by introducing complex and slow-digesting carbohydrates into the diet – of soluble dietary fibers found in legumes, fruits, vegetables, and whole grains [105]. Soluble dietary fibers include pectins (black currant, apples, plums), gums, dextrans, mucus (oatmeal and pearl barley, rice, flax seeds and plantain), hemicellulose fractions, inulin. Dietary fiber helps to reduce the cholesterol concentration, absorbs TG, LDL-C and VLDL-C in the intestine, preventing their absorption. Food with fiber at the dose of 5–15 g/day is well-tolerated and is recommended for reducing the level of atherogenic fractions of the lipid spectrum [106]. Mucofalk (as a source of dietary fiber) has been recommended by the FDA and AHA since 1998 as a component of dietary therapy in patients with mild to moderate hypercholesterolemia.
The protein content in children’s diet with DLP should be approximately 15%. Protein deficiency in the diet leads to protein-energy deficiency, and vice versa, excess protein in the diet is often associated with a high intake of saturated fat and cholesterol. Replacing 50% of dietary protein with isolated soy protein helps to reduce the content of cholesterol and LDL-C [107]. Thus, diet therapy for DLP is a starting and mandatory component of treatment. For child’s diet planning, dietary counseling is necessary. It should be aimed not only at DLP correction, but also at ensuring optimal growth and child’s body development.
Physical exercise is an important DLP therapy component. A high rate of motor activity is directly associated with a reduced risk of developing ASCVD in children and adolescents. Regular physical training helps to control body weight, to reduce blood pressure, to adjust disorders of lipid metabolism, to promote an increase in HDL cholesterol, prevent vascular dysfunction, and reduce the thickness of the intima – media complex, as the earliest marker of subclinical AS [107]. WHO has developed recommendations in physical activity for children and adolescents: (1) the minimum total amount of physical activity for people aged 5 to 17 years should be at least 60 minutes per day; (2) the motor activity exceeding 60 minutes per day provides additional health benefits; (3) the main part of physical activity should be of the aerobic type [108]. Depending on the detected level of lipids and LP, the therapeutic tactics is established.
At the acceptable level of CH total and LDL-C, a healthy lifestyle is formed, refusal of bad habits, overcoming hypodynamia and obesity, normalization of blood pressure, monitoring the glycemia level, the lipid spectrum indicators – once every 5 years.
At the borderline level of CH total and LDL-C, Level 1 hypolipidemic diet is added (the caloric content is provided by 55% carbohydrates, 15% proteins and 30% fats; fats are 2/3 unsaturated fats, the daily CH intake is not more than 300 mg/day). The examination of blood serum lipid spectrum is done annually.
With an elevated content of CH total and LDL-C, the secondary and primary DLP genesis shall be excluded. Level 1 diet therapy, and in case of its inefficiency, Level 2 diet therapy (reduction of saturated fat content down to 7%; the daily cholesterol intake not more than 200 mg/day) [109], auxiliary pharmacotherapy, and hypolipidemic therapy. The examination of lipid spectrum is done once every 3 months.
Auxiliary pharmacotherapy includes administration of membrane protectors in courses, of choleretic drugs, antioxidants, metabolites, and fatty acid drugs.
Hypolipidemic therapy in children has its own features, it is based on recommendations of the National Cholesterol Education Program/NCEP and the American Heart Association [110] (see Table6 (Ref. [111, 112, 113, 114, 115, 116]) and Table7 (Ref. [110]) below).
| Medical drug | Pediatric age | Dose, mg/day | Notes | Reference |
| (years) | ||||
| Atorvastatin | 10–17 | 10–20 | Titration for 4 weeks | [111] |
| Fluvastatin | 10–17 | 20–80 | Titration for 6 weeks | [112] |
| Lovastatin | 10–17 | 10–40 | Titration for 4 weeks, then 20 mg/day | [113] |
| Pravastatin | 8–18 | 20–40 | 8–13 years – 20 mg/day | [114] |
| 14–18 years – 40 mg/day | ||||
| Rosuvastatin | 10–17 | 5–20 | Titration for 4 weeks | [115] |
| Simvastatin | 10–17 | 10–40 | Titration for 4 weeks | [116] |
| NCEP recommendations | |
| (1) Drug therapy is recommended for children over 10 years of age after 6–12 months of dietary restrictions. | |
| (2) Drug hypolipidemic therapy is recommended if: | |
| (a) LDL-C | |
| (b) LDL-C | |
| (c) 2 or more cardiovascular RF persist after attempts of their strict control. | |
| (3) Medical activities are preferably done in specialized centers. | |
| (4) Therapy objectives: | |
| (a) Minimum LDL-C | |
| (b) Ideal LDL-C | |
| Amendments of American Heart Association | |
| (1) Patients with a complicated family history, overweight or obese should be screened with a mandatory assessment of the lipids level in blood on an empty stomach. | |
| (2) In children and adolescents with overweight and impaired lipid metabolism, other components of the metabolic syndrome shall be identified. | |
| (3) In children who are candidates for drug hypolipidemic therapy, statins should be regarded as first-line treatment drugs. | |
| (4) In children at a high risk of lipid metabolism disorders, the presence of additional risk factors or comorbid conditions may influence the need to achieve a lower target LDL-C level or contribute to deciding to start a drug therapy at the age of | |
Statins form the first line of hypolipidemic therapy in pediatrics . They contribute to the reduction of atherosclerotic vascular manifestations, thereby reducing mortality [117, 118]. The main pharmacological effects of this group are aimed at reduction of cholesterol synthesis in the liver due to the competitive inhibition of enzyme HMG-CoA reductase. They are used in children over 8–10 years of age with hereditary DLP forms and the LDL-C level exceeding 4 mmol/L, with a burdened hereditary history of cardiovascular pathology Statins reduce the LDL-C content in 20–40% of cases [101]. In the Russian Federation, the following drugs have been approved: Simvastatin since 10 years of age, Atorvastatin since 10, and Fluvastatin since 9. In the United States, Pravastatin has been approved since the age of 8, and Rosuvastatin in the European Union – since the age of 6 [78]. It is necessary to start therapy with a minimum dose, before starting the treatment, the activity of creatine phosphokinase, alanine - and aspartate aminotransferase shall be determined. The tests are repeated after 4 and 8 weeks, and then monitored once every 3–6 months. If the target values of LDL cholesterol are not reached within three months, the drug dosage is titrated to the maximum effective.
In the absence of the desired effect, cholesterol absorption inhibitor – Ezetimibe – is added to statins. It should be noted that the combination of Ezetimibe + statin is effective: against the background of combination therapy, there is a greater decrease in LDL-C level compared to statin monotherapy (54.0% vs. 38.1%). Ezetimibe is prescribed at a dose of 10 mg/day, used in children over 12 years of age [119].
Inhibitor of Subtilisin-Kexin Type 9proprotein convertase is a new drug class (PCSK9 inhibitors). They target at protein (PCSK9) involved in expression control of LDL-C receptors. It is a monoclonal antibody to PCSK9 – Evolucumab (administered at the dose of 420 mg in children over 12 years of age, subcutaneously once a month). Evolucumab is used for the treatment of homozygous FHCH. This group of drugs additionally reduces the level of LDL cholesterol up to 60% in patients who receive the most tolerable hypolipidemic therapy. This results in more than 80% of these patients being able to reach the recommended LDL-C targets [119].
For treatment of hypercholesterolemia and hyper LDL-C in children, bile acidsequestrants are also used. This group of drugs binds bile acids in the intestine, and prevents absorption of bile acids and cholesterol in the intestine (Cholestyramine, Colestipol – the initial dose is 5 mg, the maximum dose is 20 mg (unregistered in the Russian Federation).
In particularly severe cases, it is recommended to add extracorporeal treatment methods to drug therapy.
Lipoprotein apheresis: LP-apheresis is performed in specialized medical institutions. LP-apheresis is prescribed to patients who, after 6 months of combined lipid-lowering therapy in the maximum tolerated doses, have not reached the target levels of LDL-C and/or Lp(a). Apheresis is performed weekly or once every two weeks. During the procedure, up to 80% of LDL-C and/or Lp(a) are removed from patient’s blood plasma, while LDL-C and/or Lp(a) concentration in patient’s blood plasma, depending on the volume of the treated plasma, is reduced by 70–80%. Currently, there are several LP apheresis methods: cascade plasma filtration, lipid filtration, LDL (HELP) heparin-precipitation, affine plasma and hemosorption of lipoproteins, LDL and Lp(a) immunosorption.
Since more than 90% of the total amount of LDL-C is localized in hepatocytes, liver transplantation is the only treatment that normalizes the LDL-C level. So far, the long-term effect of this procedure is understudied. After liver transplantation, the LDL-C level decreases sharply, but this method of treatment should be selected before the occurrence of cardiovascular complications. In fact, despite the normalization of the lipid spectrum, cardiovascular events can progress. Due to the significant clinical problems of organ transplantation (the need for lifelong immunosuppressive therapy), it remains an acceptable option only for the treatment of patients who do not respond to traditional lipid-lowering therapy and, possibly, before the onset of significant cardiovascular disease [120, 121].
Today, AS is not only the area that children’s doctors are in charge of, but also, first and foremost, it is the problem of pediatrics in view of the need for early diagnostic, preventive and therapeutic effects. It determines the relevance and significance of purposeful DLP identification as the leading RF of atherogenesis in children, teenagers and adolescents. DLP plays a significant role in ASCVD development, while ASCVD dominates the structure of morbidity and mortality in the world.
The conducted epidemiological studies have shown a wide spread of DLP in children and adolescents – more than 70% in the population. It justifies the need for further additional examination of this contingent in order to verify the diagnosis and timely resolve the issue of management tactics.
FHCH prevalence in the general population is quite high – 1:200. At the same time, unfortunately, the diagnosis of the disease remains extremely low. No more than 1% of patients are diagnosed before the developed clinical manifestations. FHCH is an extremely severe disease with an unfavorable prognosis. Severe complications of the atherosclerotic process in the form of acute myocardial infarction are possible already in the first decade of life. The first clinical manifestation of the disease is skin xanthomas, lesions of the cardiovascular system, and the eyes. However, to date, pediatricians are not sufficiently informed about the clinical course and modern approaches to FHCH treatment. It results in late diagnosis and incorrect patient management tactics.
Children with FHCH should be monitored by pediatricians, pediatric cardiologists who have received additional training in lipidology. Normalization of lifestyle is a mandatory component in the first stage of child management. It includes normalization of the physical activity level, the elimination of additional risk factors for the development of cardiovascular diseases, and a hypocholesterolemic diet.
An important area of management of pediatric patients is the adjustment of other risk factors for the development of cardiovascular diseases. Among them, it is mandatory to stop smoking, including passive smoking, to normalize blood pressure and to eliminate excess body weight. The determination of lipoprotein(a) helps to identify an increased risk of developing cardiovascular diseases. An increase in this indicator of more than 50 mg/dL increases the risk of early development of coronary heart disease by 1.5 times. It is extremely important to treat concomitant diseases that increase the risk of developing cardiovascular diseases: Type 1 and Type 2 diabetes, chronic kidney diseases, connective tissue diseases, etc. Diet is of crucial importance in the correction of familial hypercholesterolemia in childhood. The restriction in nutrition does not apply only to children of the first year of life. In children over one year of age, the total caloric content of food is reduced by 30% due to strict restrictions on the amount of saturated fat and trans fats containing the highest amount of cholesterol. Cholesterol intake should not exceed 300 mg/day.
Timely hypolipidemic therapy can reduce the total cholesterol and LDL-C levels. Statins should be prescribed for FHCH as early as the first decade of life, to prevent the development of cardiovascular catastrophes. With insufficient effectiveness of statins, another class of drugs that prevent the absorption of cholesterol in the intestine (ezetimibe) is connected. Currently, to enhance the hypolipidemic effect, a third class of drugs for combined hypolipidemic therapy with subtilisin/kexin type 9 proprotein kinase inhibitors is proposed. In severe cases, the addition of LDL-C apheresis is possible. If hypercholesterolemia persists, liver transplantation is executed.
Today, more and more young people suffer from AS, and it has become the greatest epidemic of our era, threatening to affect the entire Earth population in the near future unless its prevention activities have been revealed by studying the pre-disease and the onset disease periods. Today, it is of no doubt that an intense AS prevention shall be started at an early age. AS is not just one of pediatric problems, but a primary pediatric problem in terms of prevention and treatment efficiency. Revealing children predisposed to AS development can be of great practical importance due to exceptional possibilities of adjusting functional disorders of organs, systems, and metabolism in childhood and adolescence.
AVB, VET, LVP, VMP conceived and designed the content; AVB, VET wrote the manuscript; LVP, VMP supervised and prepared the final version for submission.
Not applicable.
Authors express the gratitude to all the peer reviewers for their opinions and constructive suggestions.
This research received no external funding.
The authors declare no conflict of interest.