
 , Yan-Jing Liang 1, Xin-Xiao Zhang 1, Su-Hao Yang 1, Zhan-Qiong Zhong 4, Shu-Qing Liu 1, Jia-Yi Sun 5, Yong Tang 2,3,6,7, Shu-Guang Yu 1,2,3,*
, Yan-Jing Liang 1, Xin-Xiao Zhang 1, Su-Hao Yang 1, Zhan-Qiong Zhong 4, Shu-Qing Liu 1, Jia-Yi Sun 5, Yong Tang 2,3,6,7, Shu-Guang Yu 1,2,3,*1 Acupuncture and Tuina School, Chengdu University of Traditional Chinese Medicine; 611137 Chengdu, Sichuan, China
2 Sichuan Provincial Key Laboratory for Acupuncture & Chronobiology, 611137 Chengdu, Sichuan, China
3 Key Laboratory of Acupuncture for Senile Disease (Chengdu University of TCM), Ministry of Education, 611137 Chengdu, Sichuan, China
4 School of Basic Medical Sciences, Chengdu University of Traditional Chinese Medicine, 611137 Chengdu, Sichuan, China
5 Innovative Institute of Chinese Medicine and Pharmacy, Chengdu University of Traditional Chinese Medicine, 611137 Chengdu, Sichuan, China
6 International Collaborative Centre on Big Science Plan for Purinergic Signalling, Chengdu University of Traditional Chinese Medicine, 611137 Chengdu, Sichuan, China
7 School of Health and Rehabilitation, Chengdu University of Traditional Chinese Medicine, 611137 Chengdu, Sichuan, China
Abstract
Background: The purpose of this study was to
investigate the potential involvement of pyruvate kinase M2 (PKM2), an enzyme
acting as a rate-limiting enzyme in the final phase of glycolysis, in the
regulation of glial activation and brain damage of intracerebral hemorrhage
(ICH). Methods: Western blotting and immunofluorescence were
performed to investigate PKM2 expression, terminal deoxynucleotidyl transferase
deoxyurinary triphosphate (dUTP) nick end labeling staining, hematoxylin and
eosin staining, and behavioral tests were employed to evaluate the brain damage
of ICH mice, and RNA-seq and bioinformatic analyses were performed to detect gene
expression changes in ICH mice treated with TEPP-46. Results: Increased
PKM2 levels in perihematomal brain tissue were found starting from 3 days
following ICH and peaked at 5 and 7 days post ICH. The increased expression of
PKM2 was mainly co-localized with glial fibrillary acidic
protein (GFAP)
Keywords
- PKM2
- glial cell
- brain injury
- intracerebral hemorrhage
Intracerebral hemorrhage (ICH) is a leading cause of morbidity and mortality worldwide [1], with fatality rates ranging from 40–54% at 1 month to 1 year after the index event [2]. Even though current stroke guidelines recommend early, positive, goal-directed treatment for ICH patients [1], there are currently no effective neuroprotective strategies for reducing brain injury in ICH patients, due to the risk of early neurological impairment and worse clinical outcomes.
Neuroinflammation has been identified as one of the key contributors to the aggravation of brain damage after ICH [3, 4]. Furthermore, neuroinflammation is emerging as an instigator rather than an outcome in the pathogenic process of numerous brain diseases [5]. Glial cells (e.g., astrocytes and microglia) have been identified as the primary inflammatory cells in several neurological disorders, including stroke. Consequently, inhibiting glial activation may be a promising strategy for reducing the neuroinflammation-mediated brain injury caused by ICH [4]. Energy is an essential component for supporting cellular life and biofunctions, particularly for cells that transition from a resting to an active state. Accordingly, glial cell activation is mostly reliant on energy support. Mounting evidence indicates that glial cells mainly consume the energy produced by glycolysis to promote their activation [6, 7, 8]. Pyruvate kinase M2 (PKM2) is a rate-limiting, glycolytic enzyme that orchestrates the final step in the glycolytic pathway by catalyzing the conversion of phosphoenolpyruvate (PEP) and adenosine diphosphate (ADP) to pyruvate and adenosine triphosphate (ATP) [9, 10]. PKM2 is expressed in a wide range of proliferating cells, including embryonic cells, adult stem cells, tumor cells, and immune cells [11, 12, 13, 14]. Their elevated rates of nucleic acid synthesis are a shared characteristic. It is noteworthy that PKM2 has dual functionality, serving not only as a metabolic enzyme but also as a protein kinase and transcriptional coactivator [10]. The enzymatic activity of PKM2 is closely related to oligomerization states, that is tetramer form for active, dimer form for less active, and monomer form for inactive [15, 16, 17, 18]. As a direct consequence of detetramerization, PKM2 monomer translocases to the nuclear and acts as a protein kinase/transcriptional coactivator to regulate gene transcription [18, 19, 20]. However, the role of PKM2 as a protein kinase in regulating brain injury in central nervous system (CNS) diseases, particularly ICH, are poorly understood.
Therefore, we examined the kinase activity of PKM2 in regulating glial activation and brain damage following ICH in this investigation. We showed that the up-regulated expression of PKM2 and increased nuclear translocation of PKM2 in the perihematomal brain tissues coincided with glial activation following ICH. While TEPP-46, a PKM2 tetramer stabilizing activator, significantly inhibited PKM2 translocation, leading to a reduction in glial activation and subsequent brain injury following ICH. Subsequently, RNA-seq technology was employed to investigate potential mechanisms. The results revealed that about 90% of differentially expressed genes (DEGs) were downregulated in response to TEPP-46 treatment in mice with ICH. These DEGs are likely to have important roles in the modulation of autophagy, metabolic processes in neurons, endothelial cells, microglia, and astrocytes. The present work offers a unique perspective on the protein kinase functions of PKM2 in the regulation of ICH-induced brain damage, as well as prospective therapeutic targets for ICH therapy.
This study used C57BL/6 mice (male, 8 weeks, 23
We constructed the ICH mouse models according to our previously developed
methods [21, 22]. Briefly, a stereotaxic apparatus (RWD Life Science Co.,
Shenzhen, Guangdong, China) was applied to immobilize mice after they were anesthetized with
3% and 1.5% isoflurane (Cat# R510-22-10, RWD Life Science Co., Shenzhen, China), respectively, for anesthesia induction and maintenance.
A total volume of 0.5 µL type VII collagenase (0.075 IU,
C0773, Sigma Aldrich, St. Louis, MO, USA) was continuously injected into the
mouse left striatum (0.8 mm anterior, 2 mm lateral to bregma, and at a depth of
3.5 mm) using a syringe pump (Legato 130, RWD Life Science Co.,
Shenzhen, China). Control mice received saline injection (0.5 µL). Mouse
body temperature was maintained at 37 °C throughout surgery and
anesthesia resuscitation. After recovery from anesthesia, a Longa score
According to our previously reported method [23], western blots were performed for assessing the PKM2 levels in the perihematomal brain tissues of ICH mice. Briefly, the ipsilateral brain tissues and control brain tissues of mice were collected and homogenized to extract proteins using a prepared radio immunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China), and a Bicinchoninic Acid (BCA) protein assay (Cat# 23227/23225, Thermo Scientific, Waltham, MA, USA) was used to determine the protein concentration. Proteins were resolved by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotted onto polyvinylidene fluoride (PVDF; Millipore, Burlington, MA, USA) membranes after adjustment to the same final concentration. Next, after blocking the membranes for 2 hours at room temperature with 5% non-fat milk in tris-buffered saline containing 0.1% Tween-20 (TBST; Biosharp, Guangzhou, Guangdong, China), they were incubated with anti-PKM2 (#4053, 1:50, Cell Signaling Technology, Danvers, MA, USA) antibody overnight at 4 °C. The membranes were then washed four times with TBST and incubated with a secondary antibody conjugated with horseradish peroxidase (HRP) (1:2500; OR03L, Sigma-Aldrich, St. Louis, MO, USA) at room temperature for 2 hours. An enhanced chemiluminescent (ECL) substrate was used to view the immunoreactive proteins with an imaging system (GelView 6000Plus, Guangzhou Biolight Biotechnology Co., Ltd., Guangzhou, Guangdong, China). Following stripping, the membranes were incubated again with glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Cat# 60004-1-Ig, Proteintech, Wuhan, Hubei, China) acting as the loading control. The band intensities were normalized to the values for GAPDH and quantified using Image J software (version 1.46J, National Institutes of Health, Bethesda, MD, USA).
Based on our previously reported procedures [23], we performed immunohistochemical staining to investigate the PKM2 expression profiles in the brains of ICH mice. The mice were sedated and their brains were rapidly removed and preserved in 4% paraformaldehyde (PFA; Cat# P1110, Solarbio, Beijing, China) at 4 °C overnight, after being intracardially perfused with PBS and 4% paraformaldehyde. Brain sections of 15 µm were then incubated with the following antibodies: anti-PKM2 (#4053, 1:50, Cell Signaling Technology, USA), Iba-1 (ab178846, 1:200, Abcam, Cambridge, MA, USA), NeuN (#24307, 1:200, Cell Signaling Technology, USA), and glial fibrillary acidic protein (GFAP; ab7260, 1:300, Abcam, USA). Secondary antibodies, including Alexa Fluor 594 (1:200, donkey anti-rabbit), Alexa Fluor TRITC (1:200, rabbit anti-goat), and Alexa Fluor 488 (1:200, donkey anti-mouse), were purchased from Proteintech Group (Wuhan, Hubei, China). The brain slices were imaged using confocal microscopy (Olympus IXplore SpinSR10, Olympus, Tokyo, Japan). Quantification of double-labeled cells was used to calculate the ratio of co-labeled cells. Three brain slices were selected from each mouse brain at random; double-labeled cells were counted automatically, and the average data obtained by three independent researchers who were unaware of group information was used to determine the value for each brain.
After the mice were euthanized, their brains were rapidly removed and fixed in a 10% neutral-buffered formalin solution (Cat# G2161, Solarbio, Beijing, China). Following dehydration in ethanol solutions, the brain tissues were embedded in paraffin. The brain tissues were then sectioned at 5 µm thickness using a rotary microtome. Lastly, three slices of each brain tissue were selected at random, then stained with H&E, and viewed under an Olympus microscope (Olympus SLIDEVIEW VS200, Olympus, Tokyo, Japan) to evaluate the damage in these perihematomal brain tissues.
Based on the manufacturer’s instructions of a commercial One-step TUNEL In Situ
Apoptosis Kit (E-CK-A320, Elabscience, Wuhan, Hubei, China), TUNEL staining was
performed. Briefly, 10 µL 1
The corner test was conducted using previously published procedures [24, 25].
ICH mice were first placed between two cardboard pieces with a 30
Perihematomal brain tissue of ICH mice, after treatment with or without TEPP-46 at 5 days post ICH, was collected for sequencing. Brain tissue total RNA was extracted, and cDNA libraries were constructed using an Oxford Nanopore Technologies (ONT)-supplied Ligation Sequencing Kit 1D (PM) (SQK-LSK110, Oxford Nanopore Technology, Oxford, England) in accordance with the company’s protocol. Full-length cDNA was enriched using reverse transcriptase and the addition of defined PCR adapters to both ends of the first strand of cDNA, then 14-circle cDNA PCR (8 min extension time) was performed using LongAmp Tag DNA polymerase (New England Biolabs, Ipswich, MA, USA). T4 DNA ligase (New England Biolabs, Ipswich, MA, USA) was then used to bind the PCR products to the ONT adaptor ligation. DNA purification was performed using Agencourt XP beads (Beckman Coulter, Brea, CA, USA). A PromethION platform of the Biomarker Technology Company (Beijing, China) was used to analyze the final cDNA libraries after being processed by FLO-MIN109 flow cells (FLO-PRO002, Oxford Nanopore Technology, Oxford, England).
Raw reads with an average quality score of less than 7 and a length of less than
500 bp were eliminated. After the rRNA was mapped to the rRNA database, it was
discarded. Next, primers at both ends of the reads were searched to identify the
full-length, non-chimeric (FLNC) transcripts. The Mimimap2 alignment program [26]was applied to the reference genome library in order to obtain clusters of FLNC
transcripts. Full-length reads were mapped to the reference genome (Rnor_5.0).
Reads with match quality more than 5 were further used for quantitative analysis.
Counts per million (CPM) [27] was used as an indicator to measure the levels of
transcript or gene expression. Using the DESeq R package (DESeq2
1.6.3, Germany; http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html), DEGs between the two groups were analyzed. The p-values
were adjusted using Benjamini and Hochberg’s approach, and the DEGs induced by
TEPP-46 treatment were defined as those with a p-value
Conjoint analysis was performed using our published method [21, 22] by matching
the down-regulated DEGs induced by TEPP-46 with previously published
transcriptome data [29]. Considering the extensive research conducted on brain
cells in the context of stroke, including neurons, glial cells (e.g., astrocytes
and microglia), and endothelial cells, these four cell types were selected for
further analysis. Firstly, the relatively high expression of genes in the four
cells were defined using previous methods [21, 22]. For example, gene A is
defined as being highly specific to astrocytes when the ratio of the fragments
Per Kilobase of exon model per Million fragments mapped (FPKM) of gene A in
astrocytes to that in the other three neural cells (neurons, microglia, and
endothelial cells) is
The data were presented as the mean
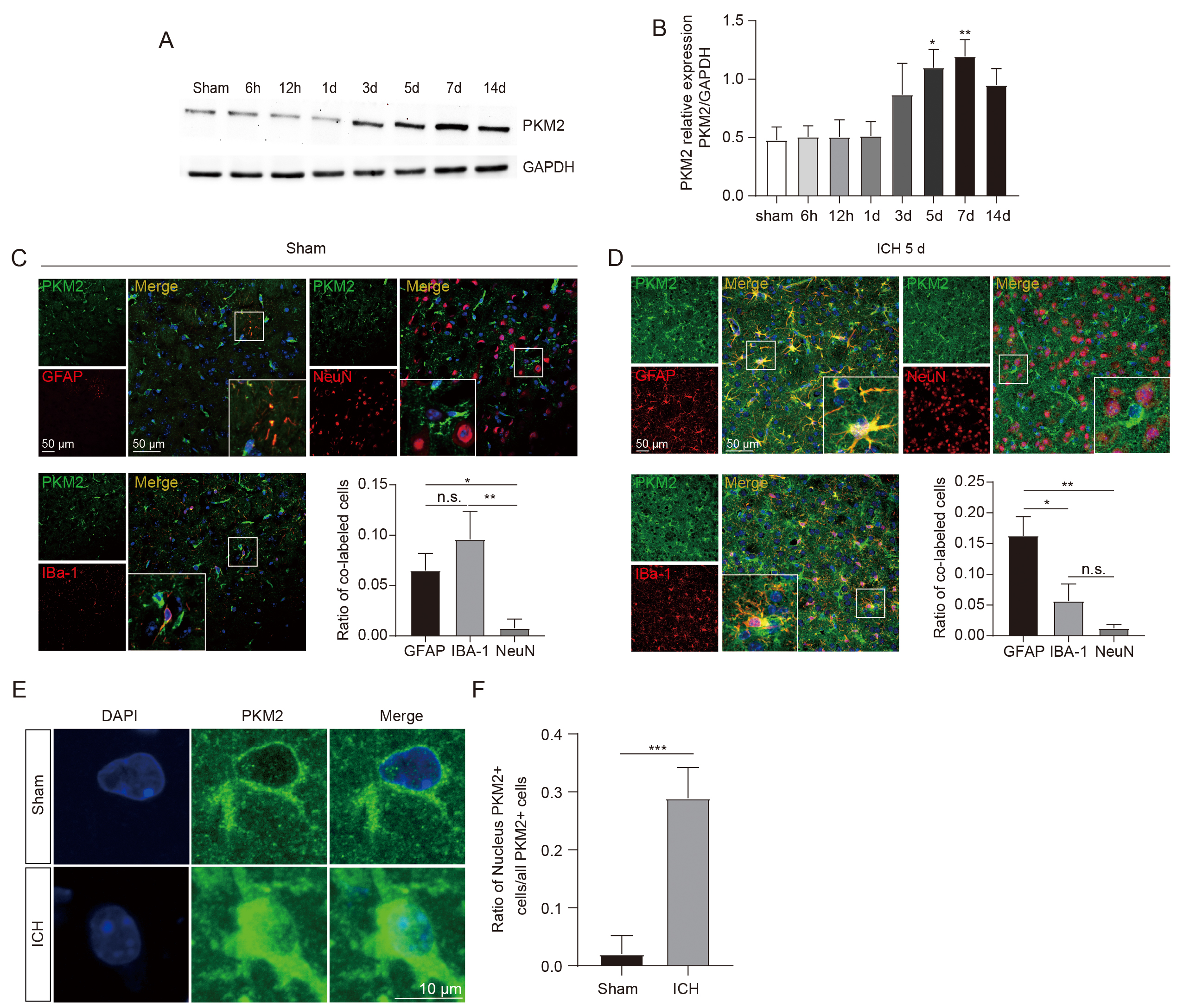
To determine the role of PKM2 in ICH-induced brain damage, we analyzed the expression profiles of PKM2 in the perihematomal brain tissues following ICH. First, we performed a western blot and observed that PKM2 levels were significantly elevated beginning 3 days after ICH, peaked at 5 and 7 days, and remained elevated at day 14 compared with sham groups (Fig. 1A,B). The expression levels of PKM2 and the cellular sources of PKM2 in ICH mouse brains were further analyzed by the immunofluorescent staining. In the sham groups, the total PKM2 expression level was low, and PKM2 expression was mostly localized in glial cells, particularly in the IBA-1-positive microglia, even though these cells were not activated (Fig. 1C). In contrast, in the perihematomal brain tissues of mice at 5 days after ICH, we found that PKM2 expression and glial activation were both significantly increased, and PKM2 was mainly co-localized with GFAP-positive astrocytes, followed by IBA-1 positive microglia, and rarely co-localized with NeuN-positive neurons (Fig. 1D). These data revealed a dynamic change feature of PKM2 expression after ICH, in which low and microglial PKM2 expression gradually switches to high and astrocytic expression in response to the brain damage caused by ICH.
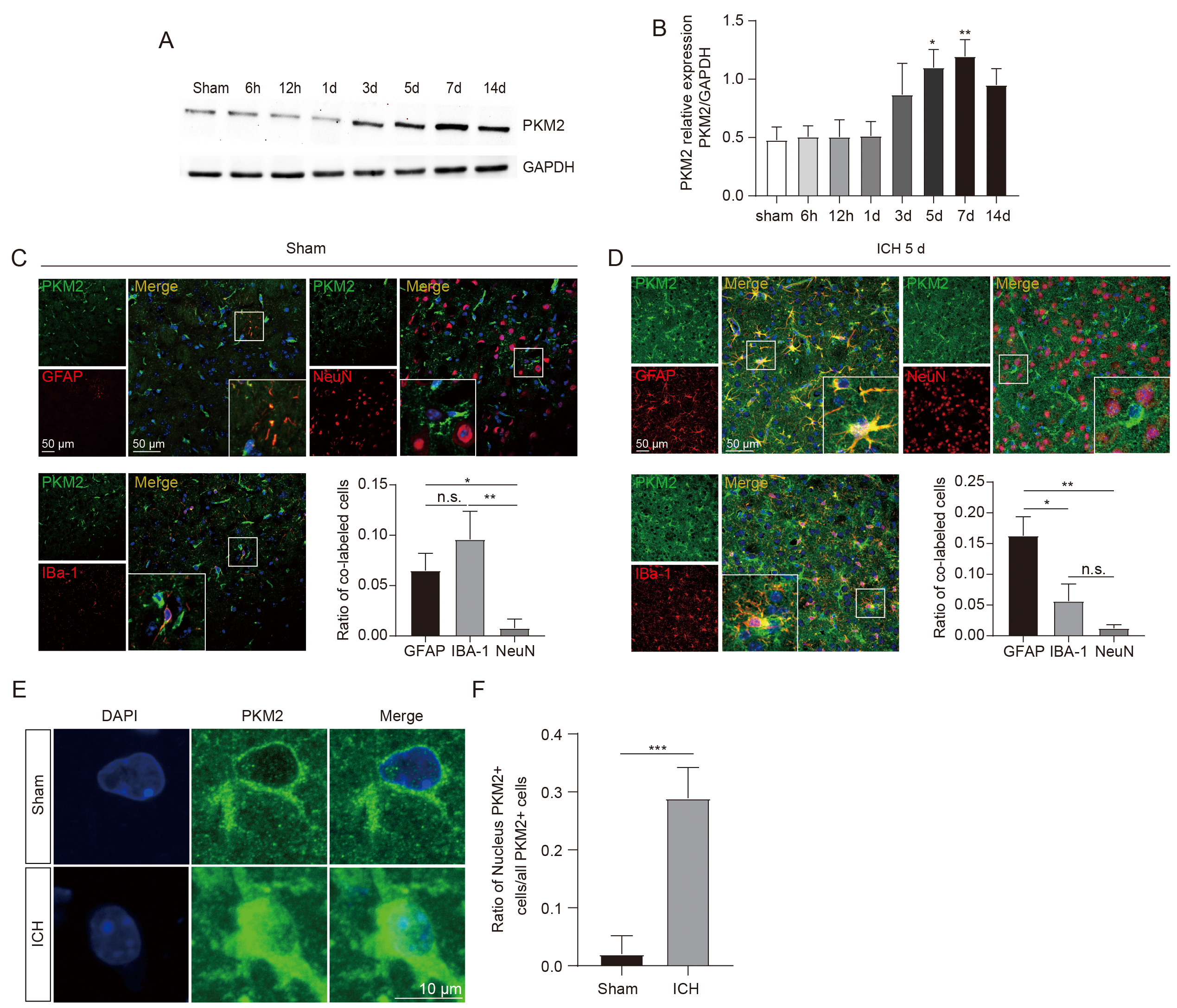 Fig. 1.
Fig. 1.PKM2 expression profiles in the perihematomal brain tissues of
ICH mice. (A,B) Representative image of western blot (A) and statistical
analysis (B) of PKM2 expression in the perihematomal brain tissues at different
time points after ICH (each time point, n = 5 for each group). (C,D)
Immunofluorescent staining of cells expressing PKM2 with NeuN+ neurons, IBA-1+ microglia, and GFAP+ astrocytes in sham (C) and ICH mice
(D), respectively (n = 4 for each group). (E) Representative immunofluorescent
image of PKM2 nuclear translocation in the perihematomal brain tissues at 5 days
following ICH. (F) Statistical results of immunofluorescent staining. n = 4 for
each group. *p
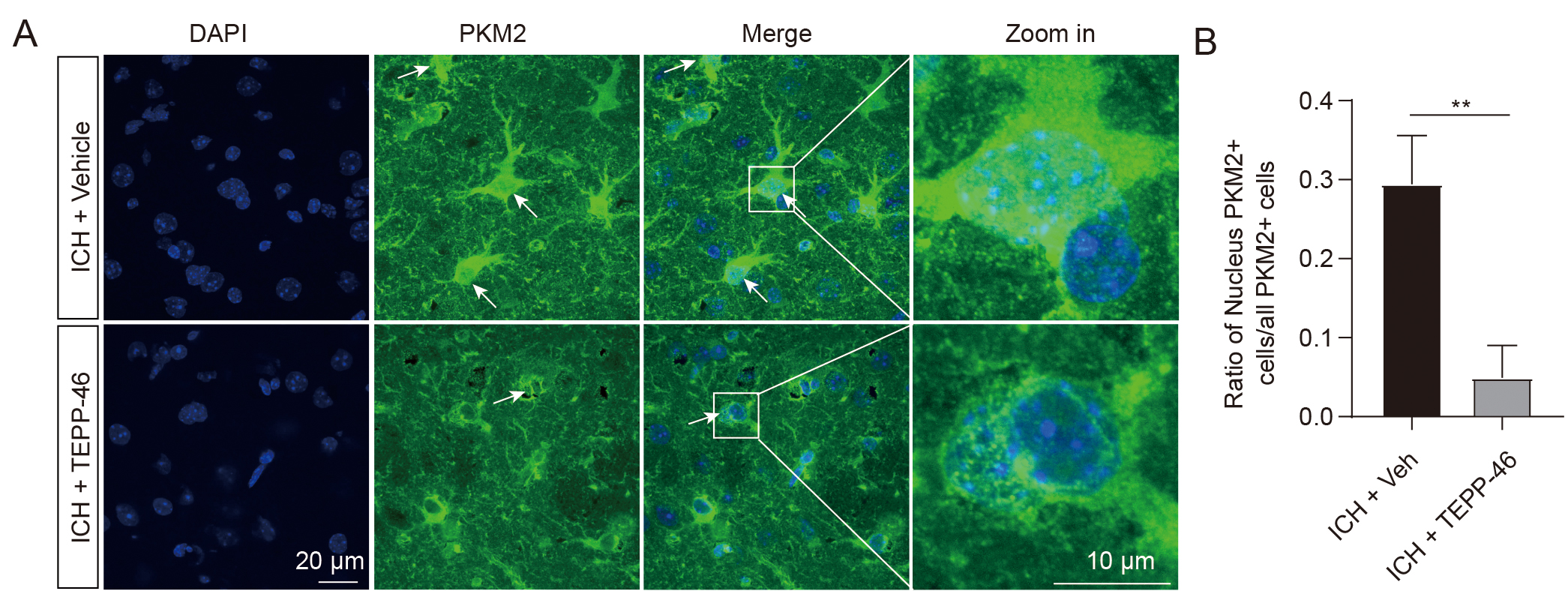
The enzymatic activity of PKM2 is closely related to its oligomerization status, including active tetramer, less active dimer, and inactive monomer [18]. PKM2 detetramerization directly leads to the nuclear translocation of its inactive monomer [20], which has been shown to regulate gene transcription by functioning as a protein kinase in the nucleus. [18, 19, 30]. Our immunofluorescent staining data was analyzed to determine the PKM2 oligomerization status after ICH by evaluating the nuclear expression levels. In the sham groups, low PKM2 expression was observed mostly in the cytoplasm, whereas 30% of PKM2-positive cells exhibited nuclear translocation in the ICH mice (Fig. 1E,F), suggesting an ICH-induced shift to PKM2 monomers. TEPP-46, a small-molecule activator that stabilizes PKM2 in its tetramer form [31], was then intraperitoneally injected into ICH mice for 5 consecutive days. The results demonstrated that TEPP-46 treatment notably prevented the nuclear translocation of PKM2 in the perihematomal brain tissues of ICH mice, compared with ICH mice treated with a vehicle (Fig. 2A,B). This indicates that increased PKM2 nuclear translocation in glial cells resulting from the PKM2 detetramerization induced by ICH may participate in the regulation of glial cell activation and brain injury, as PKM2 nuclear translocation has been shown to promote the activation of immune cells [13].
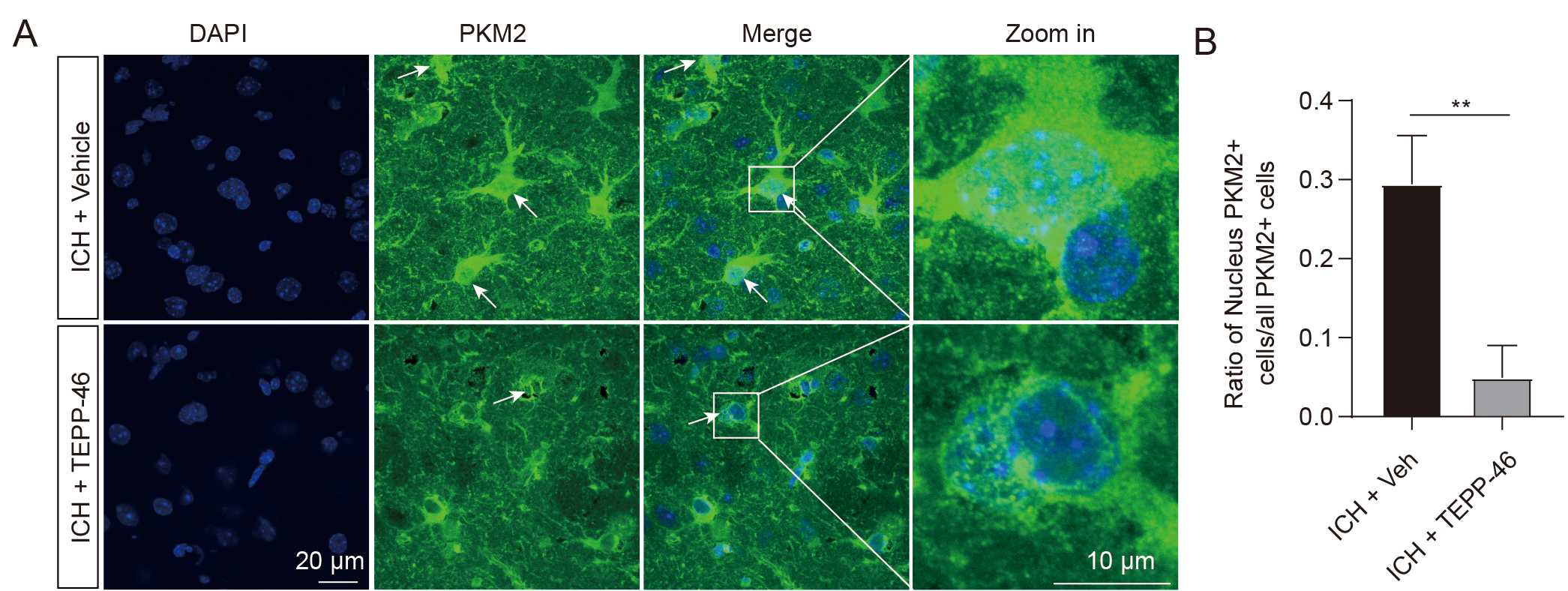 Fig. 2.
Fig. 2.TEPP-46 treatment markedly reduced the PKM2 nuclear
translocation after ICH. (A) Representative immunofluorescent image showing that
TEPP-46 induced a significant reduction of PKM2 nuclear translocation in the
perihematomal brain tissues at 5 days after ICH. (B) Statistical results of
immunofluorescent staining. n = 4 for each group, **p
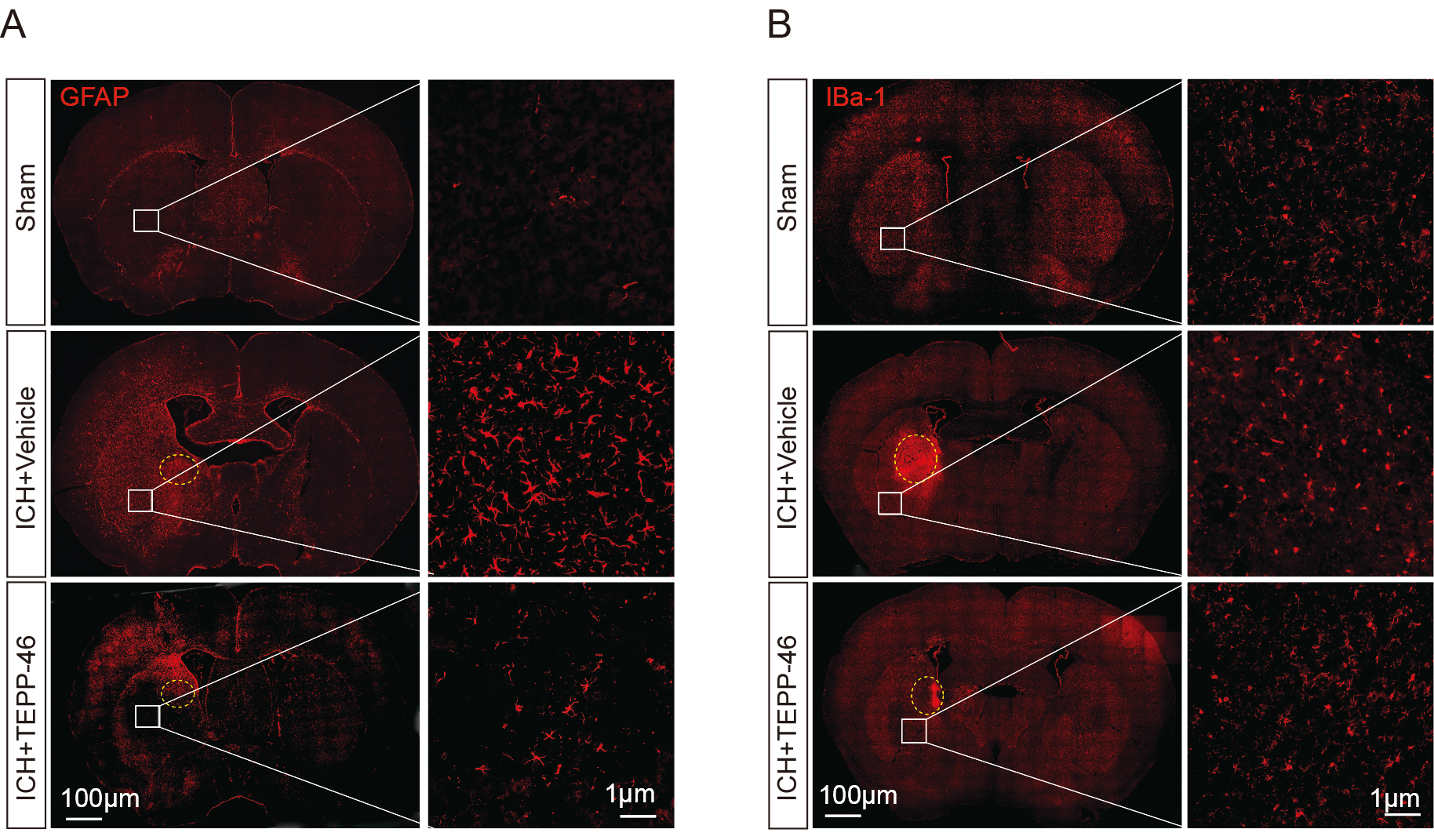
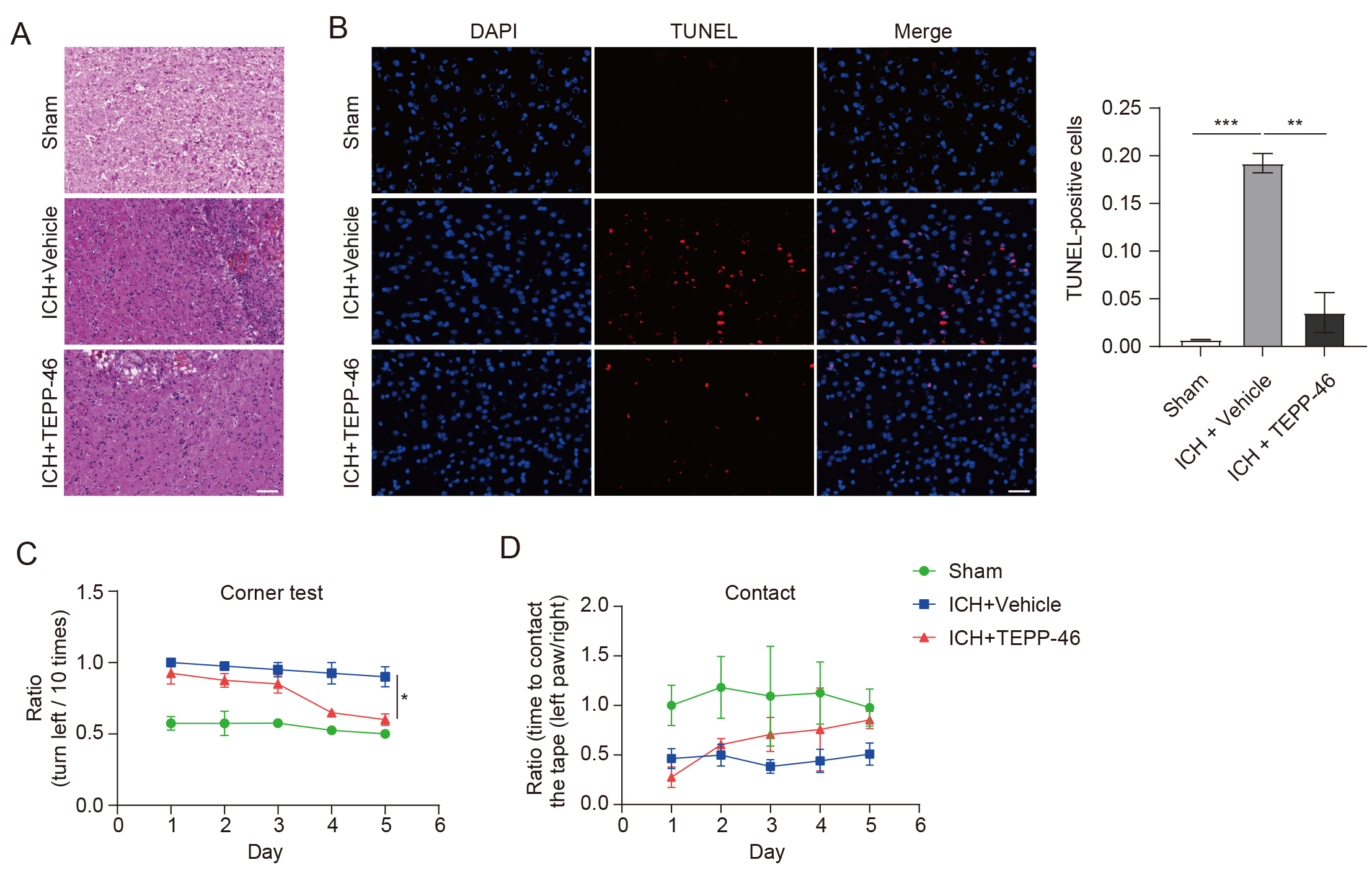
Through the evaluation of glial markers, it was shown that the mice in the vehicle-treated ICH group had a significant increase in the presence of activated astrocytes (Fig. 3A) and microglia (Fig. 3B) inside the perihematomal brain tissue, as compared with the sham group. In contrast, the TEPP-46 treatment significantly reduced both the activation of astrocytes and microglia after ICH (Fig. 3), suggesting that PKM2 detetramerization promoted glial activation after ICH. H&E staining was conducted to assess the general brain injury and we observed that TEPP-46 treatment resulted in a reduction of brain injury in the perihematomal brain tissues at 5 days post ICH. (Fig. 4A). Similarly, TEPP-46 treatment reduced the increased TUNEL+ cells in the brain tissue surrounding the hematoma (Fig. 4B). In the corner tests, the TEPP-46-treated ICH mice almost recovered from their physical disability within 5 days (Fig. 4C), which was consistent with the results of the PKM2 expression profile. However, the sensory neglect of mice with ICH was not significantly improved after TEPP-46 treatment, although there was a trend for improvement (Fig. 4D). These results strongly indicate that TEPP-46 treatment reduces brain injury and partly improves functional recovery of ICH that may be related to the inhibition of glial activation by preventing PKM2 nuclear translocation in glial cells.
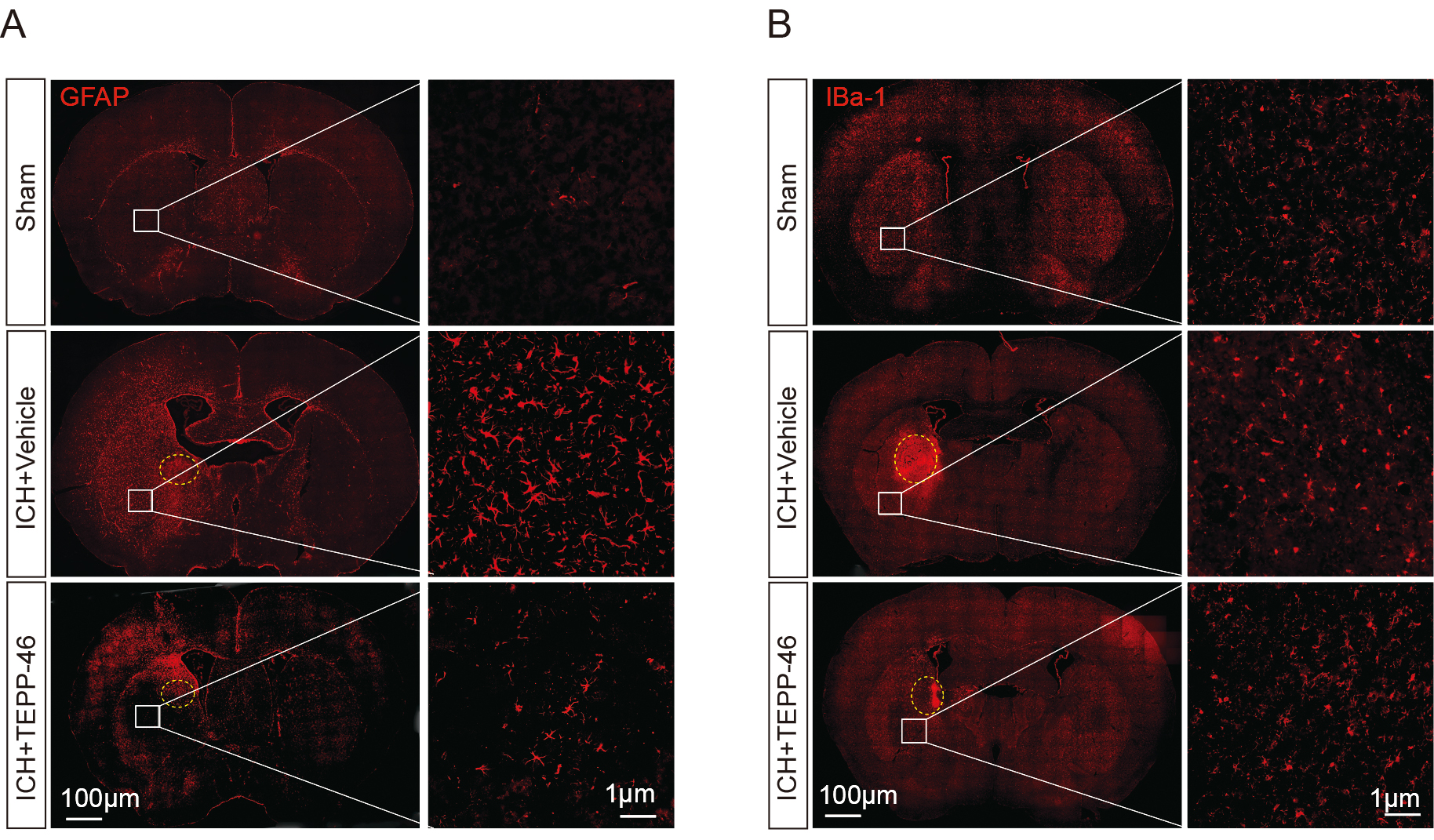 Fig. 3.
Fig. 3.Glial activation was significantly inhibited after ICH treated
by TEPP-46. The activation of GFAP
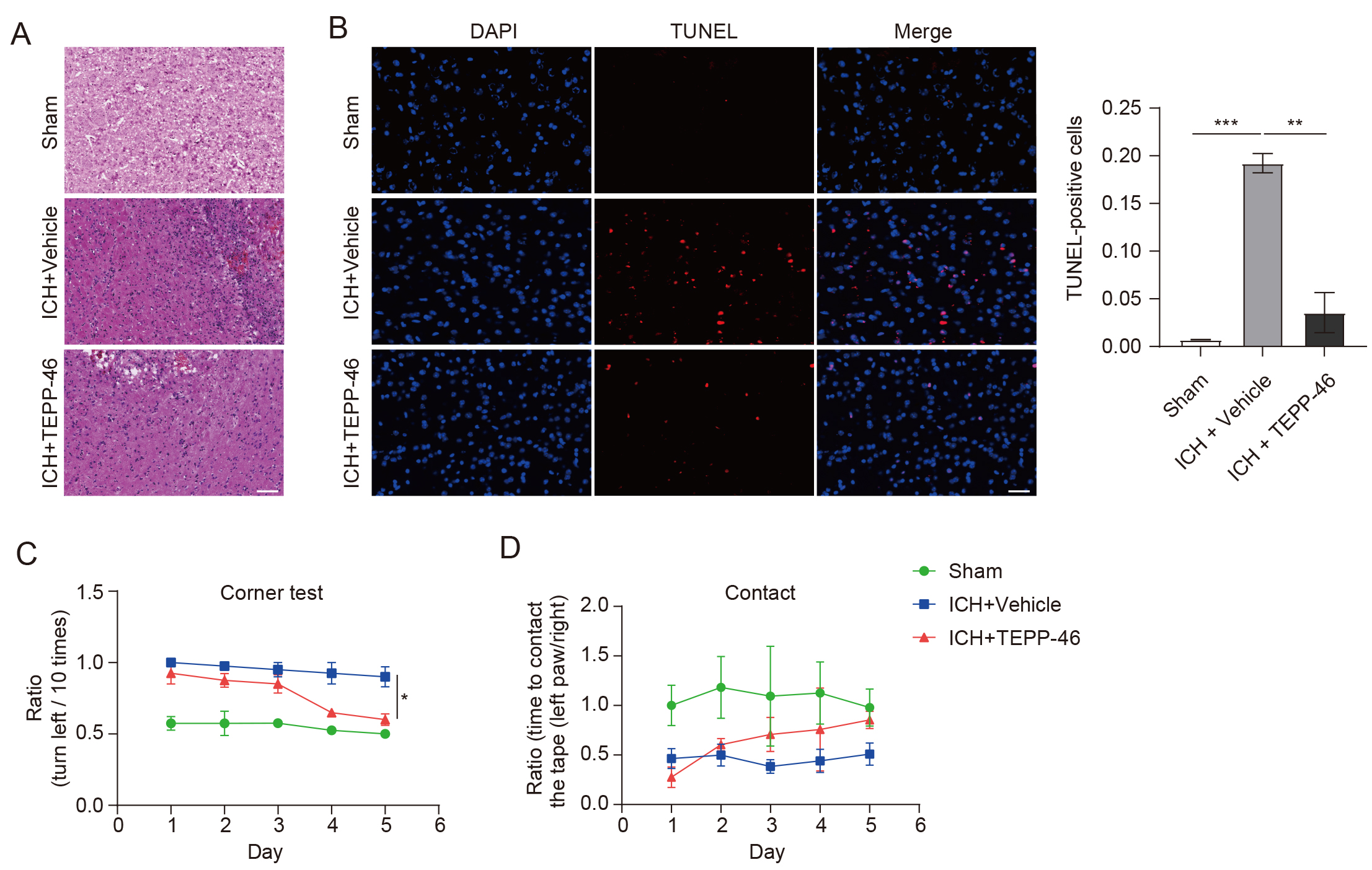 Fig. 4.
Fig. 4.TEPP-46 treatment markedly reduced brain injury and improved
functional recovery of mice with ICH. (A) H&E staining shows that brain injury
and infiltration of immune cells were markedly reduced in ICH mice treated with
TEPP-46 (n = 4 for each group). (B) TUNEL+ cells were obviously
reduced in ICH mice treated with TEPP-46 (n = 4 for each group, **p
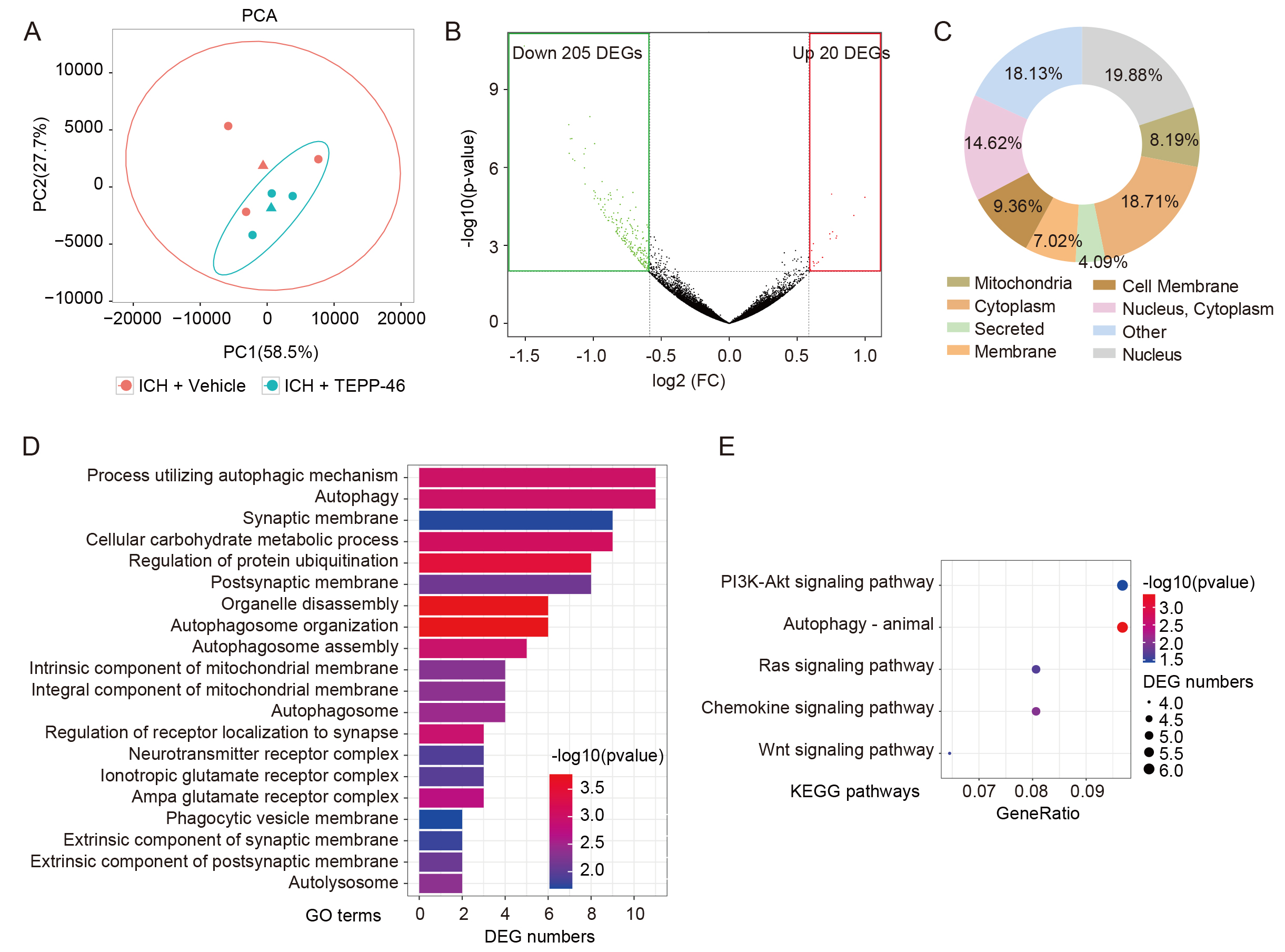
RNA-seq was then performed on perihematomal brain tissue from ICH mice treated with or without TEPP-46 to obtain insight into the molecular consequences of glial inhibition induced by PKM2 tetramerization (Fig. 5A). Surprisingly, 91.1% (205/225) of DEGs were down-regulated in the ICH mouse brains treated by TEPP-46 compared with those treated with vehicle (Fig. 5B), indicating that most of the upregulated DEGs induced by ICH may have been inhibited when PKM2 nuclear translocation was blocked. The proteins encoded by these down-regulated DEGs were widely distributed throughout the cells, including in the nucleus and cytoplasm (Fig. 5C). An analysis of these down-regulated DEGs using GO term enrichment analyses revealed that TEPP-46-treated mice, when compared with vehicle controls, had decreased responses to autophagy and its related biological processes, metabolic processes, receptor complex, and membrane-related events (Fig. 5D). In addition, KEGG pathway analysis found that the majority of these downregulated DEGs were enriched in the PI3K-Akt signaling pathway, autophagy, Ras signaling pathway, chemokine signaling pathway, and Wnt signaling pathway (Fig. 5E); all of which have been shown to be closely related to ICH brain damage.
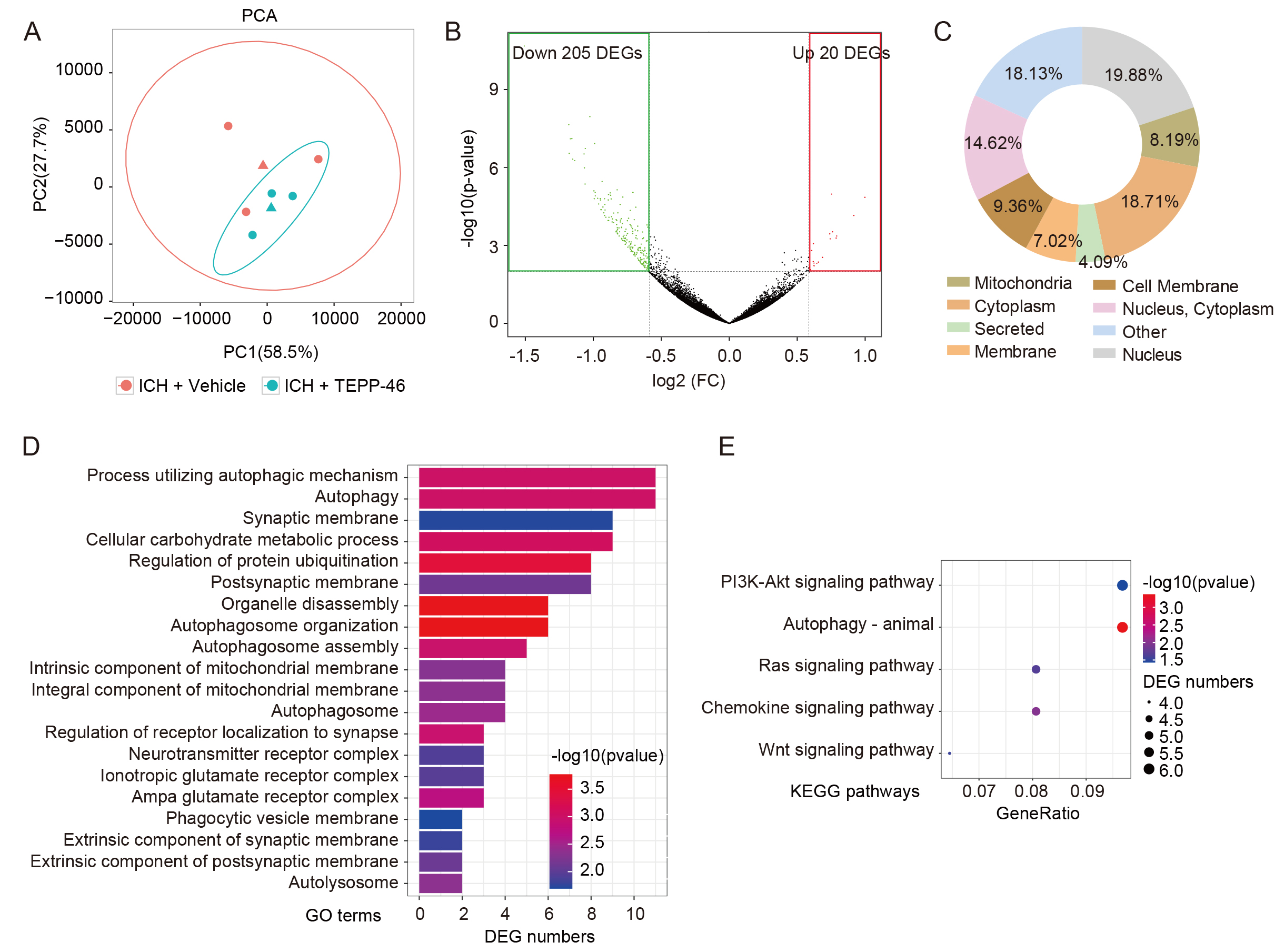 Fig. 5.
Fig. 5.Analysis of RNA-seq data of ICH brains. (A) PCA analysis reveals the dispersion degree of the transcriptome between the TEPP-46- and vehicle-treated ICH brains. (B) The volcano plot shows the down-regulated and up-regulated DEGs induced by TEPP-46 treatment for ICH brains compared with the vehicle-treated brains. (C) The subcellular localization of proteins encoded by down-regulated DEGs in (B). (D,E) GO (D) and KEGG (E) analysis of the down-regulated DEGs in (B). (n = 3 for each group). PCA, Principal Component Analysis; FC, fold change; DEGs, differentially expressed genes; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes.
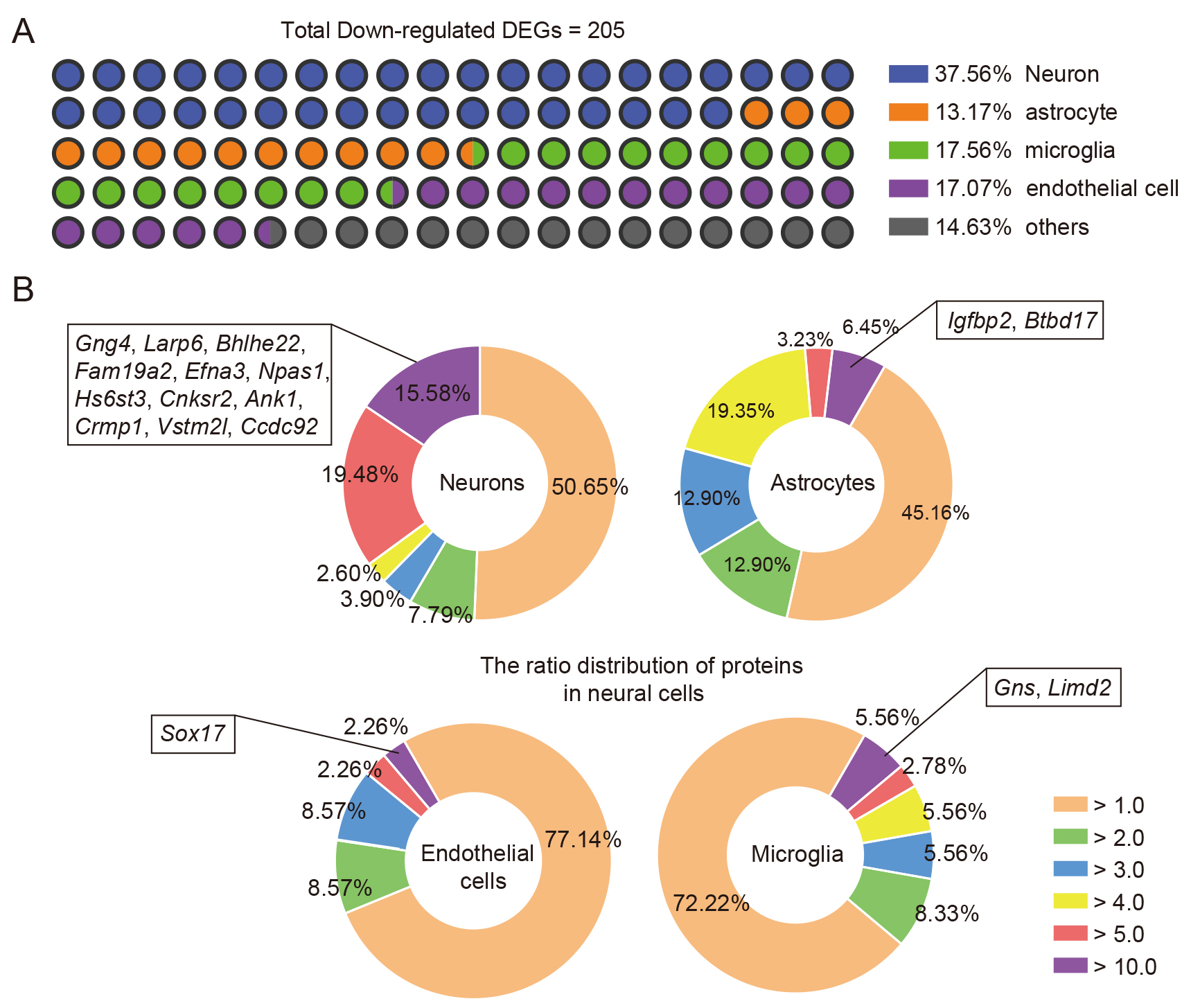
As the assessed samples were perihematomal brain tissues, cellular information
regarding changes at the transcriptome levels was lost. To demonstrate that
TEPP-46 treatment induced alterations in the transcriptome of neural cells in
response to ICH injury [29], we performed a conjoint analysis of previously
published RNA-seq data of brain cells and our down-regulated DEGs using our
previously published methodologies [21, 22]. After comparing the highly expressed
genes of brain cells [21, 22] with the down-regulated DEGs, we showed that the
majority of the DEGs were relatively highly expressed in neurons, microglia,
endothelial cells, and astrocytes (Fig. 6A); 37.56% of total down-regulated DEGs
were identified in neurons, 17.56% in microglia, 17.07% in endothelial cells,
and 13.17% in astrocytes (Fig. 6A). Considering that the levels of mRNA
expression in cells vary greatly, and that higher mRNA expression may be
important for cell function, the specificity ratio of the DEGs was further
analyzed to evaluate whether these down-regulated DEGs induced by TEPP-46
treatment could potentially influence the biofunction of neural cells. The
results depicted by pie charts indicate that 12 down-regulated DEGs
(Gng4, Larp6, Bhlhe22,
Fam19a2, Efna3, Npas1, Hs6st3,
Cnksr2, Ank1, Crmp1, Vstm2l, and
Ccdc92) with a specificity ratio
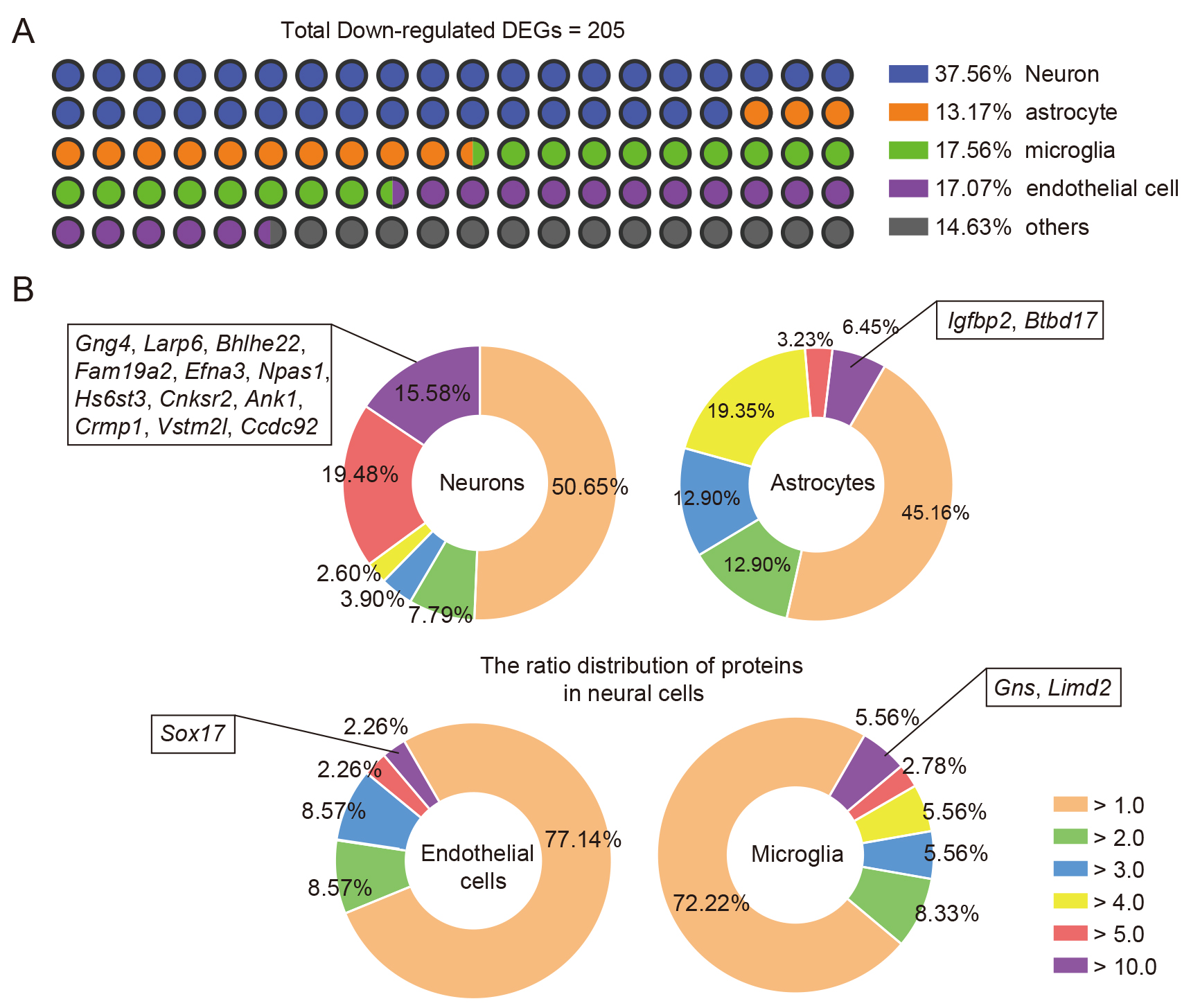 Fig. 6.
Fig. 6.Combined analysis of the down-regulated DEGs of ICH brains induced by TEPP-46 and transcriptomics data. (A) The major cellular distributions of down-regulated DEGs of ICH brains after TEPP-46 treatment compared with vehicle treatment. (B) The pie charts show the specificity ratio distribution of the down-regulated DEGs in neurons (upper left), astrocytes (upper right), microglia (lower right), and endothelial cells (lower left).
The switching of the role of PKM2 from metabolic enzyme to protein kinase is tightly related to its oligomerization states, such as tetramer for metabolic activity and monomer for kinase. The latter is a direct result of PKM2 detetramerization, which leads to nuclear translocation to exert its protein kinase activity. We investigated the role and potential mechanism of the protein kinase PKM2 in the regulation of brain injury after ICH. Our results revealed that the PKM2 expression level in perihematomal brain tissues was increased in reactive astrocytes and microglia of ICH mice, and about 30% of the PKM2-positive cells exhibited nuclear translocation. The treatment of ICH mice with TEPP-46, a small-molecule activator stabilizing PKM2 in tetramer form, resulted in decreased glial cell activation, reduced brain injury, and enhanced functional recovery, as well as the inhibition of PKM2 nuclear translocation. Mechanistically, TEPP-46 treatment induced a significant number of down-regulated DEGs, which may be associated with regulating neural cell functions after ICH, according to RNA-seq analysis. Consequently, we presented evidence for the regulation of brain injury and glial activation by PKM2 protein kinase, identifying a unique therapeutic target for inhibiting PKM2 nuclear translocation in order to alleviate brain injury and glial activation after ICH.
PKM2 is highly expressed in cancer cells and activated immune cells, which may
be attributable to the fact that PKM2 catalyzes the final and rate-limiting
reaction in the glycolytic pathway, as these cells consume much energy to support
their biological processes. Interfering with PKM2 prevents cancer cell metabolism
and proliferation by inhibiting the aerobic glycolysis of cancer cells [32, 33, 34].
Furthermore, the activation of NOD-like receptor thermal protein domain
associated protein 3 (NLRP3) and absent in melanoma 2 (AIM2)
inflammasomes [35] as well as the release of high mobility group box 1 (HMGB1)
protein [12] are exacerbated by PKM2-mediated glycolysis in macrophages. This
exacerbation contributes to the progression of lethal endotoxemia and sepsis,
hence enhancing the toxic impact of macrophages. Furthermore, a recent study
demonstrated that pharmacological inhibition of PKM2 moderated microglial
activation and enhanced spatial learning and memory in Alzheimer’s disease mice.
This was due to the exacerbation of microglial dysfunction induced by the
positive feedback loop involving glycolysis/H4K12la/PKM2 [14]. The aforementioned
studies have provided insight into the role and mechanism of PKM2 in disease
progression, with particular emphasis on its function as a metabolic enzyme. PKM2
might, on the other hand, function as a protein kinase, which usually occurs
after PKM2 nuclear translocation and is a direct consequence of its
detetramerization. PKM2 has been shown to participate in regulating gene
transcription [18, 19] and controlling the functions and activation of immune
cells such as dendritic cells and macrophages [13, 36, 37], which may be
attributable in part to the fact that PKM2 nuclear translocation regulates Hif-1
activity and interleukin-1 (IL-1) induction [38]. In addition, PKM2 nuclear
translocation can also promote tumor angiogenesis by regulating hypoxia-inducible
Factor-1
Furthermore, comprehending the glial activation upon ICH requires a knowledge of the mechanism triggering the PKM2 tetramer to monomer transition. One previous study found that the upregulation of astrocytic signal transducer and activator of transcription 3 (STAT3) signaling was related to nuclear translocation of PKM2 [40] and, combined with our and others’ findings, indicates that STAT3 signaling is increased in response to brain damage induced by ICH [23, 41]. Accordingly, we hypothesize that the increased STAT3 signaling may contribute to PKM2 detetramerization in glial cells after ICH.
Furthermore, we performed RNA-seq to investigate the potential mechanisms of inhibition of PKM2 nuclear translocation on influencing the brain injury of ICH. We found that more than 90% of DEGs were down-regulated in ICH mice treated by TEPP-46, indicating that inhibition of PKM2 nuclear translocation may reverse the highly expressed genes triggered by ICH. As TEPP-46 treatment could both decrease glial activation and reduce the brain damage of ICH, we performed a conjoint analysis of the down-regulated DEGs of perihematomal brain tissues with previously published transcriptome data of neural cells to investigate potential changes in neural cells after ICH in mice treated with or without TEPP-46. The majority of down-regulated DEGs were found in neurons, followed by microglia, endothelial cells, and astrocytes. Notably, 15.58% (12/77) of down-regulated DEGs were specifically highly expressed in neurons, further suggesting that these genes may be involved in the regulation of neuronal activity, injury, and death after ICH, either directly or indirectly influenced by the glial activation caused by TEPP-46 treatment.
Nevertheless, our study has some limitations. For instance, even though we found that TEPP-46 treatment offers neuroprotective benefits for ICH mice, given that PKM2 is extensively expressed in numerous cells and organs, it remains to be determined whether and to what extent it influences the function of peripheral organs. In addition, the specificity of TEPP-46 on stabilizing PKM2 tetramer formation in ICH brains remains to be thoroughly evaluated. Therefore, despite that we might have discovered a potential therapeutic treatment for ICH, these limitations will impact whether TEPP-46 can be applied clinically. As a result, considerably more effort is required to translate our results into effective therapy.
This study investigated the protein kinase activity of PKM2 in the regulation of glial activation and brain damage caused by ICH. The increased PKM2 nuclear translocation that we have shown provides new molecular insights into glial activation during ICH pathogenesis and offers a potential therapeutic strategy for reducing ICH-induced brain damage.
The datasets of this study are available from the corresponding authors upon request. The datasets of RNA-seq for this study can be found in the GEO accession GSE205082.
Conceptualization: YT, SGY and XYX; Investigation/Methodology: YJL, XXZ, SHY, ZQZ, SQL, and JYS; Writing - original draft: XYX; Writing - review & editing: SGY. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal study was reviewed and approved by Animal Ethics Committee of Chengdu University of Traditional Chinese Medicine (CDUTCM-2020-0116).
Not applicable.
This work was supported by the Science Foundation for Distinguished Young Scholars of the Science and Technology Department of Sichuan Province (2020JDJQ0046).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.