



1 Department of Pharmaceutical Sciences, University of Perugia, 06123 Perugia, Italy
Abstract
Great interest is aimed at understanding the inflammatory responses at the level of the central nervous system (CNS), referred to as neuroinflammatory. The environment and the duration of the inflammatory responses are essential factors for comprehending the biochemical and pathophysiological consequences induced by the inflammatory state. Specific inducers of inflammation associated with neurodegenerative disorders can activate inflammatory processes and produce mediators that potentiate neurodegeneration. Immune responders in the brain include microglial cells, astrocytes, and mast cells. A number of human pathologies are recognized to have an inflammatory component, including disorders related to brain function. Emerging evidence also attributes an important role to intestinal microorganisms in disorders related to brain function. In the gut-brain axis, the intestinal microbiota produce a variety of molecules and neurotransmitters, transform primary bile acids into secondary bile, and synthesize short-chain fatty acids. Communication within the gut-brain axis occurs through several pathways, including the immune system, the enteric nervous system, the vagus nerve, and the production of microbial metabolites. The CNS responds to this input from the gut by modulating the activity of the autonomic nervous system and the hypothalamic-pituitary-adrenal axis, which manages adrenocortical hormones. In this perspective, gut microbiota may influence neural function by influencing microglia, astroglia, and mast cells. Conversely, the relationship between neurons, microglia and synaptic alteration may also involve gut microbiota. The purpose of this review is to provide a concise overview of the mechanisms involved in communication between intestinal microbiota and the brain and how this contributes to the management of neuroinflammation.
Keywords
- neuroinflammation
- astrocyte
- microglia
- mast cells
- gut
- intestinal microbiota
- butyrate
- lipopolysaccharide
The brain and the immune system are thought to have a close biochemical bond. Neuroinflammation is an inflammatory response within the central nervous system (CNS), mediated by the production of a variety of factors, including cytokines, chemokines, reactive oxygen species (ROS), and other secondary molecules. These mediating factors are produced by glia located within the CNS, as well as by endothelial cells and peripheral immune cells [1]. The level of neuroinflammation depends on factors such as the environment of the disease, duration, and course of the insult. The neuroinflammatory cascade involves alterations in the interplay between glial cells and neurons as a consequence of the activation of microglia and astrocytes [2]. Inflammation can lead to immune cell recruitment, tissue injury, ischemia, and cell death [3]. While neuroinflammation can be beneficial, prolonged activation of inflammatory mediators can cause brain damage [1].
Historically, the brain was considered an immunologically privileged organ, as the blood-brain barrier (BBB) prevents peripheral immune cells from entering the brain. However, this concept has been revised. An innate immune response can be initiated in response to both pathogens and damage within the CNS. Glial cells, microglia, and astrocytes constitute the major components of the neuroinflammatory response and their activation correlates with peripheral immune activity [4]. Under normal conditions, microglia and astroglia are involved in guarding the brain parenchyma to maintain homeostasis, removing glutamate, and remodeling and reforming neuronal interactions [5]. They also phagocytose damaged neurons and promote tissue repair. Astrocytes clear debris from cerebrospinal fluid and exert a neuroprotective role. Glial cells have beneficial and anti-inflammatory functions under both physiological and pathological conditions [6], such as phagocytosis, steroid release, free radical depletion, and cell repair. On the other hand, cytokine release and free radical generation can induce neural cell death and synaptic transmission alteration.
Thus, brain injury can occur when an imbalance is established between the regulation of anti-inflammatory and pro-inflammatory functions [2]. In addition, mast cells (MCs) present in various brain areas take part in this delicate balance of inflammatory mediators, as an important source of pro-inflammatory cytokines [7]. Further players are the gut microbiota and its associated metabolites that can influence neuroinflammation through interactions between the CNS and the gut through the “microbiota-gut-brain axis” [8]. This review aims to give a comprehensive overview of the cellular mechanisms implicated in neuroinflammation, particularly emphasizing the essential two-way communication pathway between the CNS and the gut.
Immune responders in the brain include microglial cells, astrocytes, and mast cells. Microglial cells are innate immune cells of the CNS and play a critical role in mediating neuroinflammatory responses. Microglia perform primary immune and macrophage surveillance functions in the CNS through cytokine and chemokine release. They are resident CNS cells found in both the white and gray matter of the brain and spinal cord. Microglial cells account for about 10% of CNS cells, develop early during embryogenesis from myeloid precursor cells in the embryonic yolk sac [9], and possess the same progenitor as other macrophages [10]. According to this evidence, microglia are long-lived cells with limited turnover, not being replaced by bone marrow myeloid cells [11]. However, microglial turnover is guaranteed by a progeny source present in CNS [12]. The low turnover of microglial cells makes them sensitive to the potential pro-inflammatory effects of aging, damage, and other stressful stimuli [13]. Microglial cells are indispensable for immune monitoring and aiding neurons, as they can adapt their phenotype and activity in neurodegenerative conditions.
Imaging studies have shown that microglial cells use their processes to actively analyze their microenvironment [14]. They are also responsible for the propagation of inflammatory signals to the periphery [15]. These are critical communication signals between the immune system and the brain. Microglial cells can modulate synaptic formation and remodeling of the synaptic circuit [16]. In the event of infection, disease, abnormal stimulation, or in the presence of neurotoxins, microglial cells are prompted to act as inflammatory mediators, quickly changing their transcriptional profile to produce inflammatory molecules [17, 18]. Depending on the neuroinflammatory trigger, cytokine and chemokine production may facilitate leukocyte cell recruitment into the brain [19]. In addition, activated microglia undergo an intracellular reorganization that modifies the expression patterns of receptors present on their cell membrane. Such alterations permit microglial cells to move to the site of a lesion, where they can increase their phagocytic capacity to engulf and digest dead or damaged neurons [20].
Active microglial cells exert beneficial functions, as they promote pathogen
clearance and tissue regeneration, and facilitate neural function and survival
through the release of trophic factors. Conversely, excessive or prolonged
microglia activation may result in pathological modifications and neurobehavioral
consequences, including depressive states and cognitive deficits [21]. In
particular, the nuclear transcription factor, nuclear factor kappa-B
(NF-
Astrocytes have roles that go beyond the task of supporting nerve cells, as they can affect synaptic activity and neuronal circuits [22]. An increase in astrocyte number and size has been shown to reflect brain size and cognition [23]. Astrocytes can trigger various responses, including increasing intracellular calcium levels, assisting in repair processes, and promoting inflammatory and pathological states. As a result, astrocytes have been described as heterogeneous cells.
Studies have suggested that similar to macrophages, astrocytes exhibit two states of activation, namely A1 and A2 [24]. Studies have found that pro-inflammatory cytokines, which can activate the A1 phenotype with neurotoxic activity, are present in the central nervous systems of individuals with Alzheimer’s, Parkinson’s, and multiple sclerosis. In contrast, A2 astrocytes are believed to have a neuroprotective role [25, 26].
Studies have evidenced an important role for gut microbiota in astrocyte maturation, particularly during embryonic and postnatal development. Together with other glial cells, astrocytes increase in number during the early postnatal period. Embryonic and postnatal development is likely influenced by a variety of genetic and environmental factors. In particular, it was found in a rat astrocyte model that the administration of a lactobacilli metabolite plays a crucial role in the development of the nervous system [27, 28]. Microglia and astrocytes interact with MCs through a positive feedback loop, and this condition contributes to sustaining an inflammatory state.
Recent studies have revealed that butyrate and indole-3-propionic acid (IPA) can enter the brain after being absorbed in the intestine and display advantageous effects on astrocyte activity. In particular, they have been effective in restraining the production of cytokines and kynurenine when human primary astrocytes are exposed to lipopolysaccharide (LPS) [29]. Furthermore, sodium butyrate has been reported to improve the cognitive impairment of Alzheimer’s disease (AD) by affecting astrocyte metabolism [30].
Other cells involved in immune responses in CNS are MCs [31]. MCs develop from hematopoietic CD34+/CD117+ pluripotent progenitors in the bone marrow and circulate in the blood in small numbers as immature precursors. MCs are located in the mucous membranes and connective tissue near blood vessels, lymphatic vessels, and nerves [32, 33, 34]. The differentiation process of mature MCs is influenced by the local microenvironment, which defines mature MCs phenotype and function (Fig. 1, Ref. [35, 36, 37, 38, 39]).
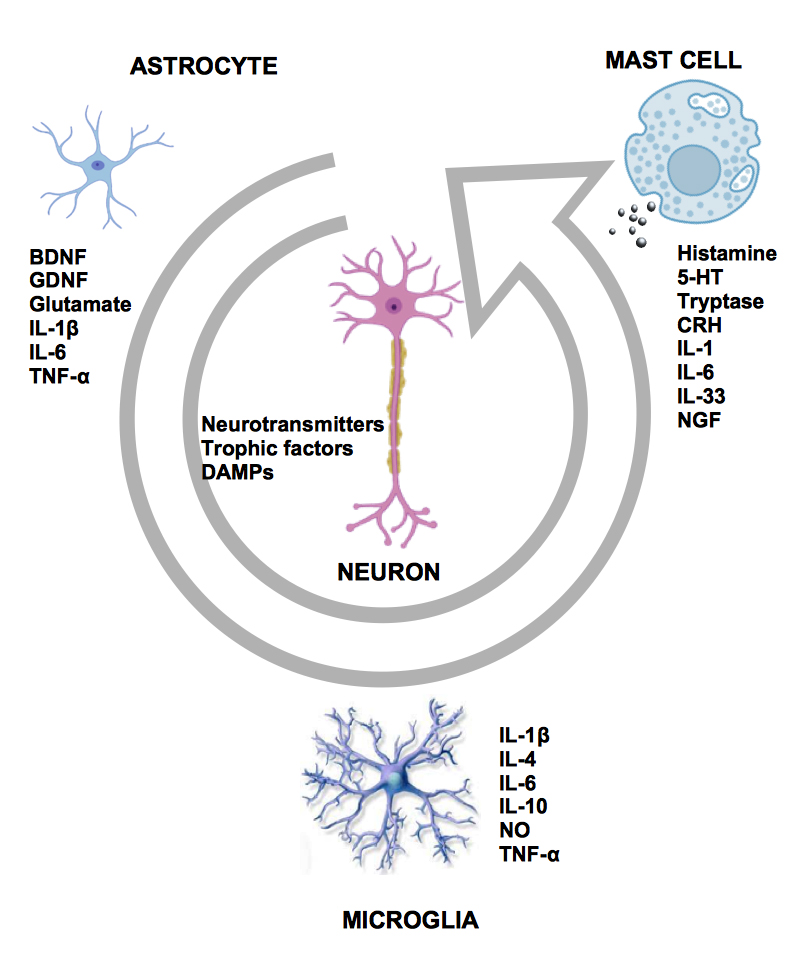 Fig. 1.
Fig. 1.Interaction between key cellular players in neuroinflammation.
Astrocytes, microglia, mast cells, and neurons communicate with each other
through a wide variety of molecules, such as pro- and anti-inflammatory
cytokines, which can have immunomodulatory. This has been especially shown for
IL-4 and IL-10 [35], IL-6 [36], IL-33 [37], tumor necrosis factor-alpha
(TNF-
Hence, the heterogeneity of MC precursors results in numerous MC subsets, each with its granule content, released mediators, reactivity, receptors, and species dependence [40]. MCs are placed near small vessels and nerves to respond to allergens, pathogens, and other agents entering the host after breaking the epithelial barrier. Although small in quantity, MCs respond first, and their location at the host/external environment interface allows them to function as sensors for invading microorganisms. MCs can detect environmental changes by communicating with other immune cells and generating an amplification signal [41]. During early development, MC progenitors enter the brain from the leptomeninges by infiltrating blood vessels and settling in the vessel-associated CNS [42]. Rat neurons can release stem cell factor, a cytokine required for MC survival, proliferation, and differentiation [43].
Neuroinflammatory responses are mediated by a variety of pro-inflammatory
cytokines (including interleukin, IL-1
A low inflammatory signal can be beneficial, such as increased neuroplasticity.
In this context, memory function can be influenced by neuroinflammation [45].
Neuroinflammation in CNS has been reported to be involved in memory and learning
processes [46, 47]. For example, prostaglandins have been reported to play a role
in hippocampal memory impairment [48]. In addition, IL-1 increases IL-6 and
TNF-
Even a medium level of transient inflammation can take on a beneficial and positive aspect when it is directed towards a remodeling free from damage, with the effect of tissue repair. Transient neuroinflammation involving the activation of glia and the production of neuroinflammatory chemical messengers (i.e., cytokines and chemokines) can occur. Microglial activation, cytokine, and chemokine production lead to physiological and behavioral responses that have positive significance without immune cell infiltration into the brain, BBB damage, or cell death [54]. A similar profile is observed for the psychological interpretation of stressors. Both of these interpretations do not result in tissue damage. Transient immune system activation has been exploited to promote immuno-conditioning and ultimately enable enhanced neuroprotection [55].
Deleterious or pathological neuroinflammation is jointly mediated by glia activation, cytokine and chemokine production, infiltration of peripheral immune cells, edema, increased permeability, and disruption of the BBB [56]. From all these, vasal occlusion, ischemia, cell death, and other inflammatory events can occur. This neuroinflammatory condition is associated with CNS infection, stroke, or disease [57] and can lead to functional alterations. The deleterious inflammation causes damage that can become chronic. It is related to autoimmune diseases, such as multiple sclerosis in humans or experimental autoimmune encephalomyelitis in mice [58]. In these disorders, there is axonal demyelination [59], with functional disability and axonal fragmentation. Chronic neuroinflammation is also associated with AD and amyotrophic lateral sclerosis [60]. The progression of AD involves protein misfolding and glia, followed by a pattern reflecting neuronal damage with infiltration of peripheral immune cells, neuronal atrophy, and eventually death.
Finally, chronic neuroinflammation observed during aging is associated with increased systemic inflammation. This condition is currently placed under the term of inflammaging. The state of fragility generally involves a critical reduction of neuroplasticity and is a risk factor for pathologies associated with cognitive decline [61, 62, 63].
The intestinal microbiota is a complex ecosystem consisting of multiple ecological niches of microorganisms, mainly bacteria, but fungi and viruses are also present. These microorganisms establish a mutualistic symbiosis with the host. An alteration of the immune system and any disturbance of the symbiotic relationship between the gut microbiota and the host can result in increased permeability of the intestinal epithelial barrier, thus facilitating bacterial translocation. Bacteria, microbial compounds, such as LPSs, and metabolites are transferred from the intestine into the bloodstream and travel to other tissues, including the brain, causing local inflammation [8, 64]. In addition, studies suggest that the gut microbiome exerts its role by modulating neuroinflammatory processes in CNS [8].
Various environmental and dietary elements can modify the bidirectional communication between the gut and the brain, leading to a transformation in the gut microbiota and consequently influencing the immune system [65, 66, 67, 68, 69, 70]. The brain controls the gut through hormone release, via the hypothalamic-pituitary-adrenal (HPA) axis, and through the autonomic nervous system (ANS) assisted by the adrenal medulla and the release of catecholamines [43, 70] (Fig. 2). Pro-inflammatory cytokines are potent stimulators of the HPA axis [33]. Neuroinflammatory mediators lead to the activation of the HPA axis and ANS. This, in turn, modulates the enteric nervous system, leading to an exacerbation of inflammatory conditions.
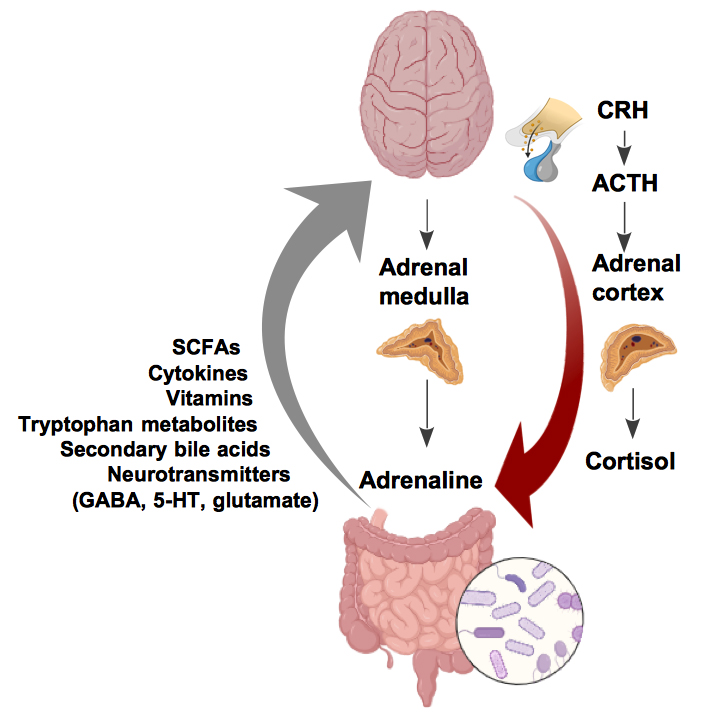 Fig. 2.
Fig. 2.The gut and brain are chemically linked through a variety of mediators, including neurotransmitters, cytokines, and microbial metabolites. ACTH, adrenal corticotropin hormone; SCFAs, short-chain fatty acids; GABA, gamma-aminobutyric acid. (Fig. 2 was created by https://www.biorender.com/).
The microbiota-gut-brain (MGB) axis develops at birth, with the colonization of the gut microbiota, and develops rapidly during the early stages of life [71, 72]. The MGB can be disrupted by various factors, including exposure to damaging substances like drugs, which can disturb the intestinal microbial balance and result in changes to the immune system and behavior linked to neurodevelopmental disorders. It has been suggested that myelination plays a role in the MGB axis influencing behavior and cognition, particularly regarding the Prefrontal Cortex (PFC). This area is responsible for the integration of signals from various brain regions to carry out complex decisions and memory retrieval [73, 74, 75, 76]. Relevant myelination occurs during neonatal life and persists into early adulthood, concurrent with the development of the MGB axis. Therefore, changes in this process can lead to adult deficits [77, 78, 79]. There is evidence of hypermyelination in the PFC of germ-free mice, an animal model born and reared under sterile conditions. As a result, these mice lack intestinal microbiota [76]. It was also reported that antibiotic administration during neonatal life results in oligodendrocyte damage, impaired myelination in the PFC, and behavioral deficits. This scenario fits into the context of intestinal microbiota dysbiosis in adulthood [61].
The functions performed by intestinal microbiota include metabolic, protective, and structural roles [70]. Gut microbiota can metabolize biologically active molecules, which are involved in multiple functions, from food. Among the different metabolites produced by the intestinal microbiota, such as tryptophan metabolites, secondary bile acids, vitamins, short-chain fatty acids (SCFAs), and butyrate (Fig. 2). Butyrate represents an important source of energy for colonocytes by stimulating turnover and differentiation. Growing evidence supports the idea that butyrate is engaged in beneficial processes in the body, including the brain [80, 81, 82]. In particular, excess butyrate not used in the colon or liver can be transported to the CNS, where it has positive neurological effects [83]. Consistent with these findings, oral administration of butyrate was reported to restore depressive and anxious states induced by chemotherapeutic agents in mice, and these conditions were related to neuroprotective and anti-inflammatory responses [84]. Moreover, butyrate can enhance CNS remyelination and neuroinflammation in the cerebral cortex induced by high-fat diet in animal models [85, 86]. The administration of a high-fat diet induces oxidative stress and hypothalamic inflammation [87]. In addition, reduced intestinal concentrations of SCFA are often found in disease models in which intestinal dysbiosis is detected [88]. Recent studies suggest that intestinal dysbiosis may influence the development and evolution of neurological disorders, as well as states of neuroinflammation [89]. Nevertheless, supplementation with SCFAs, such as butyrate, often does not restore intestinal levels despite improvement in dysbiosis outcomes [88, 90]. Thus, the mechanisms through which butyrate can affect neural physiology and behavior have not yet been fully elucidated. Changes in gut microbial composition can impact the integrity of the intestinal-epithelial barrier. An altered gut microbial composition can disrupt the intestinal epithelial barrier, allowing microbiota-derived factors such as LPS, a gram-negative bacterial outer membrane component, to enter circulation. It is an important trigger for the inflammatory cascade in response to a gram-negative bacterial infection. Physiological plasma LPS levels are low but increase during inflammatory bowel conditions, leading to systemic inflammation and BBB disruption [91]. LPS appears to translocate into the brain parenchyma and activate microglia and astrocytes to promote cytokine production and neuroinflammation. Indeed, administration of high levels of LPS to the blood or body of rodents can induce activation of microglia in most brain areas, memory deficits, and loss of brain synapses and neurons [92]. Furthermore, chronic pro-inflammatory stimuli of microglial origin have been shown to convert resting astrocytes into neurotoxic reactive cells, responsible for the death of neurons and oligodendrocytes [24].
Emerging data demonstrate the effects of secondary bile acids and tryptophan
derivatives, metabolites produced by the microbiota, on neuroinflammatory-related
disorders. In particular, the intestinal bile acid pool plays a relevant role in
attenuating neuroinflammatory processes [93]. In the small intestine, enzymes
produced by the microbiota can transform primary bile acids produced from the
liver, generating a pool of different secondary bile acids. Thus, the bile acids
processed are absorbed by enterocytes and primarily reach the liver. Low
concentrations reach extrahepatic organ systems. Bile acid receptors are
ubiquitously expressed in various organs and tissues of the body [94]. In
particular, the nuclear farnesoid X receptor and the cell-surface G
protein-coupled bile acid receptor 1 are found in the CNS, specifically in the
BBB, choroid plexus, and glial cells [94]. This suggests that bile acid may have
a role in the regulation of glial cell function. Indeed, studies in vitro have
highlighted the beneficial effects of bile acid supplementation on
neuroinflammation processes. Mouse microglial cell lines treated with acids
ursodeoxycholic acid (UDCA) secondary bile acid showed an attenuated production
of TNF-
In addition to bile acids, tryptophan, an essential amino acid, exerts
modulatory functions at multiple levels of the gut-brain axis [45, 98]. Indeed,
studies demonstrate the involvement of tryptophan metabolites in the processes of
neuroinflammation and neurodegeneration [99]. Tryptophan may be metabolized by
the microbiota resulting in various indole-containing compounds, indole-3-lactic
acid, indole-3-acrylic acid, indole-3-propionic acid, indole-3-acetic acid,
indole-3-aldehyde, and other intermediates [100]. Indoles are important
metabolites generated by the bacterial fermentation of tryptophan in the
gastrointestinal tract. Recent evidence has demonstrated their involvement in
limiting inflammatory processes in microglia [101, 102], suppressing
pro-inflammatory cytokine production in astrocytes, and alleviating
neuroinflammation in antibiotic-treated mice [64]. Tryptophan-derived metabolites
act as ligands for the xenobiotic receptors aryl hydrocarbon receptor (AhR), a
transcription factor expressed in various cell types, such as dendritic cells,
microglia, and astrocytes. The presence of AhR ligands in the cerebrospinal fluid
[103] suggests that the microbiota-derived tryptophan metabolites may act upon
the CNS-resident cells, such as microglia and astrocytes, which have the AhR
expressed on them, thus implying a direct effect. Recent data show that AhR
signaling by the microbiota-derived AhR ligands constrains the production of
inflammatory cytokines such as IL-6, TNF-
The gut microbiome is critical for maintaining neural development and myelination in the brain. Many studies have shown that alterations in the composition of the intestinal microbiota are linked to clinical conditions, including intestinal bowel disease, AD, and other neurodegenerative disorders. The relationship between gut microbiota, immunity, and CNS provides evidence that the neurological and immunological functioning of the brain may be affected by metabolites or derivatives of the gut microbiota. Consequently, certain bacterial strains, such as probiotics, may be combined with traditional treatments to restore eubiosis and potentially alleviate inflammatory and neuroinflammatory states [104, 105].
Further studies are necessary to clarify the mechanisms through which specific intestinal microbial niches and their derived molecules influence brain function and health. These studies will be important for establishing new therapeutic approaches for neural disorders, as well as tools for identifying biological markers for these diseases.
CNS, Central Nervous System; BBB, blood-brain barrier; MCs, mast cells; ROS, reactive oxygen species; AD, Alzheimer’s disease; MGB, microbiota-gut-brain; PFC, prefrontal cortex; SCFA, short-chain fatty acid; LPS, lipopolysaccharide; AhR, aryl hydrocarbon receptor; ANS, autonomic nervous system.
GT, conceptualization. GC and GT, writing-original draft. GT, writing-revision and editing. GT, visualization. GT, supervision. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Thanks to all the peer reviewers for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest. Giovanna Traina is serving as one of the Guest editors of this journal. We declare that Giovanna Traina had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Gernot Riedel.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.