

1 Department of Experimental and Clinical Physiology, Laboratory of Centre for Preclinical Research, Medical University of Warsaw, 02-097 Warsaw, Poland
Abstract
Cardiac pain is an index of cardiac ischemia that helps the detection of cardiac hypoxia and adjustment of activity in the sufferer. Drivers and thresholds of cardiac pain markedly differ in different subjects and can oscillate in the same individual, showing a distinct circadian rhythmicity and clinical picture. In patients with syndrome X or silent ischemia, cardiac pain intensity may cause neurogenic stress that potentiates the cardiac work and intensifies the cardiac hypoxia and discomfort of the patient. The reasons for individual differences in cardiac pain sensation are not fully understood. Thus far, most attention has been focused on inappropriate regulation of the heart by the autonomic nervous system, autacoids, and cardiovascular hormones. Herein, we summarize evidence showing that the autonomic nervous system regulates cardiac pain sensation in cooperation with vasopressin (AVP). AVP is an essential analgesic compound and it exerts its antinociceptive function through actions in the brain (the periaqueductal gray, caudate nucleus, nucleus raphe magnus), spinal cord, and heart and coronary vessels. Vasopressin acts directly by means of V1 and V2 receptors as well as through multiple interactions with the autonomic nervous system and cardiovascular hormones, in particular, angiotensin II and endothelin. The pain regulatory effects of the autonomic nervous system and vasopressin are significantly impaired in cardiovascular diseases.
Keywords
- pain
- cardiac hypoxia
- brain
- spinal cord
- myocardial infarction
- autacoids
- angiotensin
- endothelin
- vaptans
Strong cardiac pain often precedes myocardial infarction and may be a useful warning sign of cardiac morbidity, allowing the adjustment of activity in the suffer in order to reduce the rate of oxygenation and decrease the performance of the heart. In some cardiac patients, pain intensity is not proportional to the ischemia itself and can provoke excessive neurogenic stress, which potentiates cardiovascular responses and cardiac work, thereby intensifying cardiac hypoxia and patient discomfort. For many years, relieving pain and anxiety has been a major goal of cardiovascular therapy [1, 2, 3]. Experimental and clinical studies have shown significant differences in individuals’ subjective experience of cardiac pain. The causes for this variety in cardiac pain sensations are currently not fully understood; however, the influence of age, sex and neuroendocrine and inflammatory factors have been frequently addressed [4, 5, 6, 7, 8].
Particular attention has been given to cardiac syndrome X (CSX) and to silent ischemia. Patients with CSX experience typical chest pain although they have normal arteriogram. It has been postulated that CSX is caused by abnormal function of cardiac microvessels [9, 10]. There is also evidence for inappropriate perception and regulation of pain [8, 11, 12], and for an altered processing of pain signals in the brain, which may result in a decreased pain threshold and lack of habituation to pain stimuli [13, 14].
The mechanisms responsible for silent ischemia, in which typical ambulatory symptoms of myocardial infarction occur in the absence of pain, are not yet fully understood; however, inappropriate activation of the autonomic nervous system has been postulated [15, 16].
Growing evidence indicates that pain stimulates the release of several cardiovascular compounds that may play a role in the modulation of pain. One of these compounds is arginine vasopressin (AVP), which operates as a member of the complex vasopressinergic system (VS), for which receptors are located in the brain and peripheral organs. The purpose of this review is to summarize the current knowledge of the role of AVP in the regulation of pain in cardiac pathology. Central and systemic cooperation of AVP with the autonomic nervous system and other cardiovascular peptides is also discussed.
It is widely known that the heart receives afferent and efferent innervation from the autonomic nervous system and that heart failure significantly alters the effectiveness of the sympathetic and parasympathetic control of the heart [17, 18, 19, 20]. Studies on patients with severe angina pectoris have shown that spinal cord stimulation exerts beneficial antianginal effects and decreases ischemia, presumably due to the reduction of myocardial ischemia [21].
Transmission of pain signals from the heart to the brain. It has been shown that sensory innervation of the heart is supplied by both sympathetic and parasympathetic neurons [19, 20]. Convergence of stimuli from the somatic, nociceptive and spinothalamic tract neurons has been found in the C1-C3 segments of the spinal cord [22] that are involved in sensation of referred pain in the chest, arm, and diaphragm [22, 23, 24, 25]. Nociceptive information from the heart is transmitted to the dorsal root ganglia (DRG) and subsequently it ascends the insular cortex and the nucleus tractus solitaries (NTS) that receive input from the cardiac vagal afferents and pain processing neurons [26, 27, 28, 29]. In patients with coronary disease, stimulation of vagal afferents significantly reduces sympathetic tone and angina, and improves hemodynamic and electrocardiogram (ECG) parameters [30].
In patients with variant angina, the coronary vasospasm is preceded by stimulation of both sympathetic and parasympathetic activity [30, 31]. The sensory fibers transmitting nociceptive signals induced by cardiac ischemia have been identified as vagal afferent fibers projecting directly to the NTS, and as sympathetic afferents [17, 18].
Processing of pain signals in the brain. Several groups of neurons in the brain may participate in integration of pain signals and in modulation of cardiovascular responses. Emotional and other psychological signs modulate cardiac nociception through signals originating in the amygdala, raphe nucleus, and pons [17, 32, 33, 34].
Studies have shown that pericardial application of pain stimulating compounds or occlusion of the left anterior descending coronary artery induce activation of the caudal division of the NTS neurons [35], and that the NTS is involved in transmission of pain signals generated by chronic myocardial infarction [26]. There is also evidence for involvement of the periaqueductal grey (PAG) in the regulation of the autonomic nervous system and modulation of pain in healthy human subjects in whom brain function was investigated using resting state functional magnetic resonance imaging (fMRI) and voxel-based morphometry [36].
Cardiac pain has been also tested in patients with heart transplants. Heart transplantation is followed by sympathetic reinnervation of the heart; however, the process is slow and presumably not complete [37, 38]. Thus far there is no evidence for parasympathetic re-innervation. Presence of chest pain suggesting cardiac ischemia in recipients of heart transplants indicates the possibility of sensory reinnervation [31, 39].
Cardiac ischemic pain involves components of inflammatory pain, which is triggered by inflammatory mediators released from the injured tissue. The significance of inflammatory pain is especially important in myocardial infarction during which the inflammation is associated with tissue hypoxia, acidification, and release of several autacoids [40, 41, 42]. The activation of microglia plays an essential role in the generation of inflammatory pain. Studies on Sprague Dawley (SD) rats have shown that cardiac pain produced by chronic occlusion of the coronary artery can be alleviated by intrathecal administration of minocycline, which is an inhibitor of microglia. It has been shown that this effect can be abolished by intrathecal application of fractalkine, which is an antagonist of minocycline [43].
Ischemic hypoxia is associated with myocardial acidification, which is caused mainly by the release of lactic acid and a significant decrease in pH. Myocardial ischemia lasting 5 minutes has been shown to reduce extracellular pH to ~7.0 and severe hypoxia can even reduce the pH to 6.7 [44, 45]. Experiments on cats showed that increased release of protons plays an essential role in the activation of sympathetic afferents [44]. Stimulation of cardiac sympathetic C afferents is associated with activation of the acid sensitive ion ASIC3 channel, which belongs to the family of degenerin/epithelial sodium channels [45]. It is likely that, during myocardial ischemia, this channel detects early changes in lactic acid and ATP [46], which belong to autacoids, and the latter acts on P2 (P2X) purinergic receptors [42, 47]. It has been shown that myocardial infarction significantly elevates expression of P2X3 purinergic receptor (P2X3R) mRNA in the stellate ganglion, and P2X2R and P2X3R mRNA and protein in the nodose ganglion [48, 49].
Several other autacoids are released in the ischemic heart and may participate in the stimulation of sensory afferents. Studies have shown that cardiac chemosensitive neurons can be activated by bradykinin, substance P (SP), leukotriens, lactate, adenosine, ATP, and potassium ions [17, 35, 50]. In patients with ischemic cardiac disease, atrial pacing inducing angina enhances the release of adenosine, bradykinin, and prostaglandin (PGI2) in the heart [18, 41, 51]. Bradykinin activates cardiac afferent fibers acting on kinin B2 receptors. Release of bradykinin in the heart is enhanced by ischemic preconditioning and causes stimulation of both ischemia-sensitive and ischemia-insensitive cardiac afferents. Signals generated by bradykinin in the heart are transmitted via cardiac sensory sympathetic afferents to the dorsal root ganglion [40]. Stimulation of cardiac nociceptors by bradykinin requires activation of transient receptor potential vanilloid 1 (TRPV1) channels and activation of the 1,4,5-triphospahte (IP3) pathway [52]. It is worth noting that, in the heart, B2 kinin receptors and TRPV1 are expressed on the same ischemia-sensitive sensory fibers [53, 54].
Ischemia-sensitive cardiac afferents projecting via the left sympathetic chain and rami communicantes of T2-T3 are stimulated by histamine, which acts through H1 receptors and subsequent activation of the Phospholipase C- Protein kinase C (PLC-PKC) intracellular pathway [55]. Cardiac ischemia also induces elevation of adenosine in the heart, while adenosine intensifies ischemic pain through activation of A1 receptors [56]. Studies on patients with stable angina have shown that intracoronary infusion of adenosine provokes cardiac pain, which is frequently associated with bradycardia [56, 57]. Autacoids are locally-acting compounds [42] and it is also likely that other interleukins, growth factors, cytokines, glycolipids, and lipids synthesized in the heart modulate cardiac pain and interact with AVP and other cardiovascular peptides. However, their role in this context has thus far not been investigated.
Vasopressin is a cardiovascular neuropeptide with a wide spectrum of actions exerted in the brain and peripheral organs [58, 59, 60, 61, 62, 63]. In the brain, neurons synthetizing AVP are present mainly in the supraoptic (SON), paraventricular (PVN), and suprachiasmatic (SCN) nucleus. Axons of these neurons project mainly to the posterior pituitary where they release their product to the blood. In addition, several axonal projections innervate brain regions encompassing the nucleus ambiguous, NTS, lateral habenular nucleus, nucleus basalis of Meynert, substantia nigra, ventral hippocampus, central gray, and spinal cord [58, 59, 60, 61]. Among them are the regions controlling both the cardiovascular system and pain, such as the noradrenergic/adrenergic A2/C2 region, nucleus ambiguous, dorsal vagal complex, and subnuclei of the solitary tract [64, 65, 66]. The parvocellular neurons of the PVN send projections to the spinal cord [66, 67]. Secretion of vasopressin is stimulated by hyperosmolality, hyperthermia, hypotension, angiotensin II (Ang II), stress, pain, and inflammation. It also shows distinct diurnal rhythmicity [65, 68, 69, 70, 71, 72, 73]. Stress, pain, and inflammation play a particularly significant role in myocardial infarction and heart failure [73].
Vasopressin activates V1 receptors (V1R; subtypes: V1aR, V1bR) and V2 receptors (V2R) that are located in the brain areas involved in the regulation of pain and cardiovascular functions (Table 1). They are also present in several peripheral organs, including the cardiovascular system, lungs, kidney, liver, and gastrointestinal system [61, 74, 75, 76, 77, 78, 79, 80].
| Compound | Receptors involved in pain regulation | Site of action | Effect |
| Arginine vasopressin | V1aR, V1bR, V2R | Brain | Analgesia, increase of vagal tone |
| Sites of synthesis: brain (PVN, SON, ScN), peripheral organs | Spinal cord | Analgesia in low concentration, hyperalgesia in high concentration | |
| Stimuli: hyperosmolality, hypotension, pain, stress, inflammation, Ang II | Heart and coronary vessels | Cardiac hypertrophy, vasodilation of epicardial coronary vessels; vasoconstriction of resistance coronary vessels | |
| Agiotensin II | AT1R, AT2R | Brain | Hyperalgesia, increase of sympathetic tone |
| Sites of synthesis: brain and peripheral organs | Spinal cord | Hyperalgesia | |
| Stimuli: hypotension, pain, stress, inflammation | Heart and coronary vessels | Cardiac hypertrophy, coronary vasoconstriction | |
| Angiotensin 1-7 | MasR | Brain | Analgesia, increase of vagal tone, decrease of sympathetic tone |
| Sites of synthesis: brain and peripheral organs | Heart and coronary vessels | Cardioprotection, coronary vasodilation | |
| Stimuli: hypotension, pain, stress, inflammation | |||
| Endothelin-1 | ETAR; ETBR | Brain | Hyperalgesia, increase of sympathetic tone; decrease of parasympathetic tone |
| Sites of synthesis: brain and peripheral organs | Heart and coronary vessels | Cardiac hypertrophy, coronary vasoconstriction | |
| Stimuli: hypotension, stress, pain, inflammation |
Abbreviations: PVN, paraventricular nucleus; ScN, suprachiasmatic nucleus; SON, supraoptic nucleus; AT1R, AT2R, angiotensin receptors; ETAR, ETBR, endothelin receptors; V1aR, V1bR, V2R, vasopressin receptors; Ang II, angiotensin II; MasR, angiotensin-(1-7) receptor.
Strong evidence indicates that vasopressin has multiple associations with the regulation of pain. Studies have shown that, in the rat, mechanical or thermal stimuli inducing pain cause activation of vasopressin neurons in the hypothalamo-neurohypophyseal system and release of AVP. Furthermore, it has been shown that pain can be eliminated by pre-application of a V1aR antagonist [68]. Studies on rats suggest that the nociceptive ascending pathway to the SON encompasses the caudal ventrolateral medulla and noradrenergic A1 region [70].
Centrally mediated analgesic effects of AVP. Systemic or intracerebroventricular (ICV) application of AVP elevates the pain threshold in the rat, and this effect can be eliminated by pretreatment with a V1R antagonist [(dPTyr(Me)AVP] but not the opiate antagonist naloxone [81, 82]. Similarly, intrathecal (it) administration of AVP to the spinal cord produces antinociception, which is not mediated by opiates, but can be eliminated by blockade of V1R and is absent in V1aR knock-out mice [82, 83, 84, 85]. Experiments on rats showed that intravenously applied AVP elicits dose-related effects on nociceptive processing in the spinal cord. Namely, it exerts an antinociceptive effect, expressed by reduction of action potentials mediated by C-type nociceptive fibers, although it elicits an opposite action when it is applied at higher doses. Both effects can be abolished by administration of a V1aR antagonist [86].
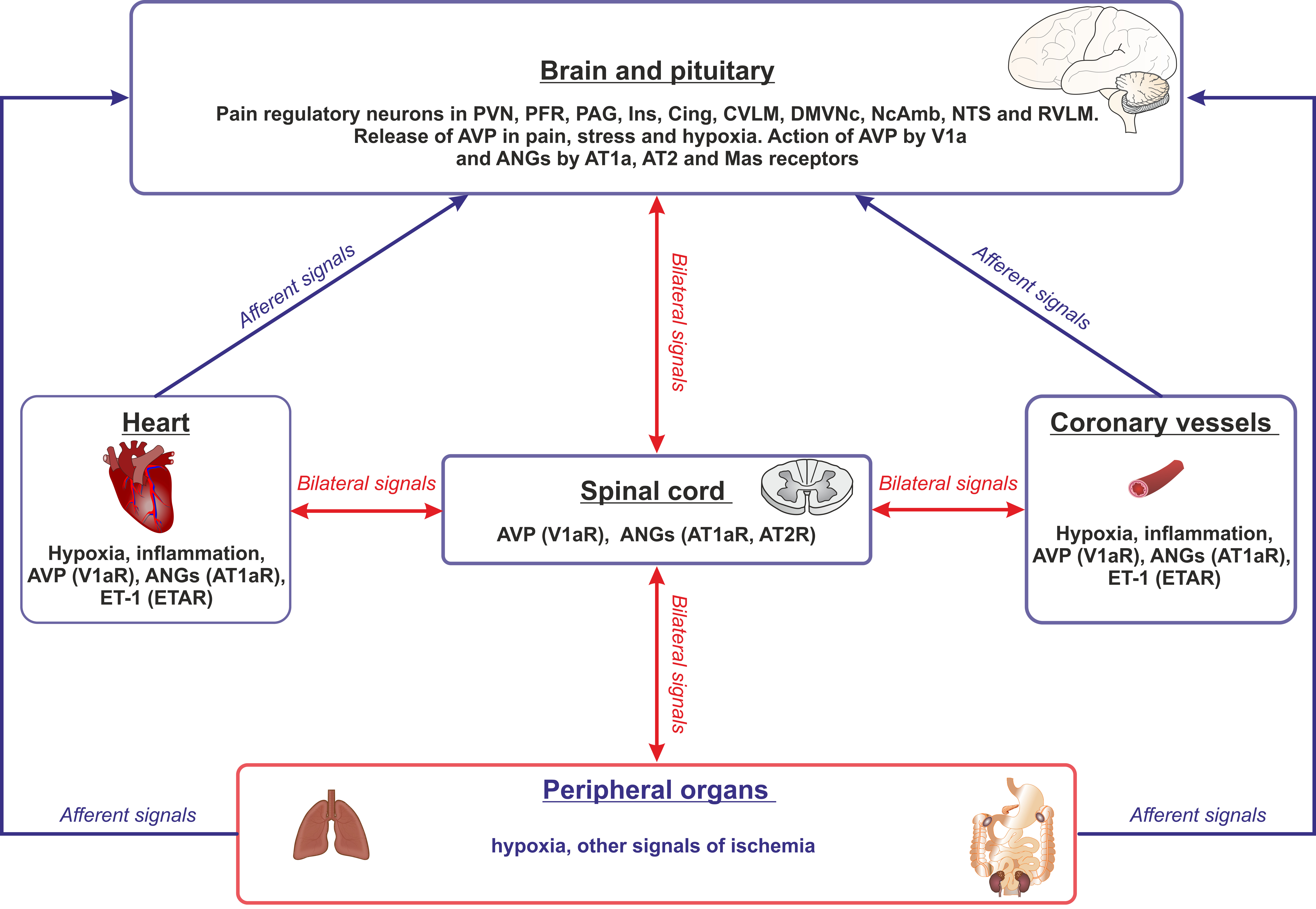
Strong evidence indicates that there are several sites in the brain where AVP may interact with neurons engaged in regulation of pain (Fig. 1; Table 1). Thus far, the most essential role has been attributed to the amygdala, PAG, and caudate nucleus (CdN). In the amygdala, vasopressin acting on V1a receptors also enhances the nocifensive reflexes related to pain [87, 88]. Experiments on rats showed that administration of AVP to the PAG increases pain threshold through stimulation of V1aR and also induces local elevation of leucine-enkephalin (L-Elk), methionine-enkephalin, and serotonin (5-HT). Moreover, the analgesic effects of AVP in PAG could be abolished by blockade of opiate receptors with naloxone [89], which indicates that, in specific brain regions, AVP interacts with opiates in regulation of pain. It should be noted that the PAG plays a significant role in regulation of cardiovascular function [90].
 Fig. 1.
Fig. 1.Associations of vasopressin, angiotensins and endothelin with regulation of pain. PVN, paraventricular nucleus; AVP, arginine vasopressin; ANGs, angiotensins; ET, endothelin; Ins, insula; Cing, cingulate gyrus; CVLM, caudal ventrolateral medulla; DMVNc, dorsal ventromedial nucleus of the vagus; NcAmb, nucleus ambiguous; NTS, nucleus tractus solitarius; PAG, periaqueductal grey; PFR, periphornical region; RVLM, rostral ventrolateral medulla; V1aR, vasopressin receptor; AT1aR, AT2R, angiotensin II receptors; ETAR, endothelin receptor; V1a, vasopressin V1a of type 1a; AT1a, angiotensin II of type 1a; AT2, angiotensin II of type 2; Mas, angiotensin-(1-7).
Pain increases release of AVP in the CdN and this effect is associated with stimulation of Ach release, which can be eliminated by application of AVP antagonists (V1ant, V2ant) [91, 92]. There is also evidence that AVP released from PVN neurons may regulate pain through the action exerted in the nucleus raphe magnus [92].
Systemically mediated effects of vasopressin. Systemically released AVP reaches the heart and coronary vessels and can regulate cardiac metabolism and blood oxygenation [59]. V1a receptors have been identified in the human heart, and their expression increases in patients with heart failure [93]. Earlier experiments performed on dogs have demonstrated that AVP acting on coronary vessels causes vasodilation of the epicardial vessels that is mediated by nitric oxide; however, it also elicits vasoconstriction and resistance of coronary vessels. Both effects require activation of V1aR (Table 1) [94].
A recent study in humans undergoing cardiopulmonary procedures revealed significant differences in responsiveness of coronary arterioles to vasopressin in patients with diabetes mellitus and in non-diabetic subjects [95]. Specifically, vessels from patients with poorly controlled diabetes mellitus responded with significantly stronger vasoconstriction than vessels from non-diabetic subjects. The difference could be eliminated by application of a V1aR antagonist (SR49059). In addition, cardioplegic arrest was followed by greater elevation of V1aR in vessels of diabetic patients and resulted in elevation of V1bR in the atrial myocardium of the diabetic subjects, while it was not effective in the atria of non-diabetic subjects [95].
Clinical evidence for the importance of AVP in pain regulation. Several years ago, it was reported that patients in the emergency department complaining of pain have significantly higher plasma AVP levels than control subjects [96]. Subsequently, other studies provided evidence that blood AVP and copeptin concentrations are significantly elevated in patients with acute myocardial infarction. Accordingly, it has been proposed that measuring copeptin concentration may help to identify patients with low and high risk of mortality [97, 98, 99].
Interaction of vasopressin with angiotensin, endothelin and other cardiovascular
peptides. Cardiovascular disorders generated by cardiac infarction
and/or cardiac failure cause release of several other cardiovascular compounds,
which presumably interact with vasopressin in the regulation of pain. In this
review, we have focused mainly on components of the renin-angiotensin system
(RAS) and endothelin because the knowledge of their role in regulation of pain is
relatively well advanced. Several components of RAS, such as angiotensinogen,
angiotensin converting enzyme (ACE), and angiotensin receptors (AT1aR, AT2R),
have been identified in the sensory dorsal root ganglia and in the brain
structures involved in the regulation of pain [71, 100, 101, 102, 103, 104, 105]. Some of them are
co-localized with substance P and calcitonin-gene related peptide, which are
involved in the regulation of pain [105]. Studies on mice have shown that
administration of angiotensin II intensifies nociceptive behavior through
activation of angiotensin receptors (AT1R) and activation of p38MAPK [104]. In the rat model of
peripheral neuropathy, administration of losartan attenuates allodynia and
reduces expression of inflammatory proteins (Tumor necrosis factor-
Cooperation of AVP, Ang II, and endothelin-1 (ET-1) with the autonomic nervous system in the regulation of cardiac pain is summarized in Fig. 1 Beneficial antinociceptive effect of stimulation of AT2R has been described in inflammatory and neuropathic pain [108].
Although substantial evidence shows that acute myocardial infarction causes strong activation of RAS and elevation of plasma Ang II concentration [71, 109, 110], not many studies have assessed the role of RAS in regulation of pain in cardiac pathology. Thus far, it has been shown that administration of ACE inhibitors intensifies pain in fibromyalgia; however, it is likely that this effect is mediated by decreased degradation of bradykinin [101]. ACE inhibitors proved to be effective in the treatment of cardiac syndrome X [9, 111] and were able to reduce pain during exercise [112].
Plasma ET-1 level is significantly elevated in patients with angina and myocardial infarction [113, 114, 115] and its level markedly increases during coronary artery spasm [116, 117]. There is also evidence that acute myocardial infarction induces release of endothelin from the heart, as elevation of ET-1 is higher in coronary blood than in systemic blood [118]. Experiments on cats suggest that myocardial infarction causes activation of endothelin receptor of type 1A (ET1A) receptors on cardiac sensory neurons of the thoracic root ganglia [119]. Patients suffering from chest pain associated with normal coronary arteriography have higher levels of plasma endothelin during the treadmill exercise test than control subjects. The latter finding suggests that endothelin may influence intensity of chest pain in patients with syndrome X [9, 120].
Systemic levels of other factors, such as orexin [121, 122], brain natriuretic peptide [123], and insulin [124], are also elevated during pain and it is likely that they may participate in the regulation of cardiac ischemic pain.
Multiple studies draw attention to a diurnal periodicity of cardiac pain sensation. Onset of angina pain in patients with myocardial infarction shows a circadian rhythm with peaks between 06:00 h and 12:00 h and 20:00 h and 23:00 h [125, 126, 127, 128]. At present, the origin of the circadian periodicity of pain sensation is not fully elucidated, but there is evidence that the periodicity may follow the circadian rhythmicity of other brain functions, including secretion of neuroendocrine factors such as melatonin, vasopressin, components of RAS, and endothelin. Secretion and action of cardiovascular hormones shows circadian variability and effectiveness of a pharmacological medication may depend on the time of application of the treatment [128, 129, 130, 131, 132, 133].
There is strong evidence that vasopressin plays an essential role in synchronization of pain sensation and cardiovascular parameters with environmental light [60, 134]. Vasopressin is present in retinal cells projecting to the suprachiasmatic nucleus (SCN) and application of light evokes its release in this nucleus. Furthermore, secretion of AVP in the SCN manifests circadian rhythmicity, showing a peak in the early morning and a decline in the late afternoon [135, 136]. There is also evidence that vasopressin significantly contributes to the pacemaker function of the SCN. Neurons expressing AVP form a large population of SCN neurons, while the AVP promoter, which is a target for CLOCK/BMAL1 transcription factors, participates in generation of the circadian rhythm [134, 137, 138]. Experiments on mice provided evidence that AVP resets the clock function of the SCN through actions exerted on V1a/V1b receptors [139]. In humans, plasma vasopressin concentration shows a nocturnal increase with a peak between 24:00 h and 04:00 h [67, 140]. The nocturnal elevation of plasma AVP concentration is markedly attenuated in elderly subjects [141]. In post-stroke patients and patients with cardiac failure, the circadian rhythm of AVP secretion is abolished [141].
It is likely that vasopressin cooperates with Ang II and endothelin in the regulation of circadian rhythmicity. For instance, the circadian pattern of blood pressure fluctuations and pressor responses to restrain stress are significantly affected by systemic infusion of Ang II [142]. It appears that Ang II, acting on angiotensin II receptor of type 2 (AT2R), exerts a short-lasting antinociceptive effect, whereas prolonged stimulation of these receptors increases nociception at the beginning and end of the light phase. Both effects are attenuated by application of an AT2 antagonist (ditrifluoroacetate (PD 123319)) [143]. Inverted diurnal pain sensation was found in spontaneously hypertensive rats (SHR) and this dissimilarity could be corrected by blockade of AT1R with losartan [144].
Evidence is emerging that ET-1 may also belong to a family of cardiovascular peptides showing clear circadian rhythmicity. In the rat, expression of ET-1 mRNA in the SCN shows a peak around 04:00 h, whereas in the heart and lungs, it culminates between 12:00 h and 20:00 h [145]. Studies in mice suggest that plasma ET-1 level peaks during the night active period [146]. In human subjects, circulating ET-1 circadian peaks are present in the morning and afternoon [146, 147, 148]. The vasodilatory responses of endothelin and acetylcholine to iontophoresis show two peaks at 20:00 h and 08:00 h [147].
Summary. Cardiac pain often signals hypoxia and/or damage of the heart. However, in some patients (for instance in cardiac syndrome X or in silent ischemia), its intensity is not proportional to the disturbance affecting the heart. Thus far, the reasons for significant divergence in susceptibility to cardiac pain signals have not been well established. This review summarized evidence showing that cardiac pain is regulated jointly by the autonomic nervous system, autacoids, vasopressin, and other cardiovascular peptides. The attention was focused mainly on vasopressin, because its role in cardiovascular pathology in association with the regulation of pain, and its interactions with other systems activated by cardiac ischemia, are relatively well recognized. A survey of studies published in recent years shows that the mechanisms controlling cardiac pain by cardiovascular peptides cooperate in the brain, spinal cord, cardiac muscle, and coronary vessels. The review reported new evidence that the joint regulation of pain by the autonomic nervous system and the vasopressinergic system is significantly altered in cardiovascular disease and can be significantly affected by implementation of pharmacological treatment.
Limitations. Pain regulation studies face several limitations resulting from interspecies differences and lack of appropriate methods allowing the evaluation of pain intensity in animals. The transfer of data from animal species to humans requires comparative studies that are often limited on ethical grounds. Furthermore, in patients with heart failure, testing of pain may provoke excessive negative cardiovascular responses requiring termination of the test. Interpretation of the results may be also impeded by the application of another pharmacological treatment which interferes with the metabolism or action of cardiovascular peptides. This review is a many-faceted study and, although it is based on a comprehensive survey of the literature, it cannot be excluded that some valuable articles were omitted.
Outlook. A complete understanding of the role of vasopressin and other cardiovascular peptides in the physiology and pathophysiology of pain is not yet possible; however, existing evidence encourages further studies to investigate mechanisms of action. It is very likely that such studies may help to identify a new specific pain relieving treatment that may be beneficial in the management of cardiovascular diseases.
AVP, arginine vasopressin; ANGs, angiotensins; ET, endothelin; Ins, insula; Cing, cingulate gyrus; CVLM, caudal ventrolateral medulla; DMVNc, dorsal ventromedial nucleus of the vagus; NcAmb, nucleus ambiguous; NTS, nucleus tractus solitarius; PAG, periaqueductal grey; PFR, periphornical region; PVN, paraventricular nucleus; RVLM, rostral ventrolateral medulla; V1aR, vasopressin receptor; AT1aR, AT2R, angiotensin II receptors; MasR, angiotensin-(1-7) receptor; ETAR, endothelin receptor.
ESz-S performed literature search and had written the review. The author contributed to editorial changes in the manuscript. The author read and approved the final manuscript. The author had participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The author wishes to thank to Marcin Kumosa from the Department of Experimental
and Clinical Physiology of the Medical University of Warsaw for technical
preparation of the Figure, and to Agnieszka Cudnoch-J
This work was supported by the Medical University of Warsaw Scientific Projects (1MA/N/2022). The work did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.