






1 Department of Biology, McMaster University, Hamilton, ON L8S 4L8, Canada
2 Division of Medical Sciences, University of Victoria, Victoria, BC V8P 5C2, Canada
3 Department of Biology, University of Ottawa, Ottawa, ON K1N 6N5, Canada
Abstract
Neurodevelopment is a highly regulated process that relies on the precise regulation of gene expression. Numerous epigenetic mechanisms contribute and cooperate to ensure the proper execution of developmental gene expression programs. Indeed, disruption of the molecular machinery regulating the deposition or removal of epigenetics markers is associated with numerous neurodevelopmental disorders, including autism spectrum disorder and intellectual disabilities. Among the various epigenetic marks that are fundamental for brain development, research has recently begun to focus on the role of histone variants and their associated chaperone proteins. Replication-independent histone variants can replace replication-dependent canonical histones in neuronal chromatin, giving nucleosomes unique properties that allow them to influence transcription. The deposition and removal of histone variants into neuronal chromatin are controlled by chaperone proteins that are integrated into chromatin remodelling complexes. Several studies report that the deposition and removal of histone variants by chaperone proteins from genes during development is pivotal for the regulation of gene expression, suggesting they are fundamental for neurodevelopment. This review will focus on the histone variants H2A.Z and H3.3, and the exclusive chaperones that regulate their dynamics, in gene expression during neurodevelopment and the progression of neurodevelopmental disorders.
Keywords
- histone chaperone
- histone variant
- brain development
- neurodevelopmental disorders
Epigenetics is a term used to describe molecular pathways that chemically modify histones or DNA to regulate gene transcription without altering the DNA sequence. Initially, epigenetics were thought of as relatively immutable mechanisms that helped to confer cell identity. It is now understood that epigenetic mechanisms are highly dynamic, such that molecular effectors can “write” or “erase” marks in response to extracellular stimuli. Of particular interest are epigenetic modifications affecting histones, due to the broad range of effects they can generate on transcription. Histone proteins assemble in octamers (2 H2A-H2B dimers + 1 H3-H4 tetramer) that bind to 147 bp sections of DNA to form nucleosomes [1] (Fig. 1A). These histones are considered “canonical”, and not only serve a structural function but are also potent regulators of transcription. While nucleosomes can prevent transcriptional machinery from accessing DNA for gene expression, histone post-translational modifications (PTMs) can further limit or permit DNA access [2] (Fig. 1B). Numerous PTMs have been linked to active or repressed gene states. Furthermore, numerous proteins involved in the deposition (writers) and removal (erasers) of PTMs have also been extensively characterized.
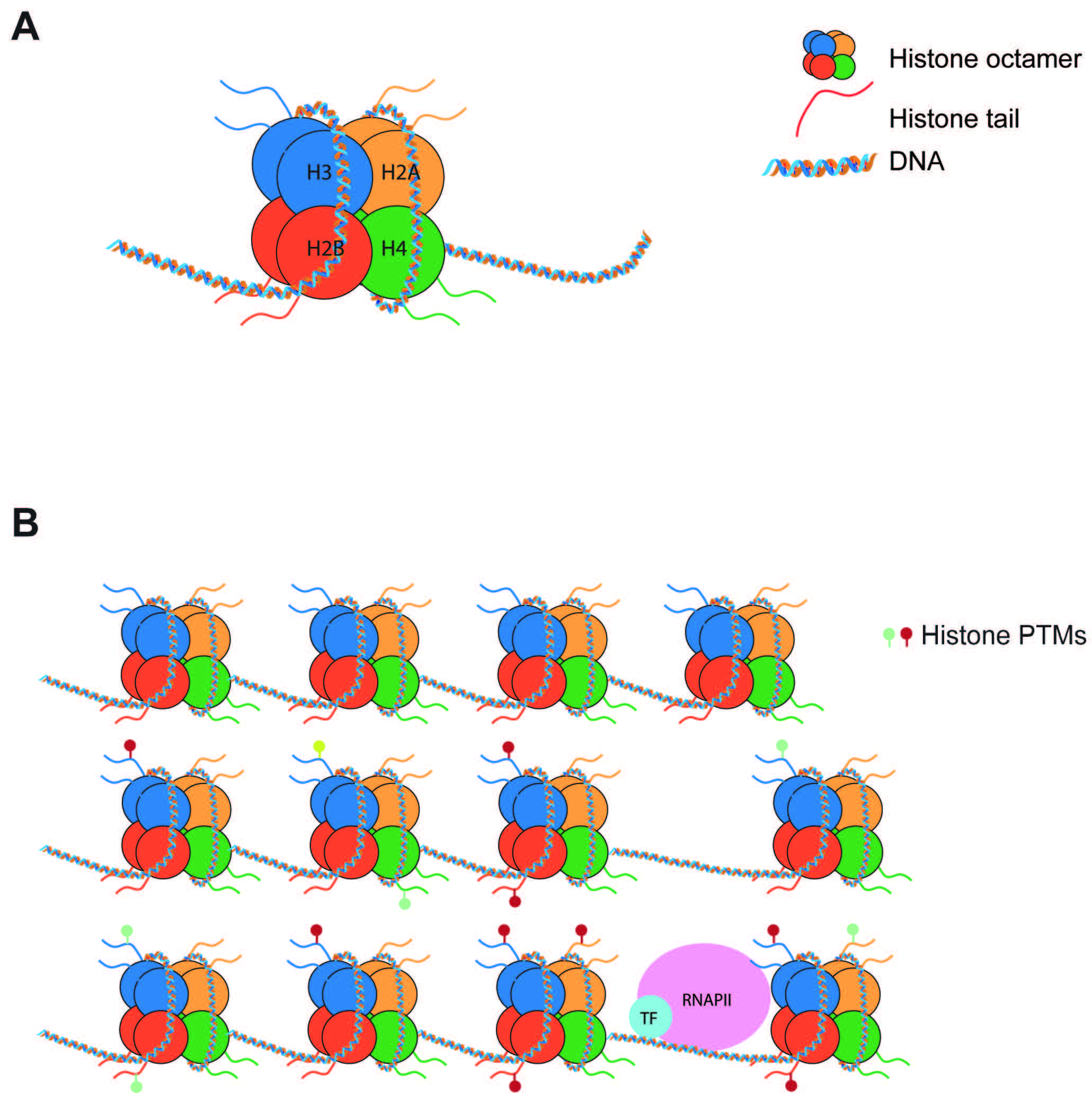 Fig. 1.
Fig. 1.Nucleosomes regulate gene expression. (A) Nucleosomes are composed of 147 bp of DNA wrapped around a histone octamer made of 2 copies of each histone (H2A, H2B, H3, H4). (B) Nucleosomes pose a barrier to transcription. Post-translational modifications of histone tails can increase or decrease this barrier to tightly regulate transcription. PTMs, post-translational modifications.
In metazoans, variants have been identified for all canonical histones (Table 1, Ref. [3, 4, 5, 6, 7, 8]). Despite having high similarity to their canonical counterparts, histone variants are encoded by separate genes and have different properties. Thus, they influence transcription in unique ways [9, 10, 11, 12] and represent a distinct epigenetic mark. Histone variants can be incorporated or removed from chromatin by specialized proteins called histone chaperones that function as writers and erasers [13, 14]. Two novel epigenetic mechanisms have been described that involve histone variants and their chaperones; histone variant exchange, which involves the substitution of a canonical histone with a variant histone [15], and histone variant turnover [16], a process whereby a histone variant is removed from a nucleosome and subsequently reincorporated (Fig. 2). Both mechanisms involve histone variants, and not canonical histones, as most histone variants can be incorporated into chromatin independently of the cell cycle. In contrast, canonical histones deposition is coupled to the S phase of mitosis [17]. This property of histone variants makes them of particular interest for post-mitotic cells, such as neurons, where they represent the only source of histone replacement [16]. Importantly, some histone variants have exclusive histone chaperones that confer high specificity to their genomic location and molecular properties [14, 18, 19] (Table 1).
| Variant | Role | Tissue | Exclusive chaperone | Role in neurodevelopment/NDD |
| H2A family | ||||
| H2A.Z (H2A.Z.1, H2A.Z.2) | Chromatin dynamics and transcription | Global | SRCAP (incorporation) | YES [3] |
| Anp32e (removal) | ||||
| MacroH2A (macroH2A1, macroH2A2) | Gene silencing | Global | YES [4] | |
| H2A.X | DNA damage response | Global | ||
| H2A.B | Transcription and RNA splicing | Testis and brain | ||
| H3 family | ||||
| H3.3 | Chromatin dynamics and transcription | Global | HIRA (incorporation) | YES [5, 6, 7, 8] |
| ATRX-DAXX (incorporation) | ||||
| CEN-H3 | Centromere identity and genome stability | Global | HJURP | |
| H2B family | ||||
| TH2B | Histone to protamine transition | Testes | ||
| H2B.W | Histone to protamine transition | Testes | ||
| H2BE | Cell survival | Olfactory neurons | ||
| H4 family | ||||
| H4G | Upregulation of rDNA transcription | Global | ||
NDD, neurodevelopmental disorders; SRCAP, Snf2-Related CREBBP Activator Protein; HIRA, histone cell cycle regulator; ATRX-DAXX, Alpha thalassemia and mental retardation X-linked/Death domain associated protein, HJURP, Holliday junction recognition protein.
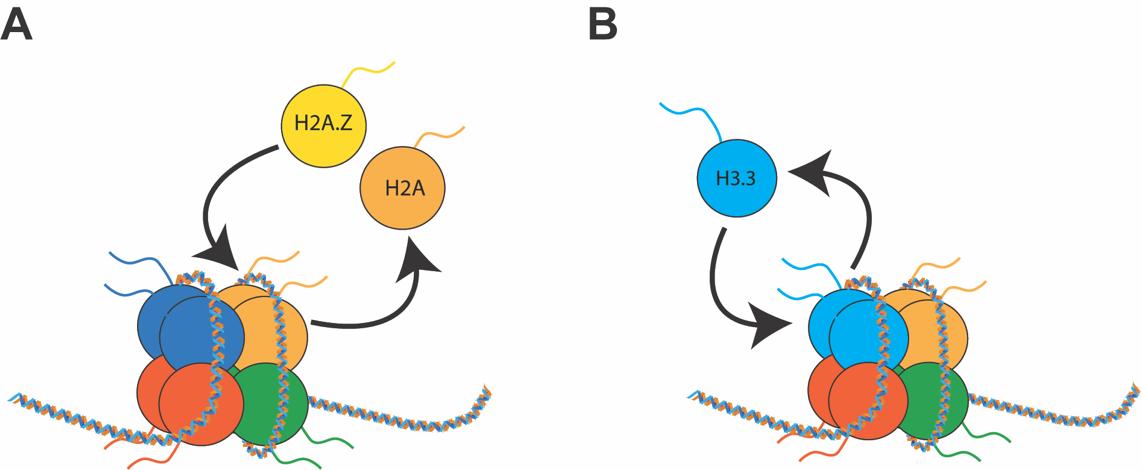 Fig. 2.
Fig. 2.Dynamics of histone variants. (A) In histone variant exchange, one canonical histone is removed and replaced with a histone variant. (B) In histone variant turnover, the same histone variant is continuously going through cycles of removal and reincorporation. Both processes are mediated by RNA Pol II and specific chaperones.
In this review, we will focus on histone variants H2A.Z and H3.3. These are the only variants for which exclusive chaperones have been identified and implicated in neurodevelopmental disorders. First, we will briefly describe their general functions and the network of specific chaperones they are bound by. Then, we will describe the role of these variants and their chaperones in neurodevelopment. Finally, we will review the role of histone variants and their chaperones in neurodevelopmental disorders.
The H2A histone family is exceptionally diverse. It is comprised of four replication independent histone variants as well as their isoforms and splice variants (Table 1). H2A.Z is one variant of H2A and is highly conserved between organisms, including yeast and humans [20]. Despite H2A.Z sharing 60% identity with H2A, fundamental differences allow H2A.Z to control nucleosome stability and transcription in unique ways [20]. H2A.Z exists in two isoforms, H2A.Z.1 and H2A.Z.2 [21], which are encoded by two different genes (H2afz and H2afv, respectively) [22, 23] and only differ by 3 amino-acid residues. Despite being similar, the two isoforms have been shown to play different roles [24].
At the molecular level, H2A.Z has been implicated in numerous cellular processes, including the maintenance of genome integrity [25], cell cycle progression [26], DNA repair and recombination [27, 28], and regulation of transcription [29]. H2A.Z has been implicated in both the activation and repression of transcription [29], indicating that the role of H2A.Z is mediated by other factors. H2A.Z also has a critical role in biological processes like cell fate specification [30], epithelial mesenchymal transition (EMT) [31], and memory formation [32]. Nevertheless, the mechanisms by which H2A.Z acts in these different, and often contrasting, cellular processes remain unclear. It has been proposed that the unique functions of H2A.Z may be dependent on factors including the organism in which it is found, binding partners, PTMs, and genomic location.
A key finding regarding the function of H2A.Z was the identification of mechanisms responsible for its deposition and removal into chromatin. Studies in yeast demonstrated that the SWI2/SNF2-Related 1 Chromatin Remodeling Complex (SWR1 complex) was responsible for H2A.Z deposition [33]. Mammalian homologs of the SWR1 complex form a family of Inositol auxotrophy 80/Sucrose Non-Fermentable (INO80/SNF2) chromatin remodelling complexes, which include INO80, Snf2-Related CREBBP Activator Protein (SRCAP), and E1A-binding protein 400 (p400) (Fig. 3). Both SRCAP and p400 complexes have been shown to mediate H2A.Z incorporation in mammalian cells [34, 35]. These complexes are comprised of various proteins, some of which can be shared among different complexes (Fig. 3). H2A.Z-H2B dimers have been shown to associate with the ATPase subunit. However, YL-1, a shared component of SRCAP and p400, is essential to bind H2A.Z-H2B dimers for their incorporation [36]. Hence, YL-1 shares chaperone duties with the ATPase subunit. Depletion of p400 or SRCAP in cell lines leads to reduced H2A.Z genomic occupancy [37, 38], indicating that both complexes are necessary to maintain H2A.Z levels. However, loss of p400 alone does not lead to loss of H2A.Z in all tissue or cell types [39], suggesting that the two complexes may have different roles depending on the type of tissue they are located in. In addition, p400 activity has been implicated in other chromatin processes, including histone acetylation [39, 40]. However, the SRCAP complex has not been found to have molecular functions other than H2A.Z incorporation, suggesting that p400 activity is not always linked to H2A.Z genomic regulation.
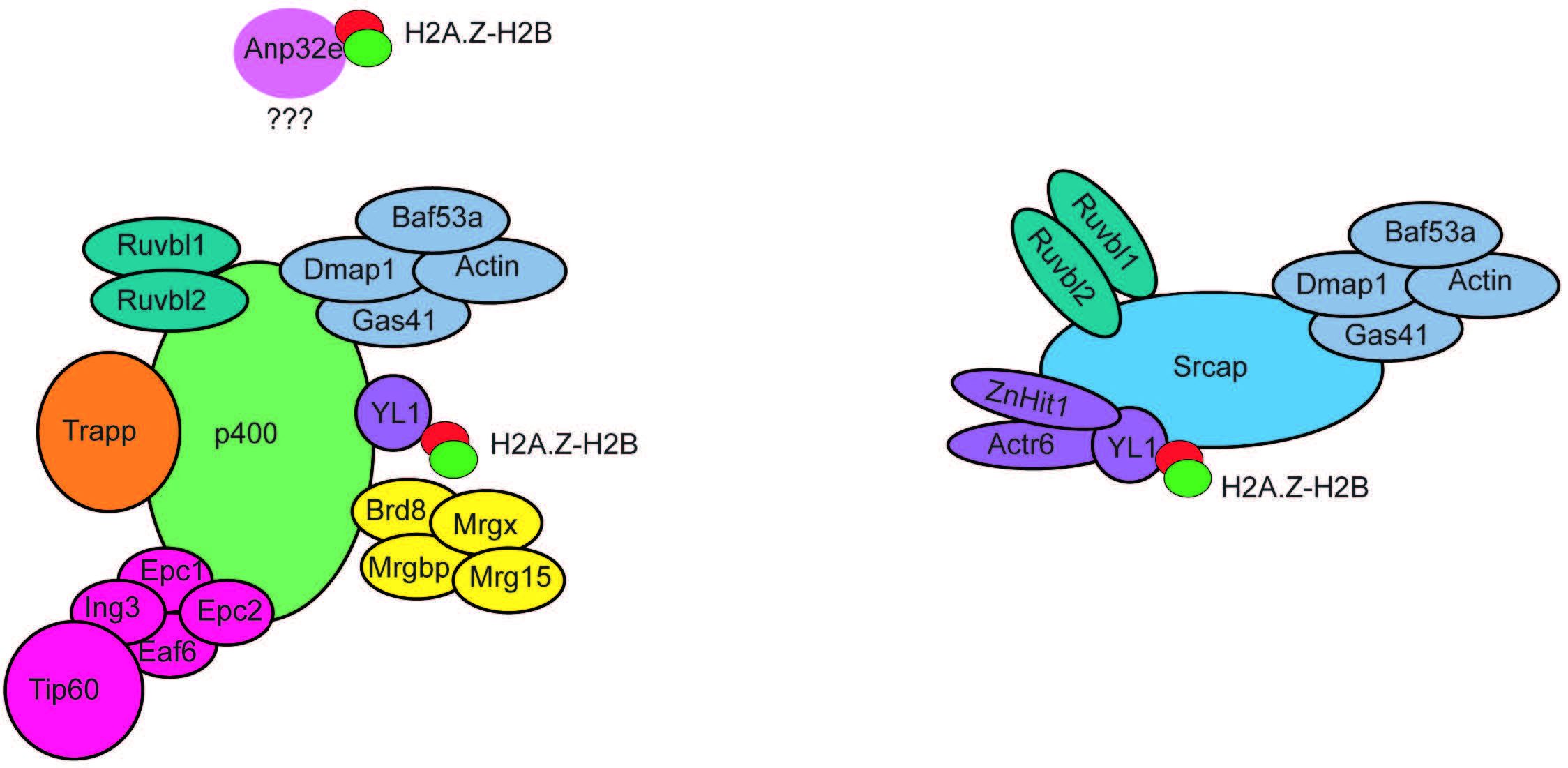 Fig. 3.
Fig. 3.Subunit composition of the p400 and SRCAP chromatin remodeling complexes. The p400 and SRCAP chromatin remodelling complexes are composed of multiple subunits organized into functional modules. Several subunits are shared between the two complexes. The subunit YL1 is fundamental to bind H2A.Z-H2B dimers to mediate their incorporation into chromatin. Anp32e associates with p400 in a still unknown way and mediates H2A.Z-H2AB removal from nucleosomes. SRCAP, Snf2-Related CREBBP Activator Protein.
Indirect evidence also suggests that other chromatin remodelling complexes may participate in H2A.Z deposition. Specifically, deletion of the embryonic stem cell specific BAF complex (esBAF) reduces nucleosome occupancy in favour of sub-nucleosome particles devoid of H2A.Z-H2B, indicating that esBAF may favour H2A.Z deposition [41]. Further, depletion of the nucleosome remodeling and deacetylase (NuRD) complex in the cerebellum leads to the loss of H2A.Z at promoters of activity-dependent genes [42].
Only one histone chaperone has been identified in mammals that can mediate H2A.Z removal from chromatin. Acidic nuclear phosphoprotein 2e (Anp32e) belongs to a family of 32 evolutionary conserved proteins containing an N-terminal domain with leucine-rich repeats (LRR) and an acidic C-terminal [43]. Anp32e was first identified in the mouse cerebellum as a protein whose expression was developmentally regulated and was found in high concentrations during synaptogenesis [44]. Anp32e is present in the cytoplasm, where it colocalizes and inhibits protein phosphatase 2A (PP2A) [44, 45]. Anp32e is also abundant in the nucleus of cerebellar granule cells, where it has been found to have chaperone activity specific to H2A.Z [18, 19]. Knockdown of Anp32e in HeLa cells results in the accumulation of H2A.Z at (promoters and/or enhancers) [18, 19]. Accordingly, overexpression of Anp32e in mouse embryonic fibroblasts (MEF), leads to a loss of H2A.Z binding a (promoters and/or enhancers) [18, 19]. Together these results suggest that Anp32e acts as a chaperone that controls the binding of H2A.Z at specific genomic locations implicated in transcription regulation.
Notably, Anp32e co-precipitates with components of the p400 chromatin remodelling complex [19], including the p400 ATPase subunit. This finding suggests that p400 complex activity linked to H2A.Z may be more complex than previously believed, where it could be involved in mediating the incorporation or removal of H2A.Z, depending on subunit composition.
The histone H3 family has one major replication-independent histone variant, H3.3 (Table 1), in addition to centromeric H3 (CenH3) [46, 47]. In mice, humans, and drosophila, H3.3 is encoded by two genes (H3f3a and H3f3b), producing the same protein with different untranslated regions [48]. H3.3 is only three amino acids different from canonical H3.2 and four from canonical H3.1. These specific residues mediate the unique properties of H3.3. In particular, mutation of the H3.3 residues abolished the ability of the histone to be deposited independently of the cell cycle and altered the genome-wide localization of H3.3, indicating that these amino acid residues determine H3.3 genomic enrichment and functional properties [49, 50]. These findings also demonstrate that histone variants’ properties depend on their genomic localization. Like H2A.Z, H3.3 is mostly enriched at active promoters and enhancers, suggesting a role for H3.3 in transcription [49, 51]. H3.3 is also highly enriched in gene bodies of actively transcribed genes, where it is speculated to contribute to the high nucleosomal turnover caused by Pol II sliding [52]. Histone H3.3 has also been found to be enriched at telomeres and pericentromeric heterochromatin [49], indicating it has a role beyond regulating active transcription.
Two highly specific H3.3 chaperone complexes have been identified, histone cell cycle regulator (HIRA) and death domain associated protein/alpha thalassemia and mental retardation X-linked (DAXX-ATRX) (Fig. 4). These complexes are exclusive for H3.3 and mediate the incorporation of H3.3 at different genomic loci. The HIRA complex contains Ubinuclein 1 or 2 (UBN-1/2) (Fig. 4) [53, 54], and mediates H3.3 incorporation at active regulatory elements (enhancers, promoters) and gene bodies [54]. Importantly, H3.3 deposition by HIRA that is dependent on UBN-1/2, is exclusive to incorporating newly synthesized H3.3 [55]. Incorporation of recycled H3.3 at the same elements also requires HIRA but relies on the activity of general H3-H4 chaperone anti-silencing factor 1 (ASF-1) [55]. The DAXX-ATRX complex (Fig. 4) mediates the incorporation of H3.3 at heterochromatin, and repetitive telomeric and pericentromeric DNA [49, 56]. The chaperone activity is carried out by death domain associated protein (DAXX), which is structurally similar to UBN-1 [57]. Depletion of HIRA leads to loss of H3.3 from gene bodies, promoters, and enhancers but not telomeres. In contrast, depletion of alpha thalassemia and mental retardation X-linked (ATRX) leads to loss of H3.3, specifically at telomeres and pericentromeres [49].
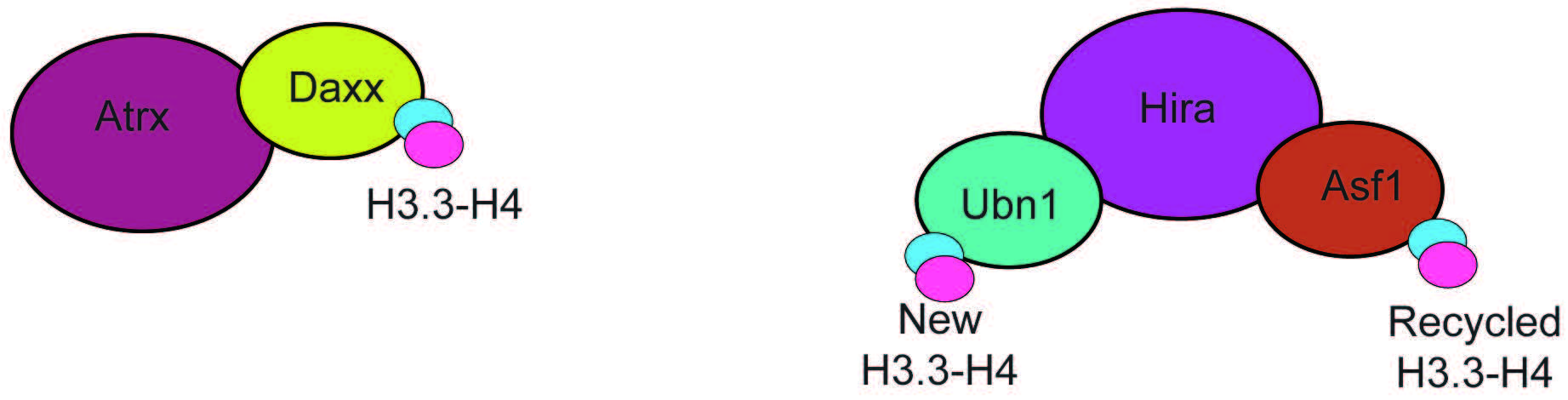 Fig. 4.
Fig. 4.Subunit composition of the ATRX-DAXX and HIRA chaperone complexes. The ATRX-DAXX and HIRA complexes are composed of few subunits and have the only function of mediating H3.3-H4 incorporation. Hira associates with two proteins that can bind H3.3, Ubn1, that mediates incorporation of newly formed H3.3-H4 dimers, and Asf1, a promiscous chaperone that binds recycled H3.3-H4. ATRX-DAXX, Alpha thalassemia and mental retardation X-linked/Death domain associated protein.
Notably, no specific chaperone that can remove H3.3 from chromatin has been identified. It is speculated that H3.3 is mainly removed by transcriptional activity [58].
The dynamic behaviour of histone variants was first identified in the adult mouse brain, where it was found that histone variant turnover was involved in regulating higher brain functions, such as memory formation [16, 32]. For example, following memory acquisition, H2A.Z was removed from the promoter region of numerous genes in the hippocampus [32, 59], a brain area involved in memory formation. Notably, H2A.Z removal mainly affected genes that became upregulated following learning [60], indicating that H2A.Z turnover is involved in the activity-dependent transcription of memory-related genes. Two hours following memory testing, H2A.Z levels returned to baseline [32]. Additionally, depletion of H2afz in the mouse hippocampus enhanced memory formation [32], implicating H2A.Z in memory suppression. H3.3 was similarly removed and reincorporated following the stimulation of primary neurons with potassium chloride to mimic neuronal activity [16]. However, loss of H3.3 in the adult mouse hippocampus impaired memory [16], indicating that different variants may have different roles in complex brain functions.
The involvement of histone chaperones in adult histone turnover to support memory formation remains unclear. It was demonstrated that neurons depleted of Anp32e displayed H2A.Z accumulation [60]; however, activity-induced H2A.Z was preserved. Remarkably, despite neurons devoid of Anp32e showing H2A.Z eviction, the genomic sites undergoing the evictions were different [60]. This could indicate that chaperone activity is still required for H2A.Z eviction, perhaps by regulating steady-state H2A.Z binding. It has also been demonstrated that RNA Pol II plays a significant role in H2A.Z removal, at promoters specifically [61], leading to the speculation that transcription could be the major driver of H2A.Z eviction. However, this possibility has yet to be investigated in brain tissue or neuron cell culture.
Conversely, H3.3 removal is mediated by RNA Pol II, and no removal chaperones have been identified so far. However, the reincorporation of the histone in neurons depends exclusively on HIRA [16]. Indeed, the knock-down of HIRA, but not DAXX, interfered with H3.3 activity-dependent turnover in neurons [16].
Numerous studies have provided evidence that suggests a role for H2A.Z and H3.3 in mammalian development. H2A.Z is present at low levels during early embryonic development. Consistent with this finding, H2afz knock-out mice develop normally until the blastocyst stage (embryonic day 3.5, E3.5) [62], but show embryonic lethality by E7.5, indicating an essential role for H2A.Z in early development. Remarkably, despite H2afv being present, H2afz knock-out mice still display embryonic lethality, suggesting that the presence of one isoform cannot compensate for the absence of the other. Similarly, the deletion of both genes encoding H3.3 in mice results in embryonic lethality by E6.5 [63]. Together these findings indicate that H2A.Z and H3.3 are crucial in early mammalian development.
Since histone variants’ activity depends on their incorporation into specific genomic loci by chaperones, the knockout of various chaperones also results in embryonic lethality (Table 2, Ref. [64, 65, 66, 67, 68]). Notably, Anp32e knock-out mice did not show embryonic lethality and appeared to develop normally into adulthood [64]. However, only 30% of Anp32e knock-out zebrafish survived to adulthood [69]. Loss of Anp32e in zebrafish embryos caused increased H2A.Z occupancy and early expression of developmental regulators [69]. This difference may indicate that in mammals, there are other mechanisms to relocate H2A.Z during early development that do not require the activity of Anp32e. One possibility is that the transcriptional activity of RNA Pol II can displace H2A.Z. Several studies have observed a link between Pol II activity and H2A.Z levels in different systems and cell lines [61, 70]; however, their activity during mammalian development has yet to be investigated.
| Mouse knockout | Embryonic lethality |
| p400 | E11.5 [65] |
| Srcap | ? |
| Anp32e | X [64] |
| Hira | E11 [66] |
| Daxx | E9.5 [67] |
| Atrx | E9.5 [68] |
Knock-out of p400 in mice results in embryonic lethality at E 11.5 [65]. It is important to note that the lethality of p400 knock-out may be independent of the chaperone activity of the complex, as p400 has been linked to numerous other pathways. While a knockout mouse model for Srcap is still lacking, truncating mutations in Srcap that abolish its ability to localize in the nucleus and bind DNA in Xenopus Laevis have been found to cause craniofacial defects and impairment in neural crest gene programs [71]. Notably, this phenotype is mimicked by the knock-down of H2A.Z.2, but not H2A.Z.1 [71]. This suggests the SRCAP complex may display isoform specificity during development. Homologous mutations in Hira caused embryonic lethality by E10 [66], with embryos showing numerous anomalies before lethality. Knock-out of Daxx also resulted in widespread apoptosis and early death in mouse embryos [67]. Finally, the knockout of Atrx gene on the X chromosome resulted in lethality in male embryos by E9.5 due to a defect in the formation of extra embryonic trophoblast [68].
Histone variants H3.3 and H2A.Z bear numerous characteristics in common in embryonic stem cells and lineage specification in genomic binding and the effects they mediate. Depletion of H2A.Z or H3.3 in mouse embryonic stem cells (mESC) induces early differentiation, a reduction in pluripotency gene expression [30], and early de-repression of developmental genes [72]. These findings suggest that H3.3 and H2A.Z are required for the maintenance of stem cell identity and regulation of lineage specification. Strikingly, H2A.Z and H3.3 display similar genomic localization in mESC, where they are enriched at highly transcribed promoters marked by the activation mark H3K4me3 [30, 72, 73] and at a subset of genes that are silent but are marked both by H3K3me3 and the repressive mark H3K27me3 [30, 72, 73]. These bivalent promoters mark key developmental genes in mESC, including members of the Pax, Hox and Dlx families [30], and become active upon the removal of H3K27me3 following differentiation stimuli.
H2A.Z and H3.3 have both been implicated in the regulation of bivalent genes. Specifically, it has been demonstrated that both variants are necessary to recruit and regulate polycomb repressive complex 2 (PRC2) activity [12], which is crucial for the deposition of the H3K27me3 mark to keep developmental genes silent in mESC. Hence, depletion of either variant can lead to the de-repression of bivalent genes caused by loss of the repressive mark, and precocious and defective differentiation [72, 73]. These findings indicate that H2A.Z and H3.3 are crucial for stem cell identity maintenance and also suggest that they are involved in the regulation of developmental genes and lineage specification.
mESC can be easily differentiated in neural precursors (NP), which are cells whose differentiation potential is restricted to cells of the neural lineage. Upon differentiation in NP, H2A.Z becomes enriched at different genes compared to mESC. Specifically, H2A.Z is lost at genes that become inactive and gained at genes that become active [30, 73]. In both lineages, H2A.Z is excluded from stably repressed genes that only bear H3K27me3, suggesting a role for H2A.Z in regulating the spatial and temporal expression of developmental and lineage-specific genes. Strikingly, upon differentiation from mESC to NP, H3.3 is also retained on the subset of bivalent genes that become active [49]. The similarities between H2A.Z and H3.3 genomic re-localization during early differentiation in neuronal lineage, together with the fact that H2A.Z and H3.3 are the only variants that have exclusive histone chaperones, suggest that these two histones and their chaperone machinery may represent a crucial yet still understudied epigenetic mechanism in development (Fig. 5).
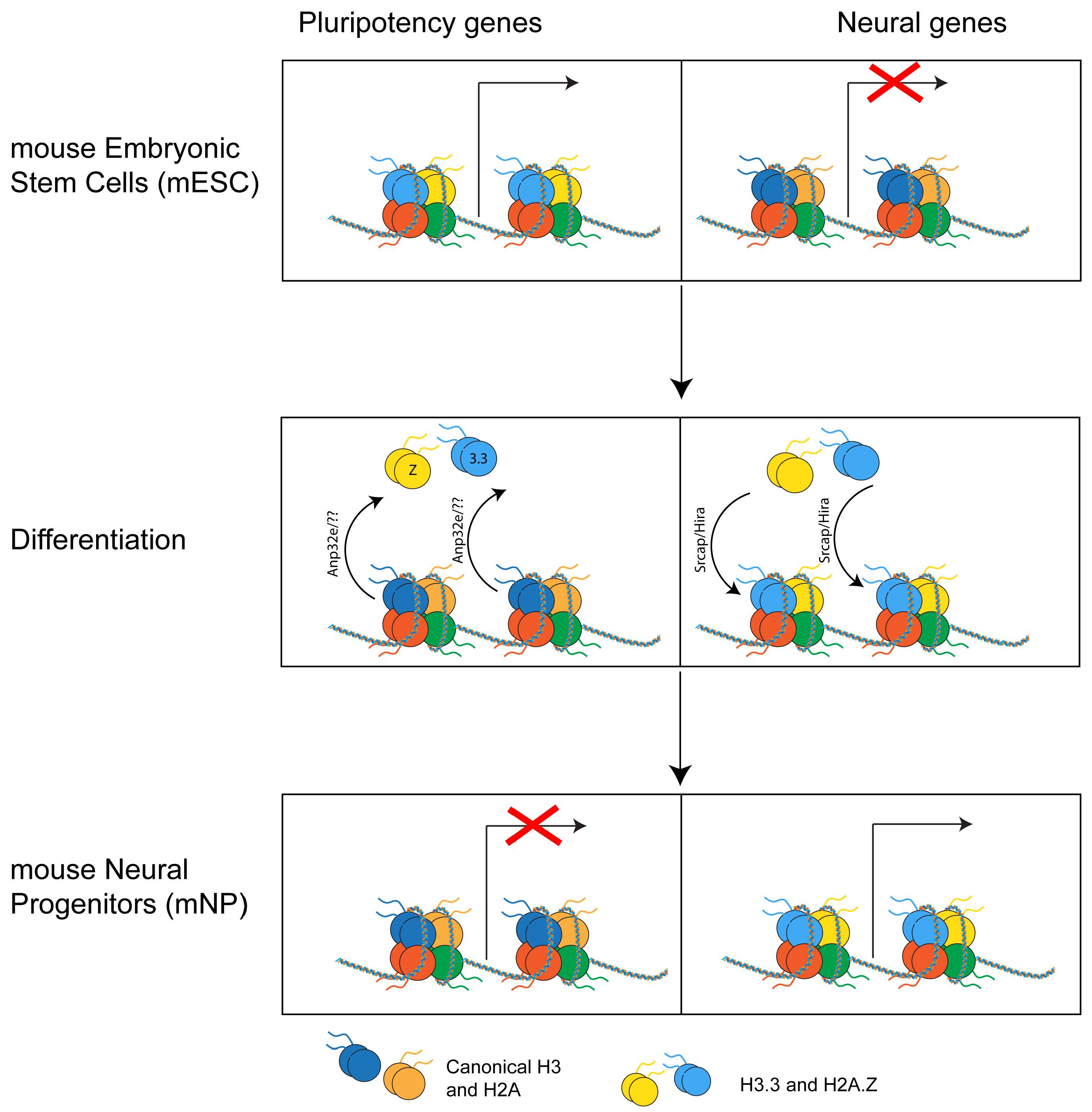 Fig. 5.
Fig. 5.Histone variants’ genomic relocation. During differentiation from mouse embryonic stem cells (mESC) to mouse neural progenitors (mNP), H2A.Z and H3.3 are removed from pluripotency genes and regulated at neural genes that become active.
Consistent with this hypothesis, manipulation of various chaperone complexes in mESC and other cell lines leads to dynamic redistribution of the altered chaperones, deregulation of bivalent genes, and impaired lineage specification. The role of Anp32e in development and lineage specification has yet to be fully explored. However, its link to the regulation of H2A.Z genomic localization is well established. A study conducted by Murphy et al. [74] compared H2A.Z localization in MEF Wild Type (WT) or knock-out for Anp32e. They found that depletion of Anp32e caused increased genome-wide chromatin accessibility and increased H2A.Z binding at promoters. Importantly, Gene Ontology (GO) analysis revealed that the increase in H2A.Z was specific for genes related to development and differentiation [74]. Notably, when comparing the enrichment of H2A.Z between mESC WT and MEF WT, the authors found that H2A.Z was relocated in MEFs at promoters of genes involved in mRNA processing and cell signalling [74]. These data suggest that Anp32e regulates H2A.Z genomic positioning to define a cellular function and differentiation.
In mESC, loss of Srcap recapitulates the effects of loss of H2A.Z, causing reduced H3K27me3 levels on bivalent domains, de-repression of developmental genes, and loss of mESC identity [39]. Importantly, the loss of Srcap also caused reduced H2A.Z occupancy [37, 39]. These data strongly indicate that Srcap and H2A.Z belong to the same molecular pathway and implicate the Srcap complex in the regulation of development through H2A.Z incorporation.
Recent findings strongly indicate that H2A.Z and H3.3 dynamics are key regulators of neurodevelopment. Knock-down of H2A.Z during cortical neurogenesis in mice increased the number of dividing intermediate progenitors and reduced the number of differentiated neurons [75], indicating that H2A.Z plays a role in neuronal differentiation and cell cycle exit. In addition, the deletion of H2A.Z during cortical development in mice also resulted in abnormal neuronal morphology [75], indicating a defect in neurodevelopment overall. The observed structural anomalies in the development of the cortex were accompanied by significant behavioural impairments. Specifically, the knock-down of H2A.Z during cortical development in mice led to increased anxiety, reduced social skills, and impaired memory [75]. This contrasts the role of H2A.Z in the adult mouse brain, where its depletion enhanced memory formation [32]. The different behavioural outcomes following H2A.Z depletion suggest a different functional role of the variant in the adult and developing brain. Further, RNA sequencing of the forebrains of mice with H2A.Z depletion indicated that at embryonic day 13 (E13) the lack of H2A.Z induced upregulation of genes involved in neuronal migration and differentiation, and downregulation of genes involved in transcription regulation and cortical development [75]. These data suggest that H2A.Z is crucial for regulating the proper spatial and temporal execution of gene expression to regulate neurodevelopment.
Similarly, the deletion of H3.3 reduced neural progenitor proliferation and enhanced differentiation [76]. Developing neurons lacking H3.3 also showed impairments in synapse maturation [16]. Recent work has also implicated H3.3 in the establishment of neuronal chromatin in early development, where H3.3 accumulation was found in the chromatin of new neurons formed at E13.5 [77]. Depletion of H3.3 before this accumulation led to the repression of genes that typically increase during development and the upregulation of genes that become silenced over the course of development [77]. Early loss of H3.3 also resulted in alteration in the landscape of H3K4me3 and H3K27me3, and perinatal lethality [77]. This suggests that H3.3 regulates essential developmental programs in the brain and is required for the establishment of neuronal transcriptomes. Interestingly, depletion of H3.3 after the initial accumulation (P0) did not cause lethality and permitted H3.3 dynamics in late development [77]. Additionally, single-cell RNA sequencing showed that cells retained their identity and only a limited number of genes showed significant alteration upon loss of H3.3 at this stage [77]. These data demonstrate that histone variants may play different roles in developing and mature cells.
The implication of H2A.Z and H3.3 in the regulation of transcriptional developmental programs raises the question of whether their genomic re-localization is necessary to permit developmental changes in transcription. While further research is required, considerable evidence supports the role of histone chaperones in neurodevelopmental programs by regulating variants’ abundance in the genome. For instance, loss of Anp32e in primary neurons caused increased H2A.Z levels at promoters, deregulation of numerous developmental genes, and impairment in dendritic development [60]. Notably, the altered dendritic phenotype observed in Anp32e depleted neurons and disruption in gene expression were rescued by the concomitant depletion of H2A.Z [60], suggesting that Anp32e regulates neuronal development through regulation of H2A.Z occupancy. Similarly, knock-down of HIRA in primary neurons caused impaired dendritic arborization [78], while loss of Atrx in the brain resulted in synapse abnormalities and impaired behavioural outcomes [79], potentially linking H3.3 turnover to neurodevelopment.
Recently, various de novo mutations in both genes encoding for H3.3 have been described in patients with neurodevelopmental delays, including altered facial features, hands, and feet [5, 6, 7]. A knock-in model of three mutations falling on residue 34 and changing the Glycine (G) to either Arginine (R), Valine (V) or Tryptophan (W), showed that each mutation presented with slightly different phenotypes [8]. At the molecular level, H3.3 G34R led to a severe reduction of H3.3K35me2/3 compared to the other two mutations, further highlighting the different outcomes that mutations can have when they affect the same residue [8].
Although no mutations in H2A.Z have been directly linked to the onset of neurodevelopmental disorders, Gretzinger et al. [3] found that fetal alcohol exposure affects hippocampal levels of H2A.Z and could therefore be involved in the pathogenesis of fetal alcohol spectrum disorder (FASD).
Additionally, mutations in a number of histone chaperones and their associated components have been observed in patients with various neurodevelopmental disorders, providing further evidence that histone chaperones play a critical role in ensuring proper neurodevelopment. For example, mutations in SRCAP have been identified in the rare genetic disease, Floating Harbor Syndrome [80]. This neurodevelopmental disorder is characterized by short stature, skeletal abnormalities, delayed speech development, and mild intellectual disability [81]. Mutations in SRCAP have also been identified in patients with bipolar disorder [82, 83], autism spectrum disorder (ASD) [84, 85], and developmental delay, hypotonia, musculoskeletal defects, and behavioural abnormalities (DEHMBA) [83]. SRCAP is also among the protein coding genes located in the 16p11.2 locus, whose copy number variation (CNV) is commonly associated with ASD [86, 87].
Mutations of other subunits of the SRCAP complex that also been found in other neurodevelopmental disorders; however, their link to H2A.Z occupancy has never been investigated. For example, heterozygous mutations affecting Baf53a (Actl6a), a shared component of SRCAP, p400, and other chromatin remodelling complexes, are associated with syndromic intellectual disability [88]. Dmap1, a shared subunit of p400 and SRCAP complexes, is mutated in autosomal dominant cerebellar ataxia, deafness, and narcolepsy (ADCA-DN) [89]. Additionally, microdeletion of 22q11.2 containing the HIRA coding gene causes DiGeorge syndrome, a condition that includes a variety of neurodevelopmental phenotypes, including intellectual disability, attention deficit/hyperactivity disorder (ADHD), and ASD [90].
Mutations in H3.3 chaperones and their subunits have also been associated with neurodevelopmental disorders. Specifically, mutations in ATRX can lead to the development of alpha thalassemia and mental retardation X-linked (ATR-X) syndrome, an intellectual disability characterized by autistic-like behaviours [91]. It has recently been demonstrated that mutations in ATRX mainly cluster in 2 domains and can have different outcomes on protein function [92]. While all mutations do not impair ATRX interaction with DAXX, they can reduce or enhance the protein’s presence at its binding site, resulting in a different transcriptional signature [92]. Finally, mutations in UBN2 have been found in patients suffering from ASD [82]. More studies are required to understand the consequences of different mutations on H3.3 deposition.
These findings indicate that the proteins and complexes regulating histone variants H3.3 and H2A.Z are involved in the etiology of neurodevelopmental disorders and may represent a possible avenue for therapeutic intervention.
In addition to H2A.Z and H3.3, other histone variants have also been identified as significant contributors to neurodevelopment and implicated in neurodevelopmental disorders. However, little is known about their chaperones and the role these histone variants play in transcription and chromatin dynamics. Here we summarize the current understanding of other histone variants’ role in neurodevelopment and neurodevelopmental disorders.
MacroH2A is a variant of canonical H2A with a unique structure. The histone domain has 60% homology with H2A; however, this variant has a large globular domain that is used to recruit several partners [93]. MacroH2A has two isoforms, macroH2A1 and 2, and can regulate gene expression mainly by exerting a repressive function [93]. Notably, macroH2A has been reported to play an essential role in regulating memory formation in the adult brain [94]. Ma et al. [4] showed that macroH2A1 is also expressed in the developing brain. They demonstrated that knock-out of macroH2A1.2, a splice variant of macroH2A1, promotes neural progenitors’ proliferation and reduces differentiation, resulting in abnormal neuronal morphology and the development of autistic-like behaviours in mice [4].
Another H2A variant, H2A.X, differs from its canonical counterpart in the C-terminus, where it has a hydrophobic residue that can be phosphorylated in response to DNA damage [95]. Phosphorylated H2A.X is expressed throughout the developing brain, where it continues to be expressed throughout the lifespan [95]. H2A.X also appears during adult neurogenesis, where it is thought to play a role in the proliferation of neural precursors and neuronal differentiation [95]. While it is clear that H2A.X is present during development, further research is needed to assess the specific role of H2A.X in the developing brain and whether it is linked to any neurodevelopmental disorders.
The linker histone H1, is not part of the histone octamer; however, it plays an essential role in chromatin regulation and transcription [96]. H1 has seven replication-dependent variants (H1-0–H1-6) that are encoded from a gene cluster and the abundance of these variants varies according to tissue and cell state [97]. Mutations in HIST1H1E, encoding for H1-4, have been found in patients with different neurodevelopmental disorders [98]. Various H1 genes have also been reported in the SFARI Gene database, which contains all identified mutations and variations associated with ASD. Similar to H1, histone H4 is also encoded by a cluster of genes (15 genes), a number of which are reported in the SFARI database.
Together these findings suggest that histone variants are involved in regulating transcription in the developing brain and that their role in chromatin regulation at this stage is required to ensure proper neurodevelopment.
In this review, we have highlighted the importance of histone variants and their chaperones in regulating chromatin dynamics, gene expression, and their role in neurodevelopment. Accumulating evidence indicates that histone variants are necessary for controlling gene expression required for proper brain development. In addition, the presence of numerous mutations and copy number variations affecting histone chaperones and variants in patients with neurodevelopmental disorders further demonstrates that they regulate vital pathways for proper neurodevelopment.
As the field continues to grow, several questions should be addressed. Further research is required to better characterize the molecular mechanisms underlying histone variant turnover and relocation. Specifically, research should address how different chaperone complexes can regulate the same variant, how they interact, what genes are regulated by specific chaperone complexes, and why they cannot compensate for one another if lost during the embryonic stage. Future efforts should also investigate the other functions linked to large chaperone complexes that often recruit other chromatin regulators and how they are affected when the chaperone activity is lost or compromised. Finally, further research is needed to identify the group of genes regulated by histone variants and their temporal dynamics.
Once the molecular pathways regulating histone variant dynamics during brain development are better understood, translational research can begin to address whether these pathways can be targeted as a possible intervention for neurodevelopmental disorders.
GS and KSJ designed and wrote the manuscript. MSC assisted in writing and designed the figures and tables. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.