
 , Masoud Nikanfar 6
, Masoud Nikanfar 61 Department of Medical Biology, Faculty of Medicine, Ege University, 35040 Izmir, Turkey
2 Department of Genetics, Tabriz Branch, Islamic Azad University, 43351-14543 Tabriz, Iran
3 Department of Basic Medical Sciences, Khoy University of Medical Sciences, 58139-68197 Khoy, Iran
4 Department of Biology, Sciences and Research Branch, Islamic Azad University, 43351-14543 Tehran, Iran
5 Neurosciences Research Center (NSRC), Tabriz University of Medical Sciences, 91656-73117 Tabriz, Iran
6 Department of Neurology, Faculty of Medicine, Tabriz University of Medical Sciences, 91656-73117 Tabriz, Iran
Abstract
Ischaemic stroke is a sudden neurological disorder caused by localised cerebral ischaemia and persistent cerebral infarction. Occlusion of large arteries due to atherothrombosis, cerebral embolism (i.e., embolic infarction), no thrombotic occlusion in small, deep cerebral arteries (i.e., lacunar infarction), and stenosis of proximal arteries due to hypotension leading to decreased cerebral blood flow in arterial supply zones are the most common causes of ischemic stroke (i.e., hemodynamic stroke). It is now known that organelles play an important role in various signaling events and cellular functions. The molecular mechanisms of mitochondria are involved in cerebral ischemia by generating and scavenging reactive oxygen species, apoptosis, biogenesis, mitochondrial dynamics, and inflammation are all examples of electron transport chain dysfunction. More knowledge about the involvement of mitochondria in ischemia-induced neuronal death and neuronal protection will contribute to the development of better treatment programs for stroke syndromes such as ischemic stroke.
Graphical Abstract
Keywords
- ischemic stroke
- mitochondrial biogenesis
- neuronal death
- electron transport chain
Stroke is the most common cause of mental and physical disability and has a high mortality rate in industrialized countries [1]. Rupture or occlusion of a blood vessel causes stroke. Stroke-related impairments always occur at onset (unlike brain tumor symptoms, which gradually worsen) and last longer than 24 hours. They are associated with visible cerebral vascular damage on neurological examination and imaging [2]. The pathophysiology of cerebral ischemic stroke consists of hypoxia and vascular damage, followed by inflammation, apoptosis, etc., resulting in neuronal damage and impaired brain function [3]. According to current guidelines, Intravenous thrombolysis (IVT) and tissue plasminogen activator (tPA) is the first choice of treatment for acute ischemic stroke [4]. Due to the limited time window of therapy, only about 8% of patients are eligible for tPA [5].
Mitochondria are multifunctional organelles involved in many metabolic activities, including energy production and synthesis of biomolecules [6, 7, 8]. Shape and distribution of mitochondria within the cell vary with time and reflect the metabolic state of a particular cell type [7]. They organize themselves into a tubular network within the cell that is constantly changing through cell division and fusion. The network breaks down into parts that spontaneously reassemble when mitochondria are removed from a cell [8, 9]. Isolated mitochondria are capable of respiration and ATP production. They retain the composition, structure, and potential of their membranes, as well as their ability to fuse and import proteins from the environment [10, 11]. Dynamic mitochondrial imbalance, free radical formation, calcium accumulation, intrinsic cell death activation, and finally cell death occurs due to negative stimuli as they do in neurodegenerative diseases [12, 13].
Mitochondrial biogenesis is a property of mitochondria that generates new mitochondria from existing mitochondria [14]. Therefore, maintaining or improving mitochondrial activity may be a therapeutic strategy for neurodegenerative diseases. Peroxisome proliferator-activated receptor-gamma (PPAR-gamma) co-activator-1alpha (PGC-1) regulates this biogenesis process. PGC-1 activates nuclear respiratory factors 1 and 2 (NRF1 and NRF2) and mitochondrial transcription factor A via phosphorylation or deacetylation (Tfam). When this PGC-1-NRF-Tfam pathway is activated, mitochondrial DNA, proteins and new mitochondria are synthesized [15]. There is evidence of a close association between neuronal cell death and excessive production of reactive oxygen species (ROS) in some neurological diseases, including Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), ischemic stroke, epilepsy, and traumatic brain injury (TBI) [16, 17]. Elevated ROS levels damage brain tissue structural and functional integrity, a process critical to the pathophysiology of cerebral ischemia [18, 19, 20].
The remarkable role of mitochondrial dysfunction and excessive oxidative stress in the ischaemic cascade has been well established. Recent research has shown that mitochondrial biogenesis and ROS detoxification are two crucial endogenous protective mechanisms in acute cerebral ischemia and chronic neurodegenerative diseases [20, 21, 22]. Therefore, preventing the adverse effects of oxidative stress and improving mitochondrial function may be promising strategies for treating ROS-related diseases, including ischaemic stroke.
According to the National Institute on Aging, stroke is the leading cause of
mortality and morbidity in individuals aged
After stroke, reperfusion occurs in various tissues including the brain, heart,
and kidneys. After reperfusion, further damage may occur due to increased
Ca
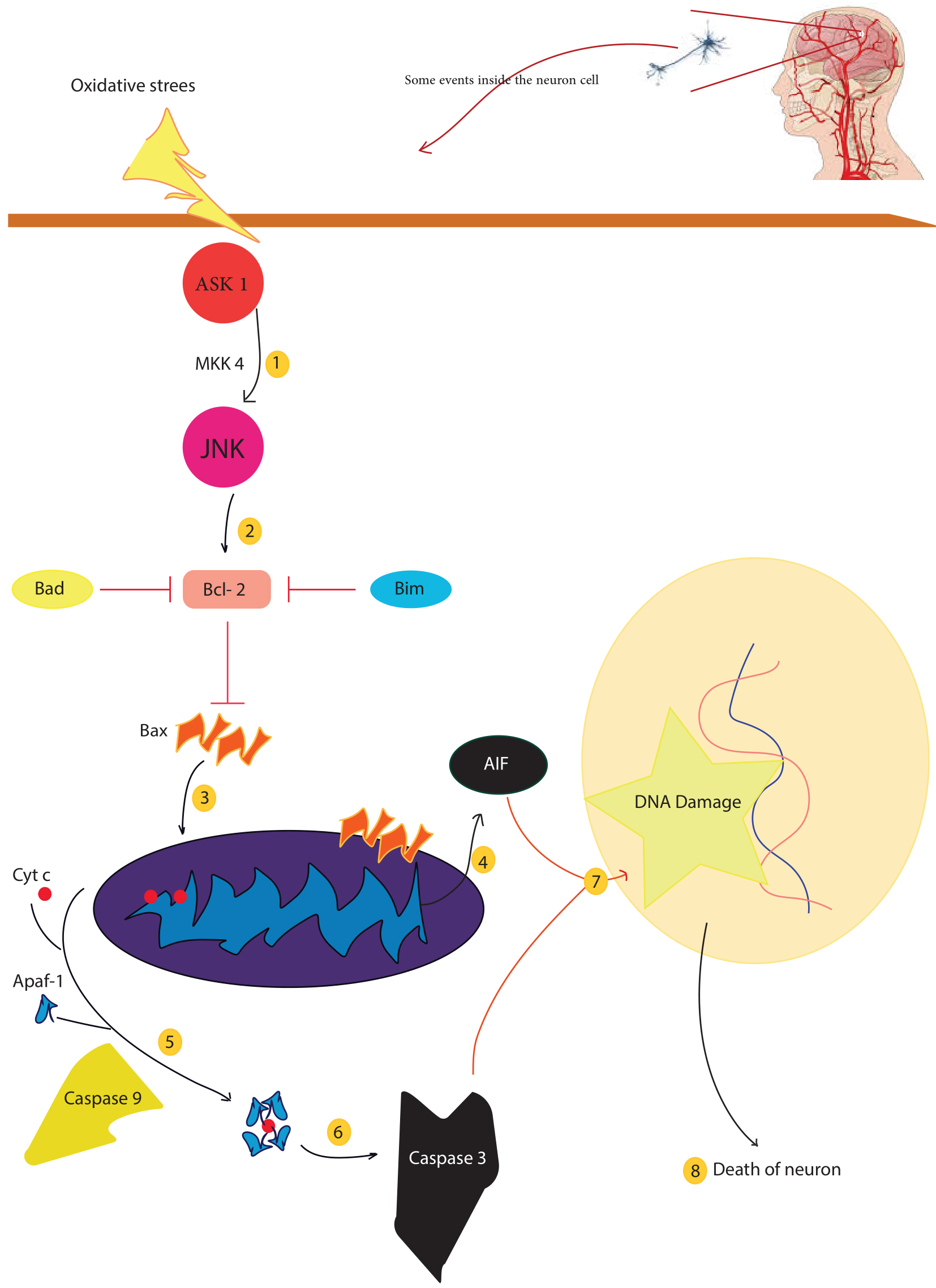
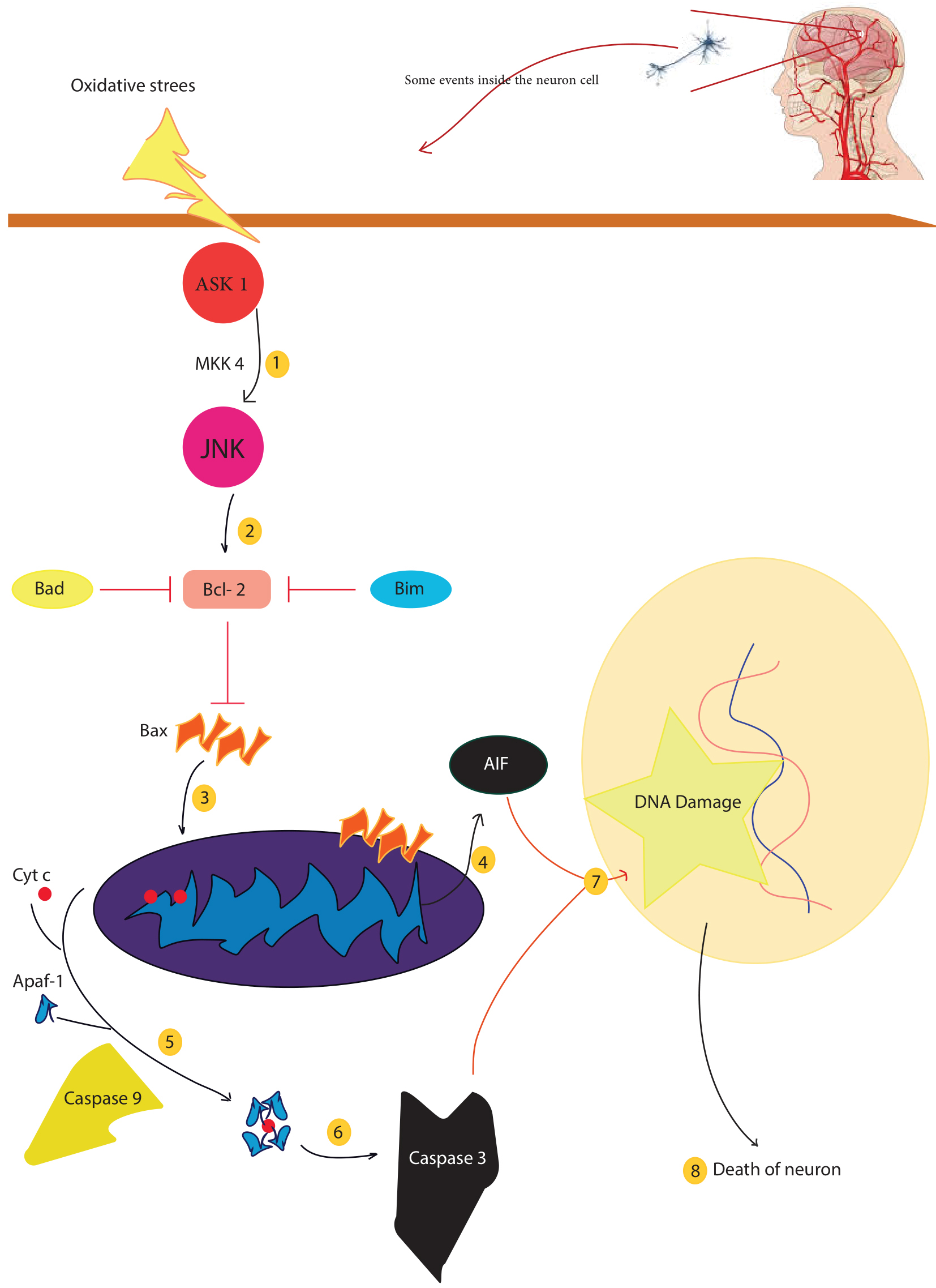 Fig. 1.
Fig. 1.There are several factors that lead to mitochondrial dysfunction, including mitochondrial fission and fusion, mitochondrial-induced apoptosis, and mitochondrial phagocytosis. Oxidative stress caused by cerebral ischemia activates mitochondrial death signals, ASK1 activates JNK via MKK4. Activated JNK disrupts the balance of Bcl-2 proteins and mitochondrial membranes, leading to DNA damage and eventually neuronal death. In addition, mitochondrial cytochrome c can induce apoptosis and lead to neuronal cell death. Injury to neurons leads to ischemia, hypoxia, acidosis, and abnormal concentrations of ions, such as calcium and iron. Apaf-1, apoptosis-activating factor-1; ASK1, apoptosis signal-regulating kinase 1; cyt c, cytochrome c; FasL, Fas ligand; JNK, c-Jun–terminal kinase; NAD, nicotinamide adenine dinucleotide; PARP, poly (ADP-ribose) polymerase; ROS, reactive oxygen species; AIF, Apoptosis-inducing factor.
Many processes are associated with neuronal loss and cell death after stroke, including mitochondrial dysfunction, excitotoxic cell damage, free radical production, protein misfolding, acid toxicity, and inflammation [33, 34].
Regulated cell death, such as apoptosis, autophagy, pyroptosis, and necroptosis, is induced by ischemia and contributes to brain damage after stroke. The cell death mechanisms may regulate each other to some extent [35, 36]. Mitochondria are an essential regulator of stress signals [37]. Ischemia impairs mitochondrial respiratory activity and membrane potential, triggering a cascade that ultimately leads to neuronal death (Fig. 2) [38]. Depolarized mitochondria produce an excess of ROS due to their depolarization. The accumulation of ROS and calcium causes the membrane permeability transition to open. This releases cytochrome c, which activates effector caspases and eventually terminates apoptosis [39]. Disruption of signaling pathways may gradually interrupt or delay malignant responses and eventually lead to cell death. Therefore, you should understand mitochondrial biogenesis.
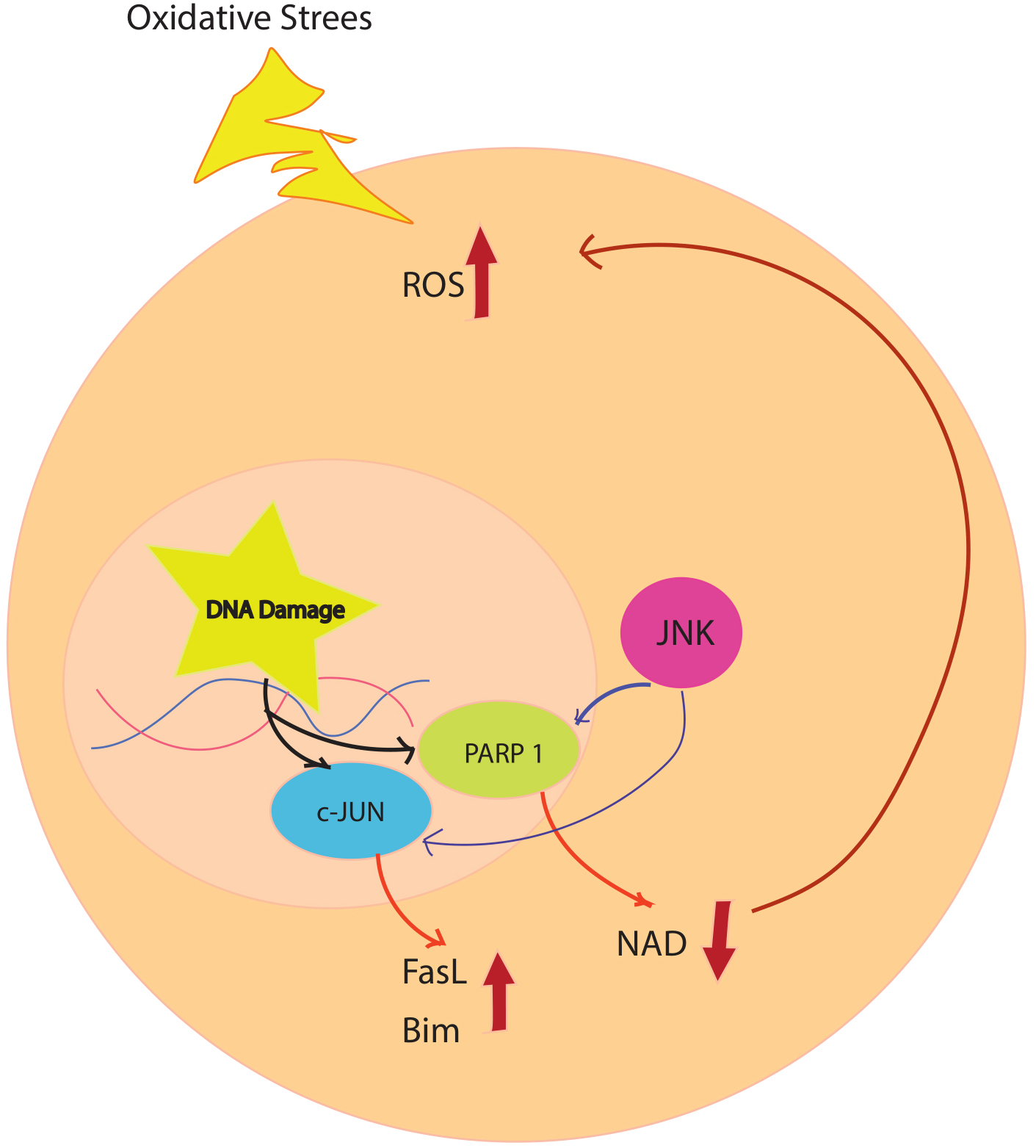
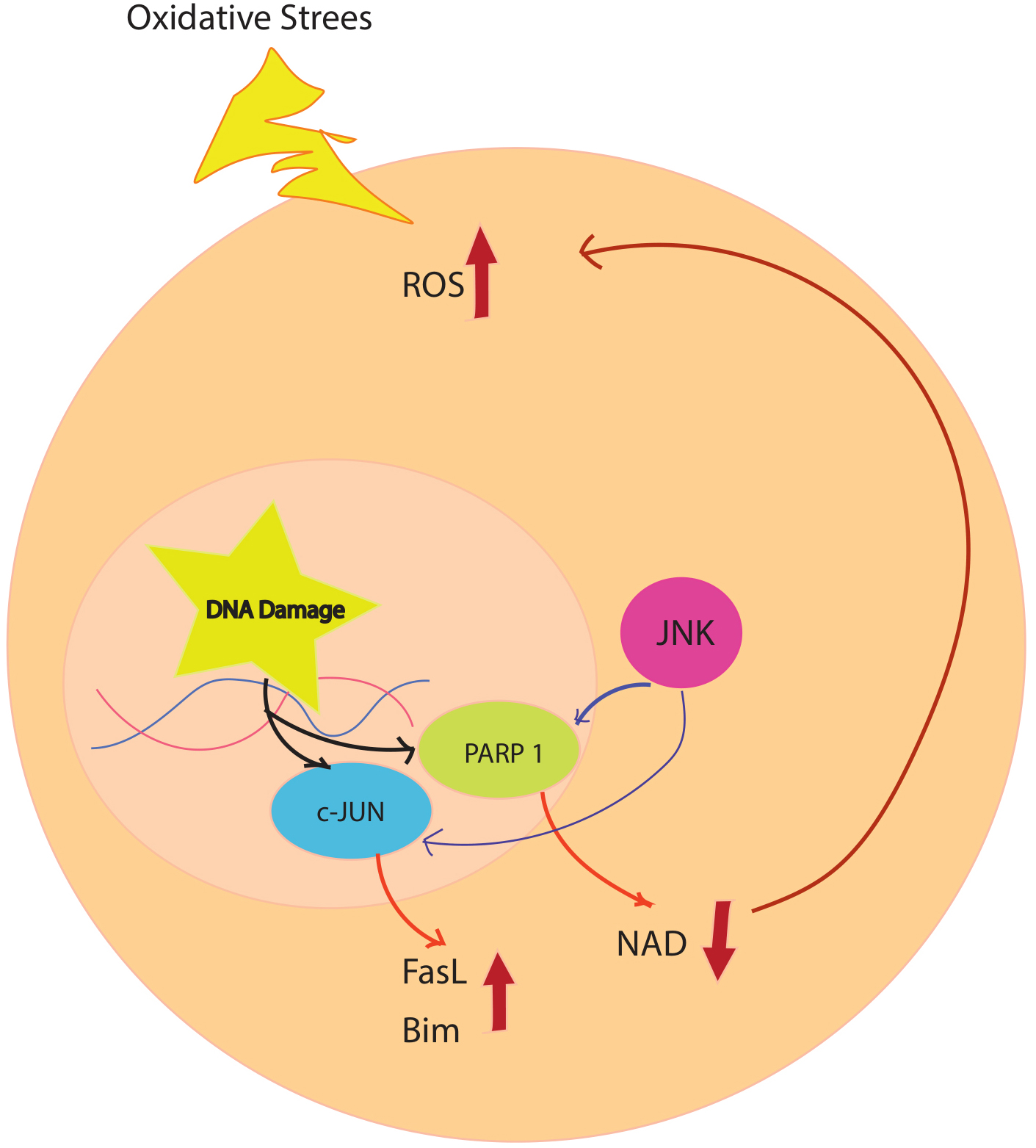 Fig. 2.
Fig. 2.Oxidative stress simultaneously causes DNA damage and activation of ASK1. DNA damage activates PARP1, which leads to degradation of NAD and an increase in ROS. This cycle repeats because PARP1 activation can activate ASK1, and JNK activation can activate PARP after ischemia. Progressive accumulation of ROS increases JNK activation and damage to multiple mitochondria, leading to neuronal death. ASK1, apoptosis signal-regulating kinase 1; FasL, Fas ligand; JNK, c-Jun–terminal kinase; NAD, nicotinamide adenine dinucleotide; PARP, poly (ADP-ribose) polymerase; ROS, reactive oxygen species.
Double-membraned intracellular organelles such as mitochondria are found in all cells and are widely distributed. The outer membrane of the phospholipid bilayer contains protein channel structures that allow molecules with a maximum molecular weight of 10 kDa to pass through, including water, ions, nutrient molecules, ADP, and ATP. The inner membrane layer acts as a reactive apparatus for energy metabolism in the mitochondria. It contains electron transport protein complexes, ATP synthesis complexes, and ATP/ADP transport proteins. Water, oxygen, and carbon dioxide can pass through the inner membrane. The mitochondrial ETC (electron transport chain) plays an essential role in cellular energy production because it most strongly influences the process of oxidative phosphorylation, which involves several enzyme complexes (i.e., complexes I to V) in the inner mitochondrial membrane [40, 41]. As biochemically demonstrated, the central part of ATP is used for neuronal electrical activity [42]. For neurons to survive and be excitable, mitochondria must provide sufficient energy. In addition to energy production, mitochondria are an important source of ROS and regulate apoptosis and neurodegenerative diseases [43, 44, 45].
Neurons are highly dependent on mitochondria for various functions, including ATP generation and calcium signaling. Therefore, they are sensitive to hypoxia and ischemia during stroke. Therefore, damage to mitochondria has destructive effects on the central nervous system (CNS). Mitochondria have been thought to spend their entire lives inside cells. Recently, there is evidence of intercellular exchange of organelles and membrane vesicles using structures such as microvesicles or tunnel nanotubes (TNT) [46, 47]. Several models of cardiovascular injury, including experimental stroke, have shown that transfer of mitochondria occurs [48, 49]. Numerous studies have shown that intercellular mitochondrial transfer can act as a protective mechanism that preserves injured cells from the effects of stress-induced mitochondrial dysfunction [48, 50, 51]. By restoring alveolar bioenergetics via mitochondrial transfer and Connexin 43 (Cx43)-dependent alveolar attachment, Bone marrow (BM)-derived stromal cells protect lungs from acute lung injury [52]. The data show that there is an exchange between healthy and injured cells by mitochondrial transfer. Recipient cells can take up and reprogram the released mitochondria to activate cell survival signals.
Mitochondria are highly active organelles that divide purposefully, fuse, and
migrate through axons and dendrites [53]. Mitochondria in neurons arise from a
highly dynamic compartment in form and function, including the movement of
mitochondria through neuronal segments and the strategic placement of
mitochondria at sites where ATP supply and Calcium ion (Ca
At different stages of neuronal development, the expression patterns of metabolic genes were studied. As NPCs differentiate, the expression of mitofusin 2 (MFN2), an important component of mitochondrial fusion, increases [57]. MFN2 deficiency inhibits neuron formation in NPCs, whereas overexpression promotes neuronal development, suggesting a crucial function of mitochondrial dynamics in human neurogenesis. During differentiation of NPCs into neurons, expression of the essential glycolytic genes Hexokinase 2 (HK2) and Lactate Dehydrogenase A (LDHA) is reduced. However, transcript levels of most oxidative phosphorylation (OXPHOS) genes remain constant, as during neurogenesis in the adult hippocampus [58]. Mitochondria are concentrated in the pre- and post-synapses of the formed neurons, where there is a substantial energy demand [59, 60]. At synaptic terminals, mitochondria supply local ATP to neurons and regulate cytosolic calcium levels. Neuron function would be impaired if mitochondria could not communicate with the center throughout development and beyond [61]. Astrocytes are essential components of the central nervous system as they are involved in neurodevelopment, neurotransmission, cerebral metabolism, and control of blood flow [62]. Normal astrocytes protect neurons from oxidative stress and excitotoxicity [63, 64]. Dysfunctional astrocytes can produce harmful substances that cause neuronal damage [65]. Although mitochondria are derived from internal nuclei for energy production and survival, they can also be released into the extracellular environment [66]. The ability of neuroglia to maintain these protective mechanisms may depend on the integrity of their mitochondria, as inhibition of astrocytic mitochondria provides a source of energy to neurons threatened with cell death [65]. Examples include retinal neurons that transport mitochondria to astrocytes for recycling and disposal and bone marrow-derived stromal cells that transport mitochondria to alveoli to protect them from acute lung injury [67, 68]. Recently, it was hypothesized that neurons discharge damaged mitochondria and recycle them into astrocytes [67]. Because of this ability to exchange mitochondria, it is conceivable that cells in the CNS communicate with each other.
Additionally, astrocytes can provide functional mitochondria for neurons. A calcium-dependent mechanism involving the cluster of differentiation 38 (CD38)/cyclic ADP-ribose signaling pathway regulates the ability of an astrocyte to release extracellular mitochondrial particles. In mice, transient local cerebral ischaemia causes astrocytic mitochondria to invade neighbouring neurones and enhances cell survival signals. Simultaneous suppression of CD38 signaling decreased extracellular mitochondrial transfer and worsened neurological status [48]. Astrocytes, another subset of glial cells in the brain, perform important physiological roles, including neuroprotective effects in the A2 state and neuroinflammatory functions in the A1 state. In animal models, active microglia rapidly generate the A1 state of astrocytes after brain injury, leading to further neuronal damage and cell death [69]. Drp1/Fis1-triggered enhanced fission of mitochondria in activated microglia results in the production of fragmented and damaged mitochondria that are rejected by these cells. Defective extracellular mitochondria, therefore, contribute to the propagation of neurodegenerative signals from microglia [70]. This study shows that extracellularly damaged mitochondria release more damaged mitochondria from microglia when they lose their immune-privileged state and that damaged extracellular mitochondria play a direct role in disease propagation by acting as effectors of the innate immune response on nearby astrocytes and neurons. Fragmented mitochondria have been detected in clinical and experimental studies of biofluids from individuals with subarachnoid hemorrhage and stroke [48, 71]. While extracellularly injured mitochondria are harmful, the mitochondrial transfer is beneficial, as demonstrated in a mouse model of acute lung injury and a stroke model [48, 68]. Joshi et al. [70] show that the ratio of damaged to functional mitochondria in the extracellular environment, determined by the extent of pathological fission in donor microglia, affects the fate of neurons.
The gradual loss of neurones and tissue damage in the brain is known as neurodegeneration. Because of their high energy requirements, neurons are susceptible to damage and death from dysfunctional mitochondria. Severe damage to mitochondria leads to cell death because cells can no longer generate sufficient energy. Many clinical and physiological studies suggest that mitochondrial dysfunction and dynamics play a critical role in the development of neurodegenerative diseases [72]. Mitochondrial biogenesis is required to maintain mitochondrial homeostasis throughout the mitochondrial life cycle to meet the physiological needs of eukaryotic cells [73].
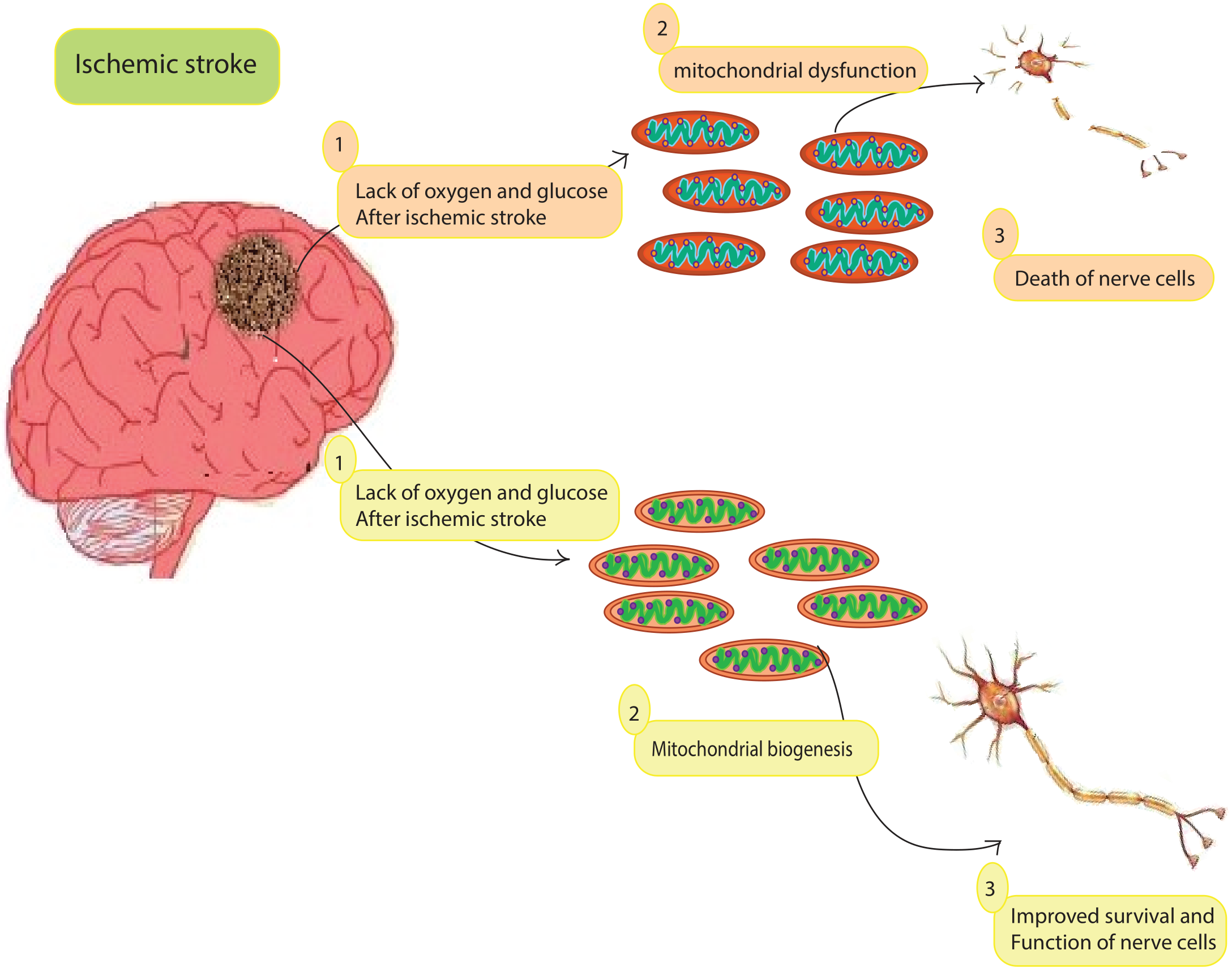
After long-term local cerebral ischemia, progressive loss of mitochondrial DNA (mtDNA) suggests mitochondrial regeneration. Several studies have shown that creating a dense population of functional mitochondria reduces ROS production [74]. Mitochondrial biogenesis is a precise process that requires both mitochondrial and nuclear genomes. It often occurs in healthy cells that are constantly dividing and fusing, whereas division (fission) after brain trauma leads to fragmentation of the mitochondrial network [75]. Increased biogenesis is associated with increased mitochondrial DNA copy number and increased mitochondrial gene expression. Peroxisome proliferator-activated receptor co-activator 1 alpha (PGC-1a), mitochondrial transcription factor A (TFAM), nuclear respiratory factors 1 and 2 (NRF1 and NRF2), and mitochondrial transcription factor B1 (TFB1M) are involved in mitochondrial biogenesis [76]. Several processes appear to be involved in the mitochondrial formation, including (1) outer and inner membrane synthesis, (2) mitochondrial protein synthesis, (3) synthesis and import of proteins encoded by the nuclear genome, (4) lipid import, (5) mitochondrial DNA replication, (6) mitochondrial Fusion and fission, and mitochondrial Fusion and fission [77, 78, 79, 80].
Due to altered physiological circumstances, such as increased ATP consumption, the cell’s ability to produce ATP is impaired. Cell division and repair, embryonic development, changes in physiological state (e.g., sympathetic stimulation, caloric restriction, exercise, cold stress, and energy deficiency), hormones (thyroid hormone, leptin, and erythropoietin), and mitochondrial disease/damage (e.g., inflammation and hypoxia/ischemia) are all major triggers of mitochondrial biogenesis [80]. Another important trigger of mitochondrial biogenesis is mitochondrial oxidative dysfunction, which contributes to various neurological disorders, as shown by numerous studies [80].
It is emphasized that decreased mitochondrial biogenesis can lead to mitochondrial dysfunction, which is necessary for the development of neurodegenerative diseases. These diseases have been linked to genes that produce proteins associated with mitochondrial biogenesis [81].
AD is a common neurodegenerative disease characterized by memory loss and
cognitive impairment and manifested by various neurological, mental, and
behavioral symptoms [82, 83]. Recent studies have shown that mitochondrial
dysfunction is an essential and early feature of AD, with reduced energy
metabolism being one of the most visible critical abnormalities of the disease
[84]. Mitochondrial intervention may help slow the progression of amyloid plaque
(A
Further studies have linked the expression of genes encoding proteins involved in mitochondrial biogenesis, such as PGC-1a, NRF1, NRF2, and TFAM, to the development of neurodegenerative diseases such as AD [74]. Significant downregulation of these genes is observed in AD and hippocampal cells and tissues, indicating a decrease in mitochondrial biogenesis [74]. However, due to the pleiotropic nature of PGC-1a, it is unclear how AD affects mitochondrial biogenesis signaling and whether these changes lead to mitochondrial dysfunction [74]. Parkinson’s disease (PD) is a degenerative neurological movement disorder of unknown etiology influenced by hereditary and environmental factors [88]. In the early and late stages of Parkinson’s disease (PD), significant reductions in putamen ATP and phosphocreatine levels and midbrain ATP levels as high-energy metabolites indicate mitochondrial dysfunction in mesostriatal dopaminergic neurons [89]. Genome-wide analysis of normal subjects and patients with PD revealed that PGC-1a, a key regulator of mitochondrial biogenesis, was reduced in patients with PD [90], suggesting that mitochondrial dysfunction plays a critical role in the cause of PD [91] has found evidence that the deficiency in PD is at or above the level of PGC-1a expression, which controls mitochondrial complex I function. Recent research has linked several proteins involved in familial PD, including a-syn, Parkin, PTEN-induced putative kinase 1 (PINK1), protein deglycase DJ–1 (Parkinson protein 7), and Leucine-Rich repeat kinase 2 (LRRK2 or Dardarin), to quality control and regulation of mitochondrial dynamics [92, 93]. Amyotrophic lateral sclerosis (ALS) is a neurological disorder that manifests as gradual paralysis in adults and is a genetic disorder. In humans, the disease is associated with degeneration of motor neurons and selective loss in the spinal cord and brain, resulting in denervation of muscles and muscle wasting after denervation [94]. Postmortem spinal cord specimens from individuals with Lou Gehrig disease (ALS) show morphological and biochemical mitochondrial abnormalities, including decreased mitochondrial DNA copy number and decreased respiratory chain activity [95]. Mutations/deletions in mitochondrial genes have been associated with specific muscle and neuronal diseases [96]. Mitochondrial dysfunction can lead to certain damage to muscles or neurons.
PGC-1a is thought to have beneficial effects on muscles, including increased mitochondrial activity and muscle hypertrophy. Mitochondrial activity and biogenesis are preserved in late-stage disease, as is muscle function, delayed muscle wasting, and improved muscle tolerance. Although lifespan could not be extended, drugs that increase PGC-1a activity in muscles are beneficial for preserving muscle functionality throughout ALS [97]. Activation of peroxisome proliferator-activated receptors (PPARs) that interact with PGC-1a and control mitochondrial biogenesis in combination with this transcriptional co-activator is another strategy to increase PGC-1a signaling and improve mitochondrial function [98]. Another study conducted in cell cultures showed that Sirtuin 3 (SIRT3) protects against mitochondrial fragmentation and neuronal cell death during developing AD [99]. The regulating PPARc transcriptional activity in the central nervous system represents a novel approach to slowing ALS progression [100]. Mitochondria, important intracellular organelles that control cell life and death, are being investigated as potential therapeutic targets in pharmaceutical research.
Mitochondria are critical for cell survival and death. Pathophysiological abnormalities transform mitochondria from essential organelles into activators of cell death [101, 102]. Mitochondria play a crucial role in cell death by ischemia by impairing ATP synthesis and participating in apoptotic and necrotic pathways [103]. The main goal of stroke research is to elucidate the process of neuronal ischemic injury to improve functional outcomes. Neurons are more susceptible to ischemic damage than other cells because they are constantly dependent on an external energy supply. As excitable cells, neurons express many receptors for excitatory transmitters, especially at synapses [104]. In stroke, oxygen and glucose deprivation are the main components of tissue damage. OGD (oxygen-glucose deprivation) models are the most commonly used stroke in vitro models. Hydrogen peroxidase exposure or glutamate-induced excitotoxicity is another in vitro stroke model. Studies on brain slices or cultured neurons have contributed significantly to our knowledge of neuroprotective and ischemic injury processes [105]. Mitochondria produce energy and are very numerous in brain cells because neurons need a lot of ATP to function properly [106].
Mitochondria use an electron transport chain (ETC) to convert electron donor
molecules such as reduced (NADH) nicotinamide adenine dinucleotide into energy
through various oxidation/reduction processes. The compounds discussed here
transfer electrons and support transmembrane proton transport, creating an
electrochemical gradient that favors mitochondrial ATP synthesis [107]. Five
enzyme complexes consisting of integral internal membrane proteins are found in
ETC: NADH-CoQ reductase, Coenzyme Q (CoQ) cytochrome c reductase, succinate CoQ
reductase, cytochrome c oxidase, and ATP synthase (complexes I–V) [108].
Cytochrome c and ubiquinone (CoQ)
The superoxide anion radical is a precursor of several other potentially
dangerous ROS, including hydroxyl, hydrogen peroxide, and peroxynitrite.
Mitochondrial dysfunction is caused by oxidative stress, energy deficiency, and
impaired calcium homeostasis in the cell. Reperfusion increases the production of
ROS in mitochondria, and Ca
Mitochondria, as receptors for a variety of stimuli, trigger both caspase-independent (AIF, Apoptosis-inducing factor) and caspase-dependent (cytochrome c) cell death [113]. Ischemia promotes these processes and leads to necrotic and apoptotic death of brain cells. Inflammation is another critical factor in the pathophysiology of cerebral ischemia. Glial cell activation, infiltration by peripheral leukocytes, and damage-associated chemicals such as high mobility group protein 1, nucleotides, nucleic acid fragments, and purines trigger the inflammatory response after ischemia [114, 115]. Acute systemic inflammatory stimuli increase the outcome of ischemic stroke, with the pro-inflammatory cytokine IL-1 playing a crucial role [116]. Recent research has shown that mitochondria alter their function in controlling the inflammatory response [117, 118, 119]. Mitochondria are the primary regulators of Neutrophil-Lymphocyte Ratio (NLR) Family Pyrin-Domain-Containing Protein 3 (NLRP3) inflammasome activation [120]: the outer mitochondrial membrane serves as an assembly platform for the inflammasome. It also induces pyroptosis and innate immune defence through caspase-1 and various pro-inflammatory cytokines [121, 122]. Recent studies indicate that the NLRP3 inflammasome contributes to renal and cardiac ischemia [123, 124, 125, 126]. This review focuses on the rapidly evolving and complex function of mitochondria in ischemic cerebral stroke. Understanding the fundamental processes behind the potentially protective activities of mitochondria may offer insights into developing new treatments.
The function of mitochondrial biogenesis in neurons is poorly understood. First, researchers investigated the transcriptional control of mitochondrial biogenesis in mice lacking the mitochondrial transcription factor peroxisome proliferator-activated receptor co-activator (PGC) 1. A ROS-mediated mechanism is involved in the progression of oxidative stress-induced neurodegeneration [127]. Hypoxic preconditioning increases PGC-1 expression in response to neuronal nitric oxide synthase (nNOS) activity. Oxidative stress is associated with widespread mitochondrial fission and neuronal death throughout the body. It has been shown that mitofusin 2, a mitochondrial Fusion protein, overexpression can reverse neuronal death [81]. For neuronal survival to occur, precise regulation of Fusion and fission must be maintained. Recent studies have also shown that hypoxia-ischemia stimulates mitochondrial production. Under hypoxia, mitochondrial DNA increases, as does the number of mitochondrial cells [128]. Hypoxia also increases the production of PGC-1, downstream mitochondrial transcription factors (mitochondrial transcription factor A and nuclear respiratory factor 1), and the mitochondrial protein heat shock protein 60 (HSP60). This breakthrough finding suggests that mitochondrial biogenesis is a novel endogenous neuroprotective response that can be activated.
Mitochondria are damaged after a fatal ischemic insult, and autophagy is triggered to remove damaged organelles from the cell. The maintenance of energy balance and calcium and zinc homeostasis is essential for neuronal function. Microsomal biogenesis is a crucial endogenous neuroprotective process that produces new functional mitochondria when the body is exposed to high-stress levels. A novel strategy for neuroprotection is to increase or accelerate mitochondrial biogenesis, a process unknown. Further studies are needed to understand the molecular basis of mitochondrial biogenesis after stroke. Other studies on the role of mitochondrial turnover, mitochondrial Fusion and fission mechanisms, and alternative transcriptional pathways after ischemia are warranted [129]. Identifying critical mediators of autophagy and mitochondrial biogenesis will lead to the identification of new therapeutic targets to enhance the beneficial effects of mitochondrial maintenance (Table 1, Ref. [3, 130, 131, 132, 133, 134, 135, 136]).
| Cell/Animal model | Conditions of research | Treat whıt | Mechanism | Results | Mitochondrial biogenesis | Improves mitochondrial function | Ref |
| OGD/R-induced SH-SY5Y cells | In vitro | PNGL | Mitochondrial protective effects of PNGL, mediated via the NAMPT-NAD+ | Inhibited mitochondrial damage, raised neuronal survival under ischemia | √ | [130] | |
| MCAO-rat model | In vivo | Selenium | Protects neurons from cerebral I/R damage by mitochondrial biogenesis | Ameliorates cerebral I/R injury | √ | √ | [131] |
| NSCs injured by OGD | In vitro | Amlodipine camsylate | Protection against mitochondria cause raised neuronal survival | Increased the viability and proliferation of NSCs | √ | √ | [132] |
| MCAO-rat model | In vivo | Sovateltide | ETBR mediated neural regeneration and repair | Activates neuronal differentiation and prevents mitochondrial dysfunction | √ | [3] | |
| C57BL/6 mice | In vivo | Taurine | Axonal Sprouting Through Mitochondrial Improvement | Recovered the motor function of focal cerebral cortical ischemic mice | √ | [133] | |
| HPPNCs | In vitro | CTRP3 | CTRP3 prevented mitochondria from OGD/R injury by activating the AMPK/SIRT1-PGC-1 |
Angiogenesis and enhancement of mitochondrial biogenesis | √ | √ | [134] |
| Human neuroblastoma SK-N-SH cells, subjected to OGD | In vitro | Ginsenoside Rg1 | Rg1 improves mitochondrial dysfunction by regulating autophagy in mitochondria | Improves mitochondrial dysfunction, protection from brain injuries | √ | [135] | |
| Bb2 KO-mice | In vivo | Drp1 | Bb2, a mitochondria-localized PP2A regulatory subunit, is a neuron-specific Drp1 activator in vivo | Mitochondrial biogenesis, maintenance of mitochondria | √ | √ | [136] |
OGD/R, oxygen and glucose deprivation and reperfusion; PNGL, Notoginseng leaf triterpenes; MCAO, Middle cerebral artery occlusion; I/R, ischemia-reperfusion; NSCs, neural stem cells; OGD, oxygen glucose deprivation; AC, amlodipine camsylate; ETBR, endothelin B receptors; HPPNCs hippocampal neuronal cells; CTRP3, C1q/tumor necrosis factor-related protein-3; Drp1, dynamin-related protein 1; PP2A, protein phosphatase 2A; Ref, Reference.
The lack of ATP and nutrients in ischemia impairs the leading role of mitochondria in stroke pathology. The rapid degeneration of penumbral neurovascular units essentially requires a new effective therapeutic strategy for stroke. Recent evidence of mitochondrial transfer through the formation of TNT, cell Fusion or extracellular vesicles (EVs) and the potential restoration of mitochondrial function in promoting cell survival in numerous preclinical studies argues for stem cell-mediated mitochondrial transfer therapies in stroke. Mitochondria are mainly in the presynaptic nerve terminals of neurons. When ischemia occurs, neurons are likely to shift mitochondria for both disposal and recycling to respond quickly to the lack of energy caused by the condition. After ischemia, mitochondria cannot be replenished promptly because they must be transported long distances. Therefore, extracellular mitochondrial translocation may provide a new opportunity for neuroprotective strategies. Further research is needed to understand better the impact of mitochondrial biogenesis and function on ischemic stroke.
BS, CBA, MN, and AN developed the concept and designed the study. VK, GJS, FE, and JBHO did a systematic search and prepared the first draft. All authors participated in the revising of the manuscript before submission. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.