



1 Orthopedics Department, Lanzhou University Second Hospital, 730000 Lanzhou, Gansu, China
2 Orthopaedics Key Laboratory of Gansu Province, 730000 Lanzhou, Gansu, China
3 Stomatology Department, Lanzhou University Second Hospital, 730000 Lanzhou, Gansu, China
4 The Second Clinical Medical College, Lanzhou University, 730000 Lanzhou, Gansu, China
Abstract
Neuropathic pain is a common pain syndrome, which seriously affects the quality of life of patients. The mechanism of neuropathic pain is complex. Peripheral tissue injury can trigger peripheral sensitization; however, what really plays a key role is the sensitization of the central nervous system. Central sensitization is a key factor in the perception of chronic pain. Central sensitization refers to the increased sensitivity of the central nervous system to pain treatment, which is related to the change of the functional connection mode of the neural network. The current study aims to reveal the basic molecular mechanisms of central sensitization, including the involvement of P2 purine X4 receptor and brain-derived neurotrophic factor. In terms of treatment, although there are drugs and physical therapy, the accuracy of targeting is limited and the efficacy needs to be further improved. Future therapeutic strategies may involve the development of new drugs designed to specifically inhibit the central sensitization process. This article focuses on the effector molecules involved in central sensitization, aiming to elucidate the pathogenesis of neuropathic pain and provide a basis for the development of more effective treatment models.
Keywords
- neuropathic pain
- central sensitization
- P2 purine X4 receptor
- brain-derived neurotrophic factor
Neuropathic pain (NP) refers to a series of diseases of the peripheral nervous system caused by injury or disease of the nervous system part that usually transmits sensory information, which seriously affects the quality of life of patients [1, 2]. A variety of diseases, such as trigeminal neuralgia, postherpetic neuralgia, diabetic neuropathy-related pain, post-stroke central pain, etc., can cause NP [1, 3]. With the aging of the global population, NP has gradually evolved into a more urgent clinical problem [4]. At present, the clinical treatment of NP encounters many obstacles: opioids provide limited analgesic effects and cause many adverse reactions [5]; non-steroidal anti-inflammatory drugs (NSAIDs) and antidepressants are facing a prominent problem of drug resistance [6]; biological agents such as monoclonal antibodies targeting recombinant nerve growth factor (NGF) have shown the advantages of targeted therapy, but concerns about their safety still exist [7]. Therefore, it is imperative to develop new, safe and effective NP therapeutic drugs.
In the pathogenesis of NP, although peripheral tissue damage can induce peripheral sensitization and enhance pain signals, more and more studies have emphasized that central sensitization is the basic mechanism for NP maintenance [8, 9]. Central sensitization refers to the increasing responsiveness of the central nervous system to pain stimuli, which is a key determinant of maintaining various pain states [10]. Under the condition of central sensitization, pain-related central regions exhibit a series of pathological changes across cellular and molecular levels, including enhanced neuronal excitability, synaptic reorganization and weakened efficacy of intrinsic pain suppression mechanisms, which eventually lead to enhanced pain response and tolerance [11, 12, 13]. The comprehensive exploration of the mechanism of central sensitization and the formulation of treatment methods for central sensitization may open up new avenues for the treatment of NP [14]. In addition, central sensitization is also an important therapeutic target in chronic, painful, inflammatory and non-inflammatory rheumatic diseases and central spondylitis [15, 16]. However, the understanding of the molecular basis of central sensitization is still limited, and its intricate cellular and molecular effect networks need to be further elucidated.
Central sensitization plays a key role in the pathogenesis of NP [17]. In recent years, the research focus of neuropathic pain has gradually shifted to the key effector molecules of central sensitization, and great progress has been made. Nevertheless, a comprehensive understanding of the precise molecular mechanism of central sensitization is still limited to ongoing research and needs to be further elucidated [18]. This article elaborates the specific mechanism of central sensitization in NP, including changes at the molecular level, changes at the cellular level, and regulation of neural networks. At the same time, the animal model of NP is summarized to promote the research in the field of NP.
NP can be manifested as spontaneous pain, hyperalgesia and malformed pain [19]. Persistent spontaneous pain seriously affects the quality of life of patients [20]. Hyperalgesia refers to peripheral and central sensitization [21]. The pain caused by deformity means the injury of pain pathway [22]. These signs suggest the presence of pain amplification and increased central sensitivity.
Spontaneous pain is the main manifestation of neuropathic pain, which is a pain sensation without obvious causal relationship. It often persists and exhibits a severe nature, seriously affecting the patient’s sleep pattern and overall quality of life [23]. According to the location of the lesion, it can be divided into central spontaneous pain and peripheral spontaneous pain [24, 25]. Central spontaneous pain often occurs after stroke or brain injury, manifested as unprovoked pain in the facial area or affecting half of the limbs [26]. Peripheral spontaneous pain is common, including trigeminal neuralgia, facial pain, postherpetic neuralgia, and limb pain caused by diabetic peripheral neuropathy [27, 28]. These pain symptoms can last for months or even years, significantly affecting the patient’s sleep patterns, emotional state and overall quality of life [29]. Drug therapy for spontaneous pain is still insufficient, and a combination of antidepressants, antiepileptics, and opioids is often required to relieve symptoms [30].
Hyperalgesia refers to the amplification response to harmful stimuli, which can increase the body’s pain response to harmful stimuli [31]. This pain often occurs in the initial stage of NP, caused by peripheral and central sensitization [32]. Hyperalgesia is particularly common after surgery or in the initial stage of nerve injury, which may last for weeks to months [33]. Allodynia refers to the reaction of ordinary or mild stimulation, such as gentle touch/pressure can cause pain response [34]. Hyperalgesia can be managed with corticosteroids, non-steroidal anti-inflammatory drugs, and nerve blockers [35]. Identifying and effectively managing hyperalgesia helps to avoid the development of central sensitization and prevent pain from progressing to a chronic state.
Phantom limb pain refers to the phenomenon that patients still feel pain or other feelings related to the limb when they lose the limb [36]. This phenomenon usually occurs in patients undergoing surgery or accidental limb amputation. Even if the limb has been amputated or lost, the patient still feels the presence of the limb, and sometimes even feels discomfort such as pain, itching, and touch [37]. Phantom Limb Pain may be related to the plasticity of the central nervous system [38]. After losing a limb, the brain may be able to adapt to the change by reorganizing neural circuits [39]. This reorganization may lead to confusion of neural signals, resulting in a sense of phantom limb pain, that is, central sensitization.
While peripheral tissue damage can induce peripheral sensitization, it is central sensitization that assumes greater significance in augmenting and sustaining pain perception [40]. Central sensitization correlates with heightened neuronal excitability, synaptic reorganization, and diminished inhibitory control [41]. Studies have shown that the signaling pathways involved in P2 purine X4 receptor (P2X4R) and brain-derived neurotrophic factor tyrosine kinase receptor B (BDNF-Trk B) are the basic components of central sensitization [42].
Peripheral nerve injury can significantly trigger the release of inflammatory
mediators such as tumor necrosis factor
Abnormal discharges from damaged nerves into the spinal cord can increase the
excitability of sensory neurons in the spinal dorsal horn [51]. This encompasses
heightened activity of N-methyl-D-aspartic acid receptors (NMDAR), elevated
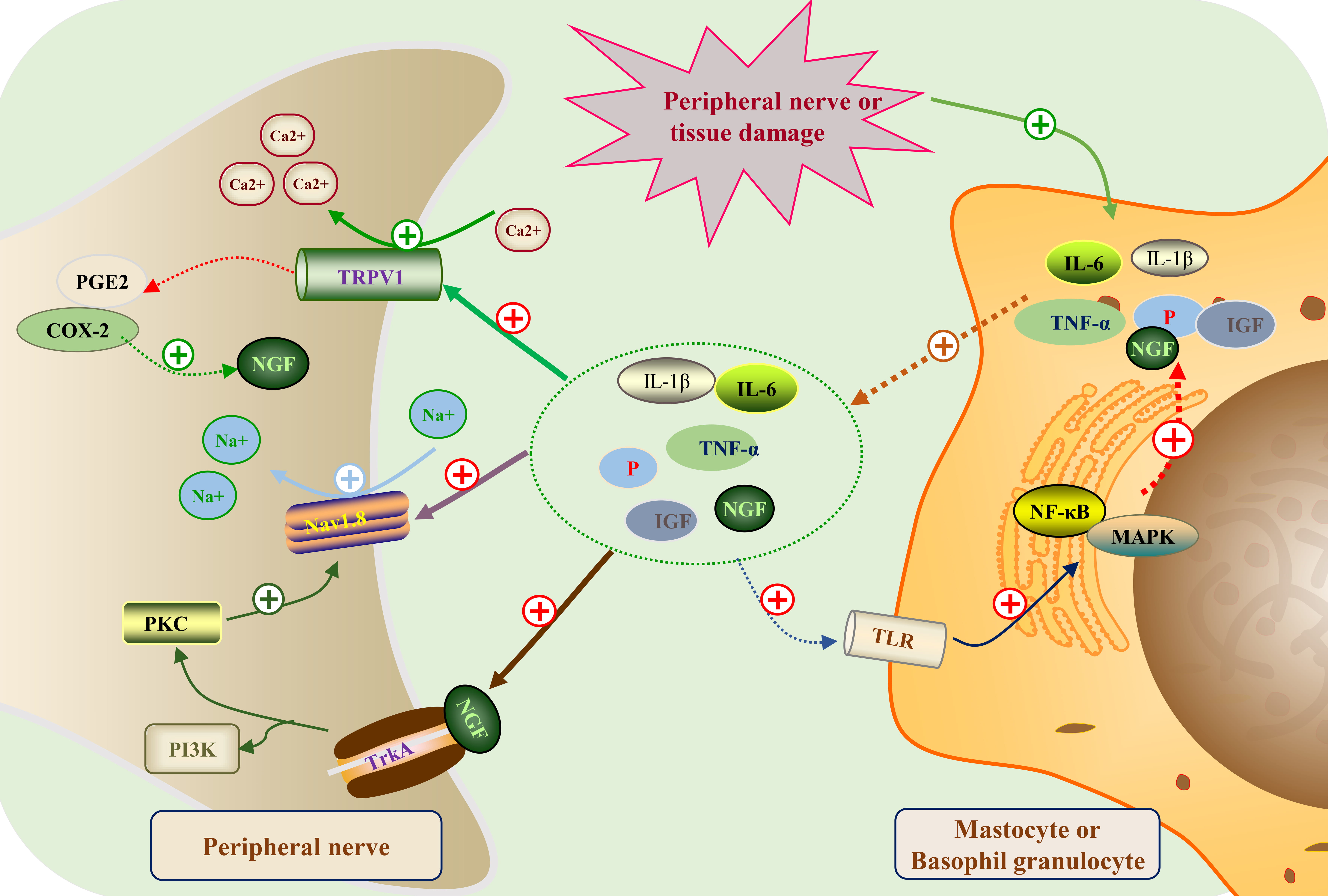 Fig. 1.
Fig. 1.Peripheral sensitization mechanism map of neuropathic pain.
Following peripheral nerve or tissue injury, immune cells (like mast cells and
basophils) liberate a range of inflammatory mediators, encompassing interleukin-6
(IL-6), interleukin-1
The enhancement of excitability of spinal dorsal horn neurons will amplify the
incoming pain stimulation information, which will eventually lead to the
establishment of pain amplification effect [59]. Increased excitability of spinal
dorsal horn neurons is associated with NMDAR activation, spinal neural network
remodeling, and decreased levels of inhibitory neurotransmitters [60]. At the
spinal level, we observed that Amyloid-
Central sensitization is the key mechanism to maintain neuropathic pain. The overactive state of spinal microglia and astrocytes is one of the key factors to activate central sensitization [65, 66]. Central sensitization mainly occurs in the pain processing center of the spinal dorsal horn and brainstem, which may last for months to years. The intensity of central sensitization is related to the amplitude and duration of pain [67]. In addition, functional magnetic resonance imaging (MRI) studies have shown that patients with chronic pain have increased and extensive activation of central regions during acupuncture pain stimulation, which means that the central nervous system is more sensitive in pain information processing [68].
Central sensitization includes cellular and molecular mechanisms, including increasing neuronal excitability, altering synaptic plasticity, and attenuating inhibitory neuromodulation [69, 70]. Activated microglia can release a large amount of neuroactive substances and pro-inflammatory cytokines [71]. Decreased release of inhibitory neurotransmitters such as GABA and glycine can also impair the inhibitory control of neural networks [72]. In addition, activated protein kinases such as extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) induce changes in downstream gene expression and cell activity [73, 74]. These factors increase the excitability of the central nervous network, thereby more effectively amplifying and transmitting pain signals. At the molecular level, during central sensitization, the concentration of excitatory amino acid neurotransmitters, such as Glutamate (Glu), increased, while the level of inhibitory neurotransmitters, such as GABA, decreased [75, 76]. The generation and maintenance of central sensitization is also related to the following mechanisms: (1) Increased release of inflammatory cytokines directly regulates the excitability of neurons. Central sensitization can increase the release of pro-inflammatory factors, which can directly act on neurons and regulate their excitability [77, 78]; (2) Activate glial cells and enhance the signal regulation of neurons [79]. In the state of central sensitization, microglia and astrocytes are activated to release cytokines and chemokines, enhance the signal regulation of neurons, and further promote central sensitization [80, 81]; (3) Calcium signaling pathway is activated, affecting neuronal excitability [82]; (4) Increased synaptic connection strength, cell structure remodeling, and changes in neural network sensitivity [83]; (5) The hypothalamus-adrenal cortex axis is activated, affecting neuroendocrine processes [84, 85]. Changes in functional connectivity between these brain regions also help to shape the effects of central sensitization. Brain imaging studies have shown that functional connectivity between different brain regions is enhanced during central sensitization, which promotes the processing and persistence of pain [86]. Although a large number of studies have conducted in-depth studies on key molecules in central sensitization, such as P2X4R, Brain-Derived Neurotrophic Factor (BDNF) and NMDAR, their precise mechanisms and complex interactions between different pain types still need to be further elucidated through recent animal experiments.
High expression of P2X4R is an important aspect of central sensitization [87]. The activation of P2X4R increases the excitability of central neurons, triggers downstream signaling cascades, induces the release of inflammatory factors, and produces an escalating hyperalgesia loop [88]. Therefore, P2X4R is a key effector molecule that connects peripheral input and central sensitization. In different animal models of neuropathic pain, the expression of P2X4R in glial cells is increased, and inhibition of P2X4R can significantly reduce hyperalgesia and spontaneous pain [89, 90]. The activation of P2X4R not only directly triggers the increase of intracellular calcium concentration and the increase of central nervous system excitability, but also initiates downstream protein kinases such as ERK and JNK, induces the release of a series of inflammatory factors and cell active substances, and finally leads to the progressive hyperalgesia loop in the central nervous system [91, 92, 93]. Activation of P2X4R in spinal microglia releases BDNF, which can convert Gamma-Aminobutyric Acid Type A Receptor (GABAA) receptor-mediated inhibition into excitation, which is related to the excessive excitability of neurons in the spinal dorsal horn [94, 95, 96]. In addition, the activation of P2X4R promotes the release of ATP and other signaling molecules in glial cells, thus expanding the amplification and regulation of neuronal signals by glial cells [97].
The downstream signaling pathway activated by P2X4R plays a multifaceted role in
central sensitization [98]. For example, P2X4R can trigger the activation of Src
tyrosine kinase, promote NMDAR phosphorylation and enhance its function [99].
When P2X4R is over-activated, Ca
BDNF is an important neurotrophic factor, which activates intracellular signaling pathways such as MAPK/ERK and phosphoinositide 3-kinase/protein Kinase B (PI3K/Akt) through its high affinity receptor tropomyosin receptor kinase B (TrkB), and regulates the growth, differentiation and synaptic connection of neurons [103, 104]. Activated glial cells secrete BDNF and lack effective neural regulation mechanisms. Excessive BDNF in the spinal cord may promote the development of maladaptive plasticity by activating the TrkB signaling pathway, promote central sensitization and strengthen the circuit, thus contributing to the maintenance of chronic pain [95, 105, 106]. Activation of BDNF-TrkB signaling induces AMPAR and NMDAR phosphorylation and amplifies Glu receptor-mediated excitatory effects [107, 108]. BDNF first promotes TrkB homodimerize and induces the self-phosphorylation of the binding site, which can activate intracellular signal transduction mediated by BDNF and promote the functional changes in synaptic connection of neurons [109, 110]. In addition, BDNF can activate downstream signaling molecules such as CREB and promote the secretion of pro-inflammatory factors [111].
Activation of the downstream effects of BDNF-TrkB signaling pathway promotes different central sensitization processes:
(1) The binding of BDNF with TrkB receptor can also result in the activation of PKC, which phosphorylates GABAA increasing their presence in the cell surface [112].
(2) BDNF can also promote the phosphorylation of ERK, and ERK can also phosphorylate potassium channels, leading to neuronal depolarization [113, 114].
(3) BDNF promotes the expression of AMPAR, which makes neurotransmitter Glu produce more significant excitatory effect [115].
(4) BDNF can up-regulate the amplification of glial cells to neuronal signals [116].
In conclusion, activation of BDNF-TrkB signaling enhances neuronal excitability, alters synaptic plasticity, and maintains central sensitization (Fig. 2).
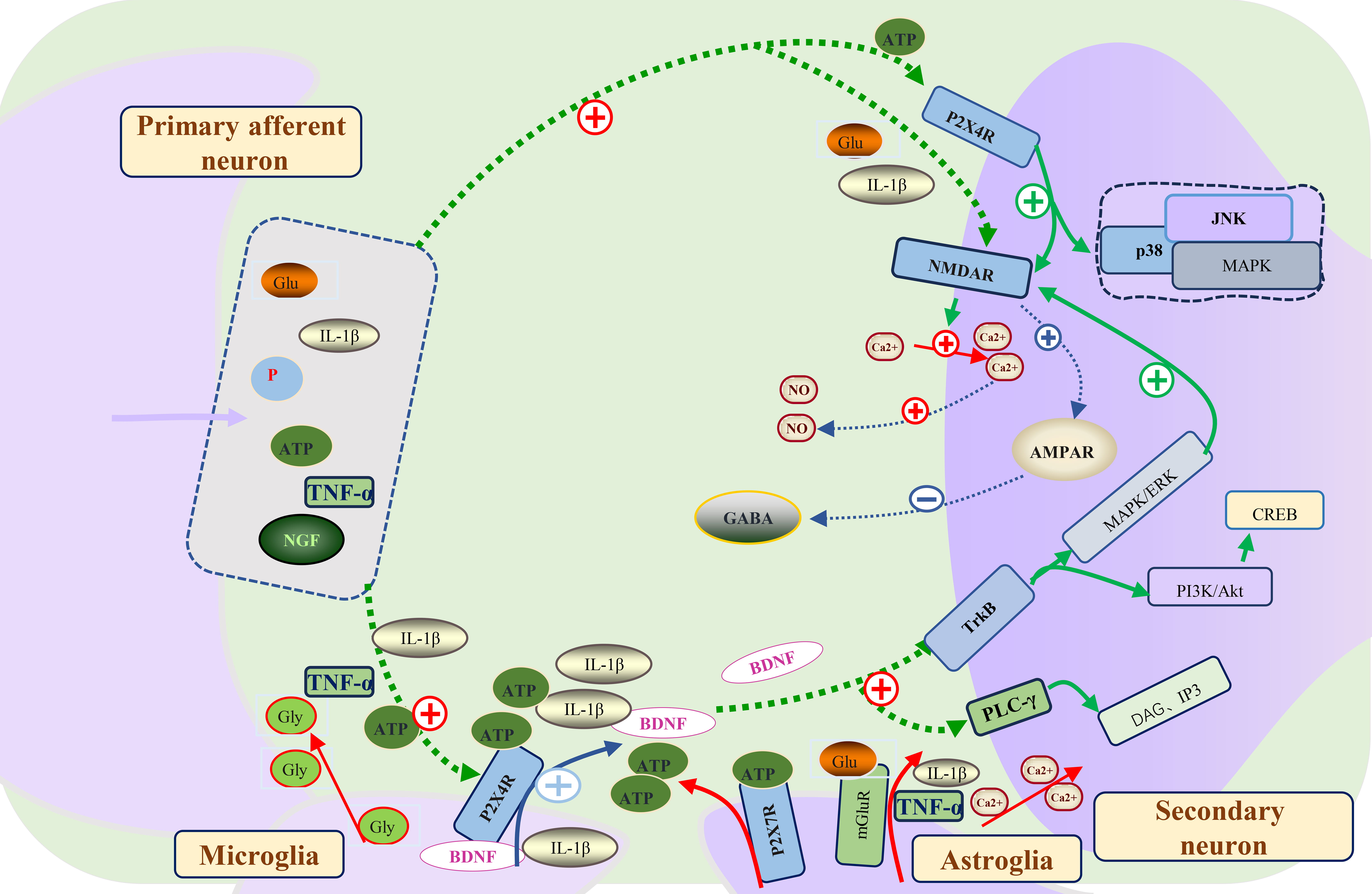 Fig. 2.
Fig. 2.The molecular mechanism of central sensitization formation and
maintenance in neuropathic pain. Primary afferent neurons in the spinal dorsal
horn release neurotransmitters including Glu, IL-1
The study of neuropathic pain mostly uses animal models, including peripheral nerve injury, spinal cord injury, central nervous injury, gene knockout model [117, 118]. However, these models have obvious advantages and limitations. In order to enhance their clinical relevance, further optimization of these models is essential.
In the field of neuropathic pain, peripheral nerve injury models are widely used and have become the main animal models for studying neuropathic pain [119, 120]. Peripheral nerve injury models mainly include:
(1) Chronic constriction injury (CCI) model of sciatic nerve. The CCI model was first proposed by Bennett et al. [121] in 1998. The model included the use of 4-0 chrome catgut to ligate four 1 mm-interval narrowing rings around the sciatic nerve trunk. In the CCI model, significant pain-related behavioral changes were observed within 24 hours after surgery, including mechanical hyperalgesia, thermal hyperalgesia and cold hyperalgesia. These are accompanied by spontaneous pain-like behaviors, including foot contraction, licking the affected limb, and raising the affected limb [122, 123]. However, the main limitation of the CCI model is the challenge of precisely controlling the shrinkage strength. At present, the best contraction intensity is considered to induce a slight tremor in the calf muscles.
(2) Sciatic nerve partial ligation model (PSL). The PSL model was proposed by Seltzer et al. [124] in 1990. The model included the use of a No.8 silk thread and a microbend needle to tightly ligature the proximal 1/3 to 1/2 of the sciatic nerve trunk. Pain behaviors associated with the PSL model can occur within 24 h after surgery and last for 9 weeks [125]. Although this model induces mild injury and usually avoids self-harm or autophagic limbs, controlling the type and number of injured nerve fibers is challenging. The model exhibits huge individual differences, limited repeatability, and significant difficulties in regulating these aspects. It is worth noting that the pain behavior induced by the PSL model tends to disappear within 3–5 weeks and cannot be observed for a long time. In addition, compared with the CCI and sciatic nerve injury (SNI) models, the pain threshold in the PSL model changed less.
(3) Spinal nerve partial ligation model (SNI). The SNI model was developed by Decosterd and Woolf [126] as an extension of the PSL model. Specifically, the SNI model included the use of 6-0 silk sutures to tightly ligature the proximal ends of the tibial and common peroneal nerves. In addition, the distal end of the nerve was sliced and ligated. The distal end of the nerve stump was removed by 2 mm to hinder the reconnection between the stumps and ensure the preservation of the sural nerve. The model has the advantages of good repeatability, consistent nerve fiber type and number of damage, and minimal individual differences [127].
(4) Spinal nerve ligation model (SNL). The SNL model proposed by Ho Kim and Mo Chung [125] includes complete ligation of L5 and L6 spinal nerves with 3-0 silk thread [128]. In this SNL model, the damaged nerve fibers showed homogeneity. It is worth noting that the change of mechanical pain threshold is more obvious than other models. The control of the ligation site and intensity is direct, individual differences are small, and autophagy will not occur [122].
(5) Chronic compressive injury model of spinal nerve root. The dorsal root ganglion chronic compression model developed by Hu and Xing [129] aims to simulate chronic low back pain caused by clinical conditions such as intervertebral foramen stenosis, intervertebral disc herniation, or root ganglion compression by tumors [130]. Other peripheral nerve injury models include L5 spinal nerve root ligation model (L5VRT) and complete sciatic nerve transection model (CST). At present, these two nerve pain models are rarely used.
Peripheral nerve injury models, including CCI, PSL, SNI, SNL and spinal nerve root chronic compression injury models, have wide application value as animal models in the field of neuropathic pain research. It is worth noting that the SNI model stands out because of its robust repeatability and strong damage consistency, and has obvious advantages. This model can effectively promote the research and long-term observation of neuropathic pain.
In view of the increased incidence of neuropathic pain after spinal cord injury, it is imperative to understand its potential pathogenesis [131]. This model, commonly known as Allen’s model, includes spinal cord exposure, fixation, and subsequent vertical strikes to induce spinal cord injury [132]. In the spinal cord injury model, it is feasible to prolong the duration of mechanical pain and thermal hyperalgesia [133]. This model provides an opportunity to study the effects of central mechanisms in pain management and central sensitization induced by spinal cord injury. Nevertheless, the creation of spinal cord injury models involves trauma and lacks precision; the resulting severe lower limb paralysis hinders post-impact behavioral assessment. At the same time, autophagy and self-mutilation in the area below the injury site tend to be manifested in the later stages of the disease.
About 8% of stroke patients experience post-stroke pain, and the incidence of pain in patients with hemorrhagic stroke is higher [134]. Therefore, the central post-stroke pain model (CPSP), especially the thalamic hemorrhagic stroke model, provides a feasible method for the study of central neuropathic pain. The CPSP model was established by injecting a specific volume of normal saline infused with type IV collagenase into the ventral posterolateral nucleus of the thalamus to induce local bleeding and simulate the clinical hemorrhagic stroke scene. The main features of the CPSP model include mechanical hyperalgesia and cold hyperalgesia, while the thermal pain threshold remains relatively unaffected, possibly due to the degree of tissue damage [135]. The central injury model helps to study the brain’s regulation of pain processing and changes in pain perception due to changes in related neural networks. However, it is more complicated to achieve accurate brain region localization within the model.
Some animal pain models have shown limitations, which makes it necessary to refine models that closely mimic human physiological characteristics and disease progression. This is still an important way of contemporary research. Although the peripheral model provides insight into the peripheral mechanism, it is not sufficient to fully simulate the clinical scenario. The spinal cord injury model allows for the observation of central effects, although the injury is more severe. The post-stroke central pain model allows direct examination of the brain’s involvement in pain, but accurate brain localization has proven to be more complex. In general, these animal models are of great value for analyzing the mechanism of neuropathic pain and designing treatment methods. However, it still needs to be further refined to enhance its applicability in clinical pain research.
In clinical practice, the efficacy of drug therapy for NP is still limited, while biological agents and physical therapy also show inherent limitations [136]. Treatment satisfaction needs to be further improved, because there is still a significant gap in achieving the desired therapeutic effect. Looking forward to the future, new drugs for key molecules of central sensitization should be developed to improve the level of treatment.
At present, drug therapy is the most important means of clinical treatment of NP. Commonly prescribed medications include tricyclic antidepressants (e.g., amitriptyline) and other antidepressants such as duloxetine, antiepileptic drugs (e.g., gabapentin and pregabalin), and opioids [137, 138]. The main mechanism of these drugs involves the regulation of various neurotransmitters and receptors in the central nervous system to produce analgesic effects. Specifically, tricyclic antidepressants can block the reuptake of norepinephrine and 5-hydroxytryptamine, thereby enhancing inhibitory signal transduction between synapses [139]; antiepileptic drugs enhance GABA-mediated inhibitory effects [140, 141]; opioid drugs mainly activate opioid receptors, inhibit calcium channels and adenylate cyclase, thereby reducing central excitability [142, 143]. In addition, some new drugs targeting key aspects of central sensitization have shown good therapeutic potential. For example, NMDAR antagonist ketamine and P2X4R antagonist oxidized ATP (oATP) [144, 145]. The treatment of NP in the elderly usually involves a variety of methods, including drug therapy, physical therapy, psychological support and behavioral therapy [146, 147]. In terms of drug selection, potential adverse event risks need to be considered, especially in view of other health problems and drug interactions that may exist in older persons. Antidepressants such as tricyclic antidepressants (such as amitriptyline) and antiepileptic drugs (such as gabapentin and pregabalin) may lead to decreased cardiac function, cognitive decline, balance problems and sleepiness in the elderly [148, 149]. In addition, cardiovascular adverse events associated with antidepressants also need to be considered. Anesthetic drugs (such as morphine or hydromorphine) can also be used to relieve neuropathic pain in the elderly, but the use of opioids can lead to cognitive decline, constipation, respiratory depression and other problems, and there is a risk of abuse and addiction [150, 151].
These emerging drugs have the ability to directly target key receptors and pathways that maintain central sensitization, making their effectiveness more pronounced. Although current drug therapy can alleviate NP, treatment satisfaction is still poor. The future development trajectory involves the creation of more targeted new drugs, specifically targeting the potential pathogenic mechanisms of central sensitization.
In recent years, biotherapy has emerged as a new strategy for the treatment of
NP. Representative therapeutic drugs include monoclonal antibodies targeting
anti-nerve growth factor (NGF) and anti-tumor necrosis factor (TNF) [152, 153].
NGF is involved in the process of peripheral and central pain sensitization.
Monoclonal antibody targeting NGF can counteract the NGF-mediated pain
amplification effect and show significant improvement in many clinical trials of
NP [154, 155]. However, long-term use of these monoclonal antibodies can lead to
adverse reactions, including bone and joint discomfort [156]. TNF-
The methods of relieving NP symptoms through physical therapy include transcutaneous electrical stimulation, low-power laser therapy, and spinal correction massage [159, 160, 161]. However, the effectiveness of these treatments is difficult to be confirmed. It is expected that precision medicine will bring more gratifying treatment results in the future. Exploring the mechanism of central sensitization and designing personalized new drugs are the key strategies to promote NP treatment in the future.
In summary, NP is a common pain syndrome caused by nervous system injury, which seriously affects the quality of life of patients. The mechanism behind it is complicated. Recent studies have shown that central sensitization plays a key role in the pathogenesis and is associated with a series of molecular and cellular mechanisms. This includes the activation of glutamatergic excitatory transmission, the involvement of P2X4R and BDNF-TrkB signaling pathways, the enhancement of neuronal excitability and the weakening of inhibitory regulation. These changes ultimately lead to enhanced and amplified effects of central pain management. Although considerable progress has been made in studying the mechanism of central sensitization, it is still crucial to translate these findings into clinical applications. For example, new drug development targeting molecules such as P2X4R or BDNF is underway. Comprehensive evaluation requires large-scale randomized controlled trials to evaluate treatment efficacy and long-term safety. Although analgesic drug therapy can provide relief of some symptoms, challenges such as patient compliance significantly affect its effectiveness.
Looking ahead, our work will continue to focus on elucidating central sensitization mechanisms and identifying precise intervention points. This work will lead to the development of new targeted therapeutic drugs and ultimately improve the treatment level of NP. In the future, continuous drug screening efforts can continue, and the use of the latest technologies, such as single-cell genomics, to design inhibitors or antagonists targeting these key molecules is expected to produce more effective treatments. In addition, it is also imperative to improve the establishment of animal models to more accurately simulate clinical conditions and evaluate efficacy. In short, the study of central sensitization mechanism has far-reaching significance. Taking it as the cornerstone of designing new drug treatment strategies will promote the continuous development of NP treatment, and more researchers are expected to contribute in this field.
WN and YM organized, designed, and wrote the article. ZK and YS designed of the work, drafted and revised this article for key intellectual content. WJ and WZ were responsible for literature collection and image creation. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank the Bone and Joint Research Institute and Cuiying Biological Center of the Second Hospital of Lanzhou University for their support.
This study was supported by the Fund Project of the Second Hospital of Lanzhou University (No. CY2022-QN-A03).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.