
 , Anwen Shao 4,5,*, Congguo Yin 1,2,*
, Anwen Shao 4,5,*, Congguo Yin 1,2,*1 The Fourth School of Clinical Medicine, Zhejiang Chinese Medical University, 310053 Hangzhou, Zhejiang, China
2 Department of Neurology, Hangzhou First People’s Hospital, 310006 Hangzhou, Zhejiang, China
3 Department of Cardiology, Hangzhou First People’s Hospital, 310006 Hangzhou, Zhejiang, China
4 Department of Neurosurgery, the Second Affiliated Hospital, School of Medicine, Zhejiang University, 310009 Hangzhou, Zhejiang, China
5 Key Laboratory of Precise Treatment and Clinical Translational Research of Neurological Disease, 310012 Hangzhou, Zhejiang, China
†These authors contributed equally.
Abstract
Ischemic stroke (IS) remains a serious threat to human health. Neuroinflammatory response is an important pathophysiological process after IS. Circular RNAs (circRNAs), a member of the non-coding RNA family, are highly expressed in the central nervous system and widely involved in regulating physiological and pathophysiological processes. This study reviews the current evidence on neuroinflammatory responses, the role of circRNAs in IS and their potential mechanisms in regulating inflammatory cells, and inflammatory factors affecting IS damage. This review lays a foundation for future clinical application of circRNAs as novel biomarkers and therapeutic targets.
Keywords
- ischemic stroke
- neuroinflammation
- circRNA
- inflammatory cytokine
- therapeutic target
According to the Global Burden of Disease Study 2019, stroke is the second leading cause of level 3 death worldwide in 2019, after ischemic heart disease, and the third leading cause of level 3 death and disability, posing a serious threat to human health and social development [1]. Ischemic stroke (IS) is a clinical syndrome characterized by cerebral tissue ischemia, hypoxia, and necrosis resulting from arterial narrowing or occlusion in the brain’s blood supply (carotid and vertebral arteries) [1, 2]. In 2019, IS accounted for 62.4% of all new strokes worldwide, making it the predominant type of stroke [1].
Following IS, brain inflammation is the major pathological event causing secondary cerebral tissue injury and poor functional recovery [3, 4, 5, 6, 7]. Post-stroke inflammation is initiated after the initial ischemia, marked by innate immune responses and the breakdown of the blood-brain barrier (BBB), releasing proinflammatory cytokines and infiltrating immune cells. Additionally, many studies on neuroinflammation have revealed that glial cells regulate the release of inflammatory cytokines and the progression of the inflammatory cascade [8, 9].
Circular RNAs (circRNAs), belonging to the non-coding RNA family, have a stable structure and high tissue-specific expression. CircRNAs are highly expressed and widely involved in regulating physiological and pathophysiological processes in the nervous system [10, 11].
CircRNAs are involved in neuronal injury and repair, glial cell protection, and BBB injury after IS [12, 13, 14, 15, 16, 17, 18]. However, the potential roles of different circRNAs in inflammatory responses remain unclear. Therefore, this review aims to provide new insights into the role of circRNAs in modulating neuroinflammatory responses in IS to enhance our understanding of its severity and prognosis.
Currently, excitotoxicity, calcium dysregulation, oxidative and nitrosative stress, cortical spreading depolarizations, and inflammation are used to explain the pathophysiology of IS [19, 20]. Ischemic neuroinflammation is a pathological process of IS that significantly affects clinical prognosis [21]. Notably, cytokines, secreted by different brain immune cells, are key players in the inflammatory response, exhibiting both proinflammatory and anti-inflammatory effects [22, 23]. Fig. 1 shows the inflammatory response of immune cells.
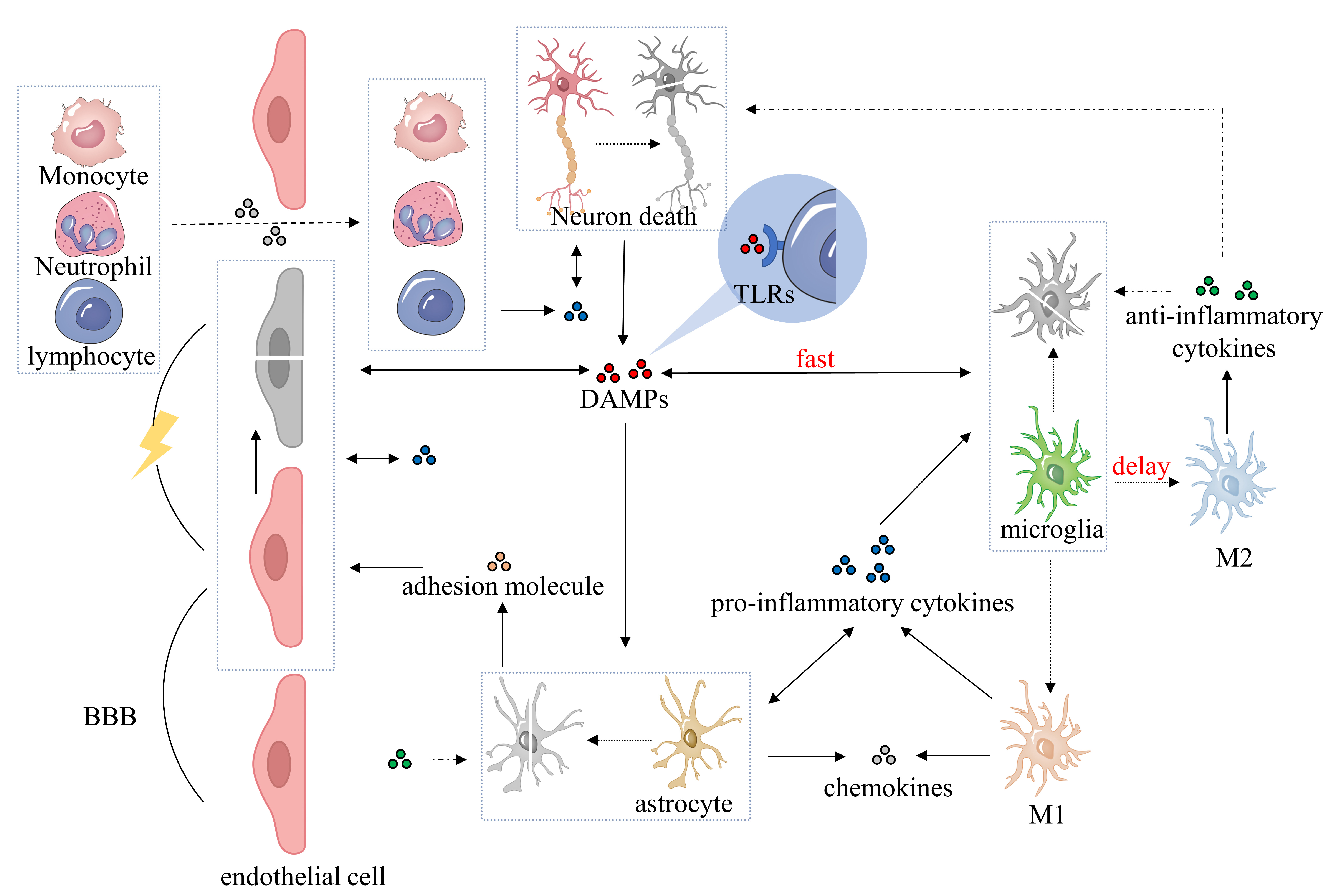 Fig. 1.
Fig. 1.Pathophysiological process of neuroinflammatory response after ischemic stroke. After ischemic brain injury, DAMPs are released during cell ischemia, hypoxia, and necrosis, promoting the activation of microglia, astrocytes, brain endothelial cells, and perivascular immune cells. Microglia polarize into classical (M1) or alternative (M2) microglia and secrete proinflammatory cytokines, anti-inflammatory cytokines, adhesion molecules, and chemokines. Activated astrocytes initially secrete proinflammatory cytokines, followed by anti-inflammatory cytokines, chemokines, and adhesion molecules. Proinflammatory cytokines contribute to the BBB breakdown and exacerbate the inflammatory response, while anti-inflammatory cytokines counteract this process and play a neuroprotective role. Adhesion molecules and chemokines facilitate the infiltration of peripheral immune cells into the BBB and promote the inflammatory response. DAMPs, damage-associated molecular patterns; BBB, blood-brain barrier; TLRs, Toll-like receptors; M1, classical microglia; M2, alternative microglia.
After ischemic brain injury, ischemia-anoxic necrosis cells release a series of danger signals commonly called damage-associated molecular patterns (DAMPs), such as the high-mobility group box 1 protein (HMGB1) [24, 25, 26]. These DAMPs are released into the extracellular environment to activate the innate immune system and further stimulate the inflammatory cascade reaction [27]. Toll-like receptors (TLRs), located on microglia, astrocytes, perivascular macrophages, and brain endothelial cells, detect DAMPs and activate corresponding cells in response [28, 29, 30, 31].
Microglia, the brain’s most predominant resident macrophages, account for
approximately 10% of all brain tissue cells that become highly activated early
after stroke [32, 33, 34]. After activation, microglia polarize into two types,
classical (M1) and alternative (M2) microglia, which play different roles,
secreting cytokines that promote or relieve inflammation and affecting tissue
repair [35, 36]. This process increases microglial phagocytosis activity [37].
Currently, microglial cells secrete a variety of inflammatory cytokines and
chemokines, including interleukin(IL)-1
In the central nervous system, astrocytes are the main type of glial cells,
accounting for approximately 50% of brain cells. In addition to playing a
supporting role, astrocytes are important regulatory factors of brain function
[41, 42, 43]. When activated by proinflammatory factors secreted by activated
microglia, astrocytes lose their neuroprotective and homeostatic ability and
induce neuron and oligodendrocyte death [44]. After brain tissue ischemia,
astrocytes reduce brain nerve damage by producing antioxidants and glutamine and
regulating extracellular potassium concentration [45]. Activated astrocytes
secrete large amounts of inflammatory cytokines and chemokines, such as IL-1,
TNF-
Neutrophils are one of the peripheral immune cells. Neutrophils also release cytokines that promote glial scarring, phagocytic debris, and edema resolution [54]. Following BBB damage, neutrophils penetrate the brain through the action of cell adhesion molecules, including selectin, integrin, and immunoglobulin [55]. Neutrophils have a strong ability to destroy brain tissue, as they generate reactive oxygen species (ROS) and proteolytic enzymes, inducing direct neurotoxic effects. Additionally, their accumulation indirectly leads to cerebral ischemia and hypoxia by blocking capillary blood flow [56, 57].
Another peripheral cells, (such as mononuclear phagocytes, natural killer cells (NK cells), and lymphocytes) also release cytokines and participate in ischemic inflammation [23, 58]. Monocytes are innate immune cells that serve as important effectors and regulatory factors in inflammation [59], crossing the BBB, accumulating in damaged brain tissue, and exhibiting microglia-like phenotypic changes [60, 61]. NK cells are important innate immune cells in peripheral blood that mobilize quickly in the initial stage of inflammatory responses, thereby promoting neuroinflammatory and inducing neuronal death [62].
A growing body of literature has begun to elucidate that lymphocytes can assume
in IS pathology. Lymphocytes cross the BBB and enter ischemic brain parenchyma
under the influence of P-selectin, VCAM-1, and ICAM-1 signaling [63]. The degree
of lymphocyte infiltration depends on changes in BBB permeability [64]. Immune
system activation mediated by lymphocyte infiltration can have both harmful
antigen-specific autoreactive responses and cellular protection in IS [65].
During this process, helper T cells are divided into Th1 cells secreting
proinflammatory cytokines (such as IFN-
Brain ischemia triggers a surge in inflammatory cytokine production by various
cell types, including microglia, astrocytes, endothelial cells, and neurons, with
contrasting outcomes. Proinflammatory cytokines (such as TNF-
| Classification | Designation | Produced by | Roles in inflammatory response | References |
| Pro-inflammatory cytokines | TNF- |
Neurons, microglia, astrocytes, monocytes, Th1 cells, and endothelial cells | Combine TNF receptors, form complex1, active caspase, and induce apoptosis | [22, 75, 76, 77] |
| Upregulate nuclear factor kappa-B (NF- |
||||
| Activate microglia and astrocytes and increase infarct volume | ||||
| Increase chemokines and adhesion molecules (ICAM and VCAM), promote coagulation and BBB breakdown, promote neutrophil infiltration | ||||
| Enhance the toxic effects of IL-1 | ||||
| IL-1 |
Neurons, microglia, astrocytes, monocytes, and endothelial cells | Increase NMDAR function and Ca |
[55, 78, 79, 80, 81] | |
| Promote the expression of adhesion molecules (ICAM-1, ICAM-2, P-selectin, E-selectin, and VCAM-1) and induce BBB breakdown | ||||
| Activate lipoprotein-associated phospholipase 2 (LP-A2), induce apoptosis, release metabolites (such as prostaglandins and leukotrienes), induce BBB breakdown, and promote leukocyte infiltration | ||||
| Activate microglia and astrocytes and increase IL-1, IL-6, TNF- |
||||
| Upregulate NF- |
||||
| IL-12 | Microglia, monocytes, NK cells, and T cells | Induce inflammatory T cell and increase infarct volume | [8, 82] | |
| IL-15 | Neurons, microglia, macrophages, astrocytes, and endothelial cells | Promote the migration of T cells | [83, 84, 85] | |
| Activate NK cells and increase infarct volumes | ||||
| IL-17 | Neurons, microglia, astrocytes, monocytes, endothelial cells, Th17 cells, |
Upregulate NF- |
[86] | |
| IL-21 | Th17 cells, B cells, and NK cells | Activate the janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway | [87, 88] | |
| Increase infarct size and lymphocyte accumulation | ||||
| IL-23 | Monocytes, microglia, and astrocytes | Promote T-cell activation to produce IL-17 | [89] | |
| IFN- |
Astrocyte, ( |
Increase the expression of IP-10 and infarct size | [69, 90] | |
| Anti-inflammatory cytokines | IL-4 | Th2 cells, microglia, and monocytes | Polarize microglia, promote angiogenesis, alleviate ischemic stroke | [91, 92] |
| Downregulate the expression of TNF- |
||||
| IL-6 | Neurons, microglia, astrocytes, monocytes, endothelial cells, lymphocytes, T cells, B cells, and neutrophils | Promote tissue recovery, reduce infarct severity, induce neutrophil apoptosis, inhibit the expression of TNF- |
[93, 94, 95, 96] | |
| IL-10 | Neurons, microglia, astrocytes, monocytes, endothelial cells, Th cells, Treg cells, and B cells | Reduce infarct size and inhibit cell apoptosis | [97, 98] | |
| Increase nerve growth factor (NGF) and l-glutathione (GSH) and decrease IL-1 |
||||
| IL-22 | Th17 cells | Activate the JAK2/STAT3 pathway | [99] | |
| Reduce infarct size and inhibit cell apoptosis | ||||
| Decrease IL-1 |
||||
| TGF- |
Microglia, astrocytes, and macrophages | Promote glial scar formation and microglia polarizing | [100, 101] | |
| Inhibit apoptosis, counteract excitotoxic neuronal loss, and reduce infarct size and neuroinflammation | ||||
| IGF-1 | Neurons, astrocytes, endothelial cells, and macrophages | Promote microglia polarizing and decrease IL-1 |
[102] | |
| Maintain astrocyte glutamate uptake and glucose homeostasis | ||||
| Reduce neurogenic death | ||||
| Chemokines | IL-8 | Microglia, monocytes, endothelial cells, and T cells | Induce neutrophil adhesion and migration | [103, 104] |
| MIF | Astrocyte | Promote leukocyte recruitment and thrombus formation | [105] | |
| MIP-1 |
Microglia | Promote macrophage and neutrophil migration, increase inflammatory mediators, and increase infarct size | [106] | |
| MCP-1 | Neurons, microglia, macrophages, astrocytes, and endothelial cells | Induce BBB breakdown, promote macrophage migration | [106] | |
| Increase infarct size | ||||
| IP-10 | Neurons, microglia, astrocytes, monocytes, endothelial cells, and NK cells | Induce NK cell accumulation and BBB breakdown | [107] | |
| Free oxygen radicals | ROS | Neurons, microglia, neutrophil, and astrocytes | Activate the NF-ĸ |
[108, 109] |
| NO | Neurons, macrophages, astrocytes, microglia, and endothelial cells | Induce mitochondrial dysfunction and energy depletion, promote angiogenesis, and increase infarct size | [110, 111, 112] | |
| Produce neurotoxic effect and promote inflammation, cell death, and BBB damage | ||||
| iNOS | Endothelial cells, astrocytes, microglia, and neutrophils | Promote NO production and vasodilation, and increase infarct volume | [113, 114] | |
| Protease | MMPs (MMP-3/9) | Neurons, microglia, astrocytes, neutrophil, endothelial cells, and macrophages | Induce BBB breakdown and neuronal apoptosis | [115, 116] |
| Promote leukocyte adherence and transmigration | ||||
| Induce cerebral edema and hemorrhagic complications | ||||
| Increase infarct size | ||||
| Adhesion molecules | ICAMs (ICAM-1, ICAM-4) | Microglia, astrocytes, macrophages, endothelial cells, and leukocytes | Promote neutrophils’ entry into the BBB | [117, 118] |
| VCAM-1 | Microglia, astrocytes, macrophages, endothelial cells, and leukocytes | Promote macrophages’ entry into the BBB and neovascularization | [119, 120] | |
| P-selectin | Microglia, astrocytes, macrophages, endothelial cells, and leukocytes | Promote leukocyte adhesion and migration | [121] | |
| E-selectin | Microglia, astrocytes, macrophages, endothelial cells, and leukocytes | Induce upregulation of endothelial cell adhesion molecules | [122, 123, 124] | |
| Promote leukocyte adhesion and migration |
TNF-
Previous studies have associated differences in the production of inflammatory to infarct size following cerebral ischemia. Proinflammatory factors can increase cell edema and death, while the expression of anti-inflammatory factors can partially alleviate nerve damage [125, 126]. IL-1 is widely produced in nerve cells, microglia, and astrocytes after brain tissue ischemia. It is an important proinflammatory cytokine in the inflammatory response [55]. IL-1 can promote neuronal death by inducing calcium ions to enter cells by acting on N-Methyl-D-aspartic acid receptors (NMDARs) [78]. IL-1 also activates phospholipase A2, disrupting cell phospholipase bilayers, inducing apoptosis, and promoting the release of metabolites such as prostaglandins and leukotrienes, which can induce BBB breakdown and promote inflammatory response [93, 127]. Furthermore, IL-1 induces BBB breakdown and promotes leukocyte infiltration through adhesion molecules on endothelial cells [55, 128, 129, 130]. Adhesion molecules act as ligands involved in leukocyte chemotaxis and inflammatory responses by binding to leukocyte integrin receptors attached to endothelial cells [20]. Moreover, inflammatory stimulation results in a significant upregulation of ICAM-1 and animal studies have shown that artificial intervention with IL-1 receptor antagonists can reduce cerebral infarct size [131, 132, 133].
IL-6 is a proinflammatory cytokine that is highly expressed after a stroke event
[134] and is associated with brain tissue necrosis and poor clinical prognosis
[55, 94, 135]. However, there is another view that IL-6 has an anti-inflammatory
effect by promoting tissue recovery and reducing infarct severity [136, 137, 138].
Currently, the known anti-inflammatory action mechanisms include inhibiting
inflammatory factors, inhibiting TNF-
TNF-
IL-10 is an anti-inflammatory cytokine widely present in astrocytes, mononuclear macrophages, Th cells, and B cells, and animal experiments have demonstrated that mice with IL-10 overexpression have smaller infarct size and limited apoptosis [97]. Previous research showed that IL-10 can reduce infarct size after focal ischemia, reduce leukocyte and monocyte/macrophage infiltration, and mitigate nerve damage caused by inflammatory cytokines [72, 147, 148].
TGF-
Nuclear factor kappa-B (NF-
MMPs are a family of proteolytic enzymes which can induce BBB breakdown [162]. During the acute phase of IS, MMP expression increases [163, 164]. MMPs are found in microglia, astrocytes, and endothelial cells. Moreover, MMPs activate microglia, macrophages, and neutrophils, further promoting MMP secretion and leading to vascular and BBB damage [165]. Experimental studies have shown that silencing MMP-Ⅸ and MMP-Ⅲ gene expression significantly alleviated BBB damage and edema [166, 167].
CircRNAs are non-coding RNA molecules that form covalent ring structures without
5
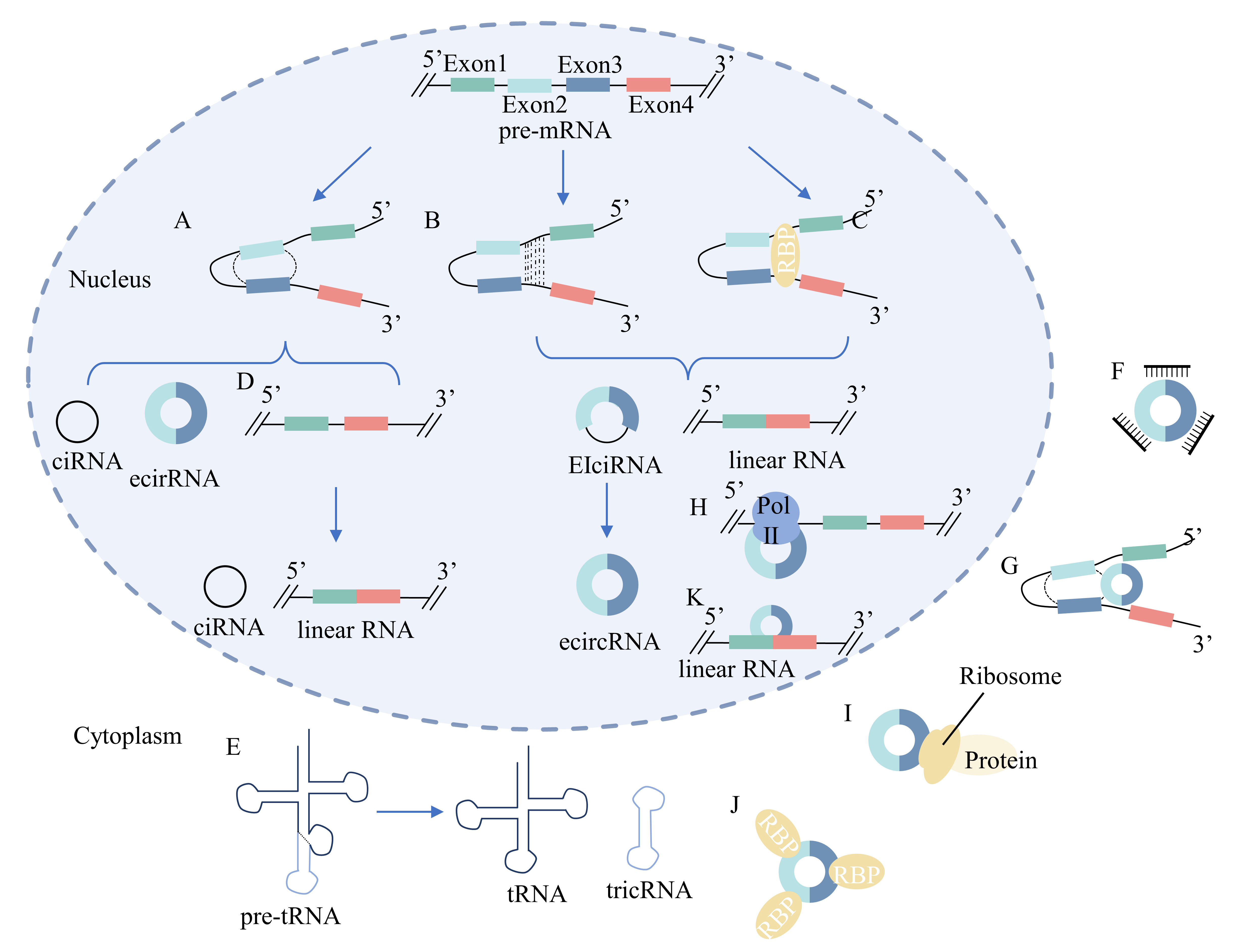 Fig. 2.
Fig. 2.Biogenesis and function of circRNAs. (A) Lariat-driven
circularization model: the 3
CircRNAs can be categorized into four groups based on their biogenesis: exonic circRNAs (ecircRNAs), synthesized from only the exon sequence, circular intronic RNAs (ciRNAs) containing introns, exon-intron circRNAs (EIciRNAs) containing both exon and intron sequences, and tRNA intronic circular RNAs (tricRNAs) [10, 173, 193, 194, 195]. The biogenesis of circRNAs is illustrated in Fig. 2.
The earliest study on circRNA expression profiles was published in STROKE in 2017 by Mehta et al. [196]. Using a transient middle cerebral artery occlusion (tMCAO) mice model, they detected 1320 circRNAs from exonic gene regions in the ischemic penumbral cortex. Among these, 283 were significantly altered after tMCAO compared to controls. This abnormal circRNA expression may play a potential pathophysiological role in the ischemic brain tissues of mice with tMCAO. In another study, Duan et al. [197] identified 14,694 differentially expressed circRNAs in MCAO rat brain tissues through high-throughput sequencing and quantitative reverse transcription-polymerase chain reaction (RT-PCR), with 87 differentially expressed circRNAs displaying significant fold changes. Moreover, Liu et al. [198] used RNA extraction and microarray assays to analyze circRNA expression in reperfused mice brain tissues after transient ischemia, revealing differential regulation of 1027 circRNAs, with 914 upregulated and 113 downregulated. These studies suggest the presence of abnormal circRNA expression in ischemic brain tissue, with potential pathophysiological implications after MCAO.
In addition to the ischemic core and penumbra, significantly expressed circRNAs were also identified in non-ischemic areas and peripheral blood from an MCAO mouse model. In one experiment, focal cortical ischemia was induced in adult male distal MCAO mice, and circRNA expression in the ipsilateral non-ischemic thalamic regions was measured using high throughput sequencing. The results revealed 2659 circRNAs with significant alterations in the non-infarct area of the ipsilateral hemisphere [199]. Another experiment validated the differential expression of blood circRNAs in tMCAO mice, confirming their role after IS. The result showed 10739 highly expressed circRNAs, and their expression across different time points showed no significant difference, indicating the non-random nature of circRNA alterations. Most circRNAs detected in blood were also present in the brain. Therefore, circRNAs may play potential functional roles after IS, and circRNAs in the blood may have the same origin as circRNAs in the ischemic brain [200]. Peripheral inflammatory cells possibly pass the BBB through chemotaxis and reach the brain tissue at the ischemic penumbra to exert an inflammatory response.
Recently, with advances in clinical trials, the relationship between circRNAs and IS has become increasingly evident. Clinical trial results are highly consistent with those of animal trials.
In a study by Dong Z et al. [201] in 2020 examining circRNA expression in
stroke, the expression profile of 164532 circRNAs was generated in peripheral
blood mononuclear cells (PBMCs) from patients with acute ischemic stroke (AIS)
and controls using high-throughput sequencing technology. Among these, 521
circRNAs were differentially expressed, with 373 upregulated and 148
downregulated. The study demonstrated significant differences in circRNAs in
PBMCs between patients with AIS and controls. Additionally, this study indicated
that circRNAs participated in the inflammatory pathways (such as the
NF-
| Pathological process | Cell line/treatment | Animal/disease model | CircRNAs and expression | Regulating axis | Overexpression role | References | |
| Angiogenesis | HBMECs | / | CircFUNDC1 | UP | miR-375/PTEN | Inhibit angiogenesis | [202] |
| OGD-treated | Detrimental | ||||||
| HBMECs/ | tMCAO | CircPDS5B | UP | hnRNPL/VEGFA | Inhibit angiogenesis | [203] | |
| OGD-treated | Mouse | Detrimental | |||||
| / | tMCAO | Circ-DLGAP4 | DOWN | miR-143/hectord1/EndoMT | Decrease BBB damage | [204] | |
| Mouse | Protective | ||||||
| Apoptosis | HT22 | tMCAO | CircCDC14A | UP | miR-23a-3p/CXCL12 | Induce apoptosis | [205, 206] |
| OGD/R | Mice | Detrimental | |||||
| astrocyte | tMCAO | Circ-CELF1 | UP | DDX54/NFAT5 | Induce apoptosis | [207] | |
| OGD/R | Mouse | Detrimental | |||||
| HT22 | MCAO | Circ-HECTD1 | DOWN | miR-125b-5p/GDF11 | Inhibit apoptosis | [208] | |
| OGD | Mice | Protective | |||||
| Inflammation | / | tMCAO | CircPUM1 | UP | miR-340-5p/DDX5/NF- |
Inhibit inflammation | [209] |
| Mouse | Protective | ||||||
| microglia | / | CircPTK2 | UP | miR-29b-SOCS-1-JAK2/STAT3-IL-1 |
Induce inflammation | [210] | |
| OGD | Detrimental | ||||||
| microglia | tMCAO | Circ-0000831 | UP | miR-16-5p/AdipoR2 | Induce inflammation | [211] | |
| OGD | Mice | Detrimental | |||||
| HCN-2 | / | Circ-0007290 | UP | miR-496/PDCD4 | Induce inflammation | [212] | |
| OGD | Detrimental | ||||||
| HBMVECs | / | Circ-Memo1 | UP | miR-17-5p/SOS1 | Induce inflammation | [213] | |
| H/R | Detrimental | ||||||
| HBMECs | / | Circ-0006459 | UP | miR-940/FOXJ2 | Induce inflammation | [214] | |
| OGD | Detrimental | ||||||
| HBMECs | / | Circ-0000566 | UP | miR-18a-5p/ACVR2B | Induce inflammation | [215] | |
| OGD/R | Detrimental | ||||||
| SK-N-SH | / | Circ-0000647 | UP | miR-126-5p/TRAF3 | Induce inflammation | [216] | |
| OGD/R | Detrimental | ||||||
| SK-N-SH | tMCAO | Circ-0101874 | UP | miR-335-5p/PDE4D | Induce inflammation | [217] | |
| OGD | Mice | Detrimental | |||||
| HT22 | MCAO | Circ-HECTD1 | UP | miR-133b/TRAF3 | Induce inflammation | [208] | |
| OGD/R | Mouse | Detrimental | |||||
| HCN-2 | / | Circ-DLGAP4 | DOWN | miR-503-3p/NEGR1 | Inhibit inflammation | [218] | |
| OGD | Protective | ||||||
| astrocyte | MCAO | Circ-CTNNB1 | DOWN | miR-96-5p/SRB1 | Inhibit inflammation | [219] | |
| OGD/R | Mouse | Protective | |||||
HBMECs, human brain microvascular endothelial cells; OGD, oxygen-glucose
deprivation; HT22, hippocampal neuronal cell line; HCN-2, cortical neuron cell
line; HBMVECs, human brain microvascular endothelial cells; H/R,
hypoxia/reoxygenation; SK-N-SH, human neuroblastoma cell line; tMCAO, transient
middle cerebral artery occlusion; PTEN, phosphatase and tensin homolog;
hnRNPL/VEGFA, heterogenous nuclear ribonucleoprotein L /vascular endothelial
growth factor-A; EndoMT, endothelial-mesenchymal transition; CXCL12, C-X-C motif
chemokine ligand-12; DDX54, DEAD-box helicase 54; NFAT5, nuclear factor of
activated T cell 5; GDF11, growth differentiation factor 11; NF-
4.1.2.1 CircFUNDC1, circPDS5B, and circCDC14A
Another study exploring the role of peripheral blood circRNAs in IS found that among the 10798 circRNAs analyzed in human blood, 68 downregulated and 10 upregulated circRNAs differed significantly between groups. Moreover, hsa_circ_0007290 (circFUNDC1), hsa_circ_0004494 (circPDS5B), and hsa_circ_0000097 (circCDC14A) levels were positively correlated with cerebral infarct volume. circPDS5B levels showed significantly higher expression in peripheral blood lymphocytes, granulocytes, and plasma in patients with stroke, whereas circCDC14A was significantly upregulated in granulocytes [220]. Bai et al. [202] demonstrated the role of circFUNDC1 in human IS and oxygen-glucose deprivation (OGD)-treated cell models. In both model types, circFUNDC1 was upregulated, resulting in low miR-375 levels. CircFUNDC1 knockdown and miR-375 overexpression enhanced the angiogenesis ability of human brain microvascular endothelial cells (HBMECs) by regulating phosphatase and tensin homolog (PTEN) expression. Previous research has shown that PTEN can regulate BBB permeability and neurogenesis [219, 221]. Jiang et al. [203] found that circPDSB5 was also upregulated in the serum of patients with IS and tMCAO mice. Moreover, circPDS5B knockdown alleviated cell necrosis and provided cerebrovascular protection via the heterogenous nuclear ribonucleoprotein L/vascular endothelial growth factor-A (hnRNPL/VEGFA) pathway. Zuo et al. [205] noted the high expression of circCDC14A in peripheral plasma, neutrophils, pericerebral infarction cortex, and astrocytes. Reducing circCDC14A levels in peripheral blood immune cells alleviated astrocyte activation in the periinfarction cortex, thus alleviating brain injury in AIS. This may be because peripheral neutrophils infiltrated ischemic brain tissue through the BBB. Additionally, Huo et al. [206] observed that circCDC14A positively modulated C-X-C motif chemokine ligand-12 (CXCL12), inducing neuronal damage and apoptosis via miR-23a-3p in tMCAO mice.
4.1.2.2 CircDLGAP4
In 2019, Zhu et al. [222] identified a correlation between has_circ_0060180
(circDLGAP4) expression and the inflammatory cascade level in patients with AIS.
CircDLGAP4 was down-regulated in PBMCs in patients with AIS and negatively
associated with miR-143 expression. circDLGAP4 downregulation and miR-143
upregulation increased serum inflammatory cytokines including IL-6, IL-8, IL-22,
and TNF-
Hu et al. [209] observed significant upregulation of hsa_circ_0000043
(circPUM1) and DEAD-box helicase 5 (DDX5) and notable miR-340-5p downregulation
in a tMCAO mouse model [209]. Abnormally low circPUM1 expression promoted the
secretion of inflammatory factors and induced apoptosis by activating microglia
and astrocytes through the miR-340-5p/DDX5/ NF-
In 2019, Wang et al. [210] found that hsa_circ_0005273 (circPTK2) regulates
neuronal apoptosis by activating microglia via miR-29b-suppressor of cytokine
signaling 1-Janus kinase 2/signal transducer and activator of transcription
3-interleukin-1
It has been reported that circ-0000831 mitigated inflammatory and apoptotic
responses in MCAO mice models, and circ-0000831 was upregulated in microglia
after OGD. circ-0000831 acted as a sponge for miR-16-5p, causing its
downregulation. Adiponectin receptor 2 (AdipoR2), a target for miR-16-5p, was upregulated and increased
peroxisome proliferator-activated receptor
Li et al. [207] observed that highly expressed circCELF1 in oxygen-glucose deprivation/reoxygenation (OGD/R) astrocytes could exacerbate astrocyte damage, and its expression is associated with inflammatory response. CircRNA hsa_circ_0000304 (circCELF1) knockdown decreased the expression of proinflammatory cytokines in astrocytes after OGD/R. By binding to the RNA binding protein DEAD-box helicase 54 (DDX54), circCELF1 enhances the stability of the nuclear factor of activated T cell 5 (NFAT5) mRNA. In another study, increased NFAT5 expression induced neuroinflammation after IS [236]. NFAT5 overexpression can restore the decline in inflammatory factor levels caused by circCELF1 knockdown [207].
Recently, circRNA catenin beta 1 (circCTNNB1) was found to play a role in the
inflammatory response in astrocytes. CircCTNNB1 and IL-10 levels were
downregulated, whereas the expression of proinflammatory factors, including
IL-1
Vascular endothelial cells, an important part of the BBB, were widely implicated
in the inflammatory response after IS. Ren et al. [213] found that circ-Memo1
and Son of Sevenless 1 (SOS1) levels were highly expressed; however, miR-17-5p expression was low in
the peripheral blood of patients with IS. After circMemo1 knockdown, the
expression of proinflammatory cytokines was low in the human brain microvascular
endothelial cell (HBMVEC) model. CircMemo1 can directly bind with miR-17-5p,
negatively regulating miR-17-5p levels, which could inhibit SOS1 expression. Cerebral ischemia stimulates SOS1 activity and disrupts
Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) phosphorylation downstream of
SOS1 [238]. Moreover, SOS1 can activate the NF-
Li et al. [214] investigated the inflammatory response of circ-0006459 in
OGD-induced HBMEC cell models. By sponging miR-940, circ-0006459
overexpression promoted proinflammatory factor levels (including IL-1
In a HBMECs model article, it has been discovered that circ-0000566 was highly expressed after OGD [244]. Another study found that circ-0000566 was upregulated after OGD/R, while miR-18a-5p was significantly downregulated. Moreover, Circ-0000566 acted as a miR-18a-5p sponge, contributing to HBMEC injury and proinflammatory cytokine release [215]. Activin receptor type IIB (ACVR2B) directly targeted miR-18a-5p, causing its upregulation and involvement in the smad2/c-jun axis in neuronal damage after cerebral ischemia in mice [245].
In 2022, Dai et al. [216] used human neuroblastoma cell line (SK-N-SH)
cells to simulate the cerebral ischemia environment and found elevated
circ-0000647 levels, with circ-0000647 acting as a miRNA sponge to bind
miR-126-5p and decrease miR-126-5p expression [216]. Circ-0000647 upregulation
and miR-126-5pc downregulation reduced the expression of TNF-
In this review, the involvement of circRNA in IS by influencing inflammatory
factors has been summarized. Pei et al. [217] observed that circ-0101874 was
upregulated in both MCAO mice and OGD-induced cell models. Circ-0101874
knockdown suppressed TNF-
The role of circular RNA HECT domain E3 ubiquitin-protein ligase 1 (circHECTD1)
in mouse MCAO and OGD cell models has been found in 2021. CircHECTD1 expression
was upregulated, and its interaction with miR-133b reduced its expression. TNF
receptor-associated factor 3, the target for miR-133b, was highly expressed and
promoted the expression of inflammatory cytokines such as NF-
In another cell model, hsa_circ_001,357 (circUCK2) inhibited miR-125b-5p
expression by acting as a miRNA sponge and increased the expression of growth
differentiation factor 11 (GDF11) [246]. GDF11 is a member of the TGF-
Wang et al. [212] identified the inflammatory effects of circ-0007290
expression in both in vitro and in vivo models. Circ-0007290 expression was
elevated, and knockdown circ-0007290 knockdown reduced the inflammatory response
after OGD. Circ-0007290 acted as a sponge to mitigate miR-496 inhibition of its
target, programmed cell death protein 4. Moreover, circ-0007290 inhibition,
miR-496 overexpression, or programmed cell death protein 4 upregulation induced
proinflammatory factor (such as TNF-
IS, a condition causing cerebral tissue death and neuronal damage, still has a high morbidity and mortality rate [249]. Inflammation is an important pathological reaction in AIS, and circRNAs may play critical roles in it [250, 251]. CircRNAs are not only potential novel biomarkers but also vital for therapeutic targets for the diagnosis and treatment of IS [252]. After IS, circRNAs are differentially expressed in neurons and inflammatory cells affecting the release of inflammatory mediators. Among them, circRNAs mainly act as sponges to combined with miRNAs so that inhibit the effect of miRNA and increase the expression level of target genes. However, only once study has focused on the function of forming circRNP complexes. Furthermore, circRNAs exhibit diverse functions through multiple pathways. As research progresses, the distinct mechanisms of regulation can be better elucidated, thereby enhancing our understanding of the inflammatory regulatory network involving circRNAs.
Currently, the potential of utilizing circRNAs as targets for early diagnosis and post-treatment analysis is yet to be determined. However, it can be concluded that circRNA is widely involved in the activation of various inflammatory cells, the secretion of inflammatory mediators, and the destruction of the BBB. By modulating circRNAs, it is possible to change the extent of inflammatory response. The regulatory mechanisms of inflammation are incredibly intricate, and the clinical application still needs to face many challenges. Moreover, because of the different etiology of the IS subtypes (such as atherothrombotic IS, cardiac cerebral embolism and so on), it’s difficult to explain the inflammatory response in the development of IS [253]. However, the ultimate result of various etiological subtypes is to lead to vascular occlusion, and the inflammatory factors and inflammatory cells involved in the process are the same. Therefore, it is meaningful to use the cell model of oxygen-glucose deprivation and the mouse model of MCAO to study the inflammatory response after IS.
Due to the complexity of etiological subtypes and the difficulty of model building, there are few studies on the involvement of circRNA in etiology. It is great significance to explore the role of circRNA in the pathogenesis of different subtypes of IS. With the advancement of technology, the establishment of more appropriate and realistic models should lead to more perfect results.
In this review, we described the inflammatory response after IS and summerized the roles of circRNAs in IS. In all, the immune response in IS is quite complex and circRNAs show dual roles to regulate the neuro-inflammation. Restricting the dentrimental neuroinflammation might be helpful to decrease ischemic volume. CircRNA light a new potential treatment target for IS. In order to determine the effective therapeutic strategy, is important to investigate the mechanisms between circRNA and other inflammatory factors.
BBB, Blood-brain barrier; circRNA, circular RNA; DAMPs, damage associated
molecular patterns; HMGB1, high-mobility group box 1 protein; TLRs, Toll-like
receptors; TNF-
LJ, AWS and CGY proposed this research direction. XG, BC, YYZ, XQL, HQS and ZZ conceptualized, wrote, and edited the manuscript. XG, YJZ, KQL, WQX, LFL, JYH and BBG designed the figures and tables, drafted and edited the manuscript. LFL, KQL, JYH, BBG and CGY helped in revision and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work is supported by the Hangzhou Science and Technology Bureau (2021WJCY009), Hangzhou Medical and Health Technology Project (0020190131), Zhejiang Provincial Public Welfare (LGF18H090017) and Zhejiang Provincial Natural Science Foundation of China (LQ21H090005).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.