

1 Department of Neurosurgery, Neuroscience Center, Integrated Hospital of Traditional Chinese Medicine, Southern Medical University, 510310 Guangzhou, Guangdong, China
2 Department of Neurology, Affiliated Zhongda Hospital, School of Medicine, Southeast University, 210009 Nanjing, Jiangsu, China
3 Department of Pathology, Integrated Hospital of Traditional Chinese Medicine, Southern Medical University, 510310 Guangzhou, Guangdong, China
4 Department of Neurosurgery, The Second People's Hospital of Pingnan, 537300 Pingnan, Guangxi, China
5 Department of Orthopedics and Traumatology, The University of Hong Kong, 999077 Hongkong, China
†These authors contributed equally.
Abstract
As a large and heterogeneous group of disorders, neurodegenerative diseases are characterized by the progressive loss of structure or function in neurons, finally leading to neuronal death. Neurodegenerative diseases cause serious threat to a patient’s quality of life and the most common are Alzheimer’s disease and Parkinson’s disease. Currently, little is known of the detailed etiology of these disorders; as such, there are no effective treatments available. Furthermore, the lack of targeted, effective, and resolvable therapy for neurodegenerative diseases, represents an expanding research field for the discovery of new therapeutic strategies. Investigations of the potential pathogenesis of neurodegenerative diseases will become the basis of preventing the occurrence and development of neurodegenerative diseases and finding effective therapies. Existing theories and mechanisms, such as genetic and environmental factors, abnormal protein accumulation, and oxidative stress, are intricately associated with each other. However, there is no molecular theory that can entirely explain the pathological processes underlying neurodegenerative diseases. Due to the development of experimental technology and the support of multidisciplinary integration, it has been possible to perform more in-depth research on potential targets for neurodegenerative diseases and there have been many exciting discoveries in terms of original theories and underlying mechanisms. With this review, we intend to review the existing literature and provide new insights into the molecular mechanisms underlying neurodegenerative diseases.
Keywords
- neurodegenerative diseases
- molecular mechanism
- Alzheimer's disease
- Parkinson's disease
- Huntington's disease
- amyotrophic lateral sclerosis
Neurodegenerative diseases (NDDs) are typically adult-onset processive disorders that affect the function and plasticity of neuron that arise through a host of one or more genetic and environmental factors [1]. Typical NDDs are Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD).The number of patients under NDDs is increasing annually. Besides, the lack of effective therapies for NDDs causes considerable burden and economic impact for society [2].
Thus far, many NDD biomarkers have been discovered around the world; these are being used to investigate the mechanisms responsible for the occurrence and development of NDDs [3, 4]. This research has generated a number of advanced theories for the factors responsible for NDDS, including genetic factors [5, 6], oxidative stress [7], accumulation of abnormal protein [8], mitochondrial dysfunction [9] etc. Besides, the association between NDDs and immunology, microbiology and other disciplines has become a new research focus [10, 11]. These factors are involved in the physiological and pathological process of NDDs and promote their occurrence and development. Furthermore, the mechanisms responsible for the transmission of NDDs are under intense investigation [12]. In this review, we provide new insight in our understanding of the pathological mechanism underlying NDDs.
Genetic Factors are important risk factors to the common NDDs. Genes, formed by nucleotides (DNA, RNA), are the fundamental unit of all biological process. DNA shows differences in terms of base pairs among individuals, thus leading to genetic variation and individual differences in specific trait [13]. The cell types of the nervous system are highly complex, with astrocytes, microglia, and oligodendrocytes present in various neuronal subtypes. The gene expression profiles of these different types of cells play an important role in neurodegenerative diseases and form complex gene expression networks. The survival and function of neurons are coordinated by these intricate gene expression networks.
AD is the most familiar disease of all neurodegenerative diseases and is
characterized by the impairment of executive function and memory followed by
progressive, global cognitive decline. In developed countries, AD is responsible
for the sixth highest death rate [14]. Early-onset AD occurs before the age of 65
years and less than 10% of AD cases represent early-onset (EOAD), a condition
that is caused by autosomal dominant mutations in APP, PS1, or PS2, each of which
increases A
PD is characterized by the presence of Lewy bodies and Lewy neurons featuring
the accumulation of
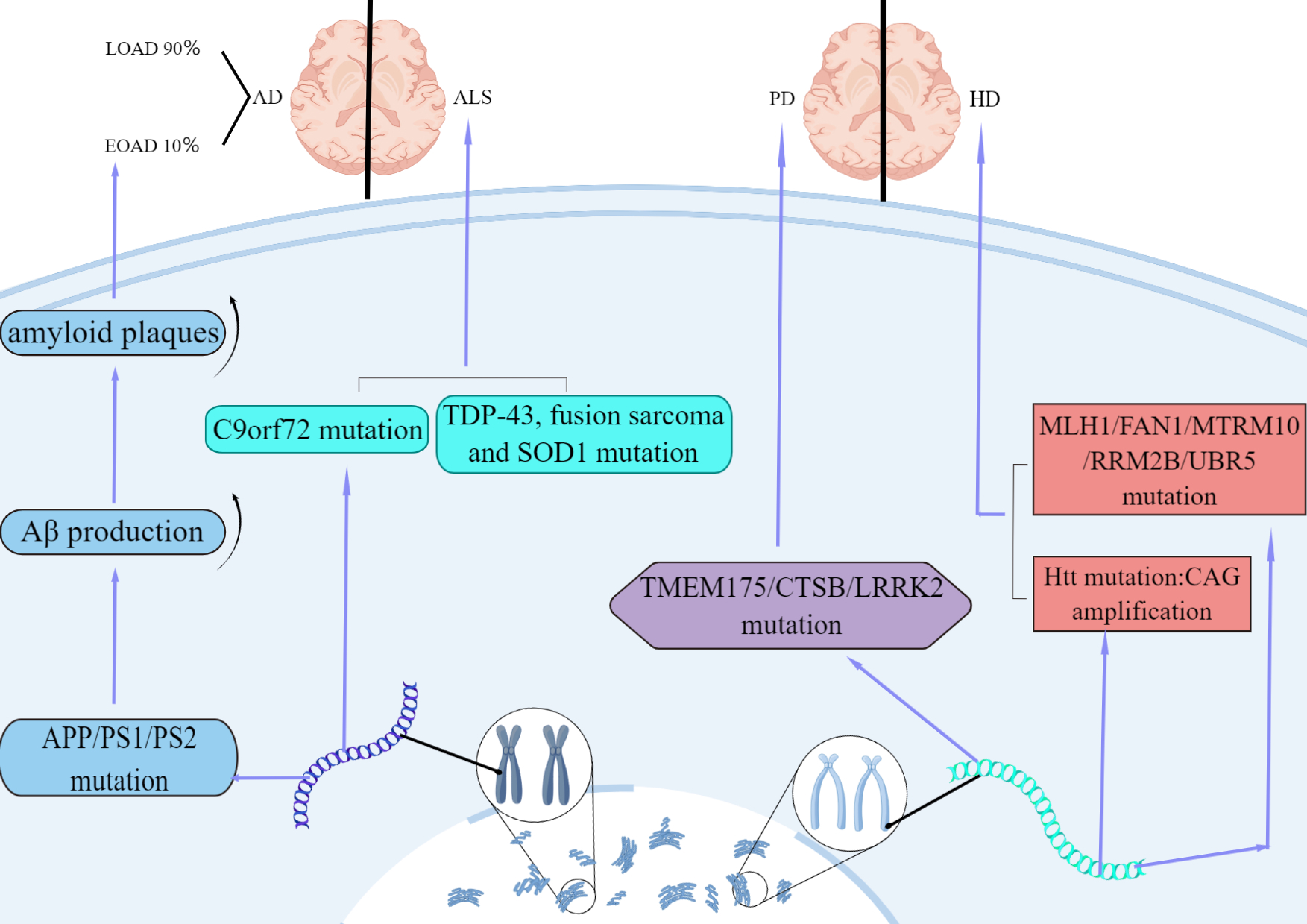
HD is an autosomal dominant disorder and is caused by a cytosine-adenine-guanine (CAG) trinucleotide repeat amplification in mutant Huntington genes [22]. Some gene variants, which are related to HD, are similar to the UBR5, FAN1, RRM2B, MTRM10 and MLH1 genes, and are associated with structure-specific DNA handling, DNA mismatch repair, oxidative stress and mitochondrial energetics [23]. Studies have shown that Brain-derived neurotrophic factor (BDNF) plays a role in the development of HD degeneration. A mutant gene product known as huntingtin expresses an extended polyglutamine chain and results in the suppression of BDNF synthesis via dysregulation of translation. Transgenic mice expressing mutant huntingtin have very similar expression impairments to those observed in BDNF knockout mice [24].
ALS can attack the motor neurons in the brain and spinal cord cells; this can lead to paralysis in patients. More than 90% of ALS cases are sporadic. The loss of C9orf72 function may affect the communication between motor neurons and muscles in patients. Mutations in the C9orf72 gene represent the main genetic cause of ALS. A hexanucleotide repeat expansion in the C9orf72 gene is the most common genetic alteration associated with ALS [25]. Mutations in the C9orf72 gene usually include six DNA base (GGGGCC) expansions, typically ranging from a few copies to more than 1000 copies; this compares to less than 20 copies in healthy individuals. Mutations in this gene cause a gain of toxicity including RNA aggregates and toxic dipeptides and are responsible for part of inherited ALS cases without a family history [26]. In the past few years, new breakthroughs in the genetics of ALS have focused on mutations in several malfunctional proteins, such as TAR DNA-binding protein 43 (TDP-43), fusion sarcoma, and SOD1. Family-superoxide dismutase 1 (SOD1) is the main enzyme that scavenges reactive oxygen species (ROS). In fact, SOD1 mutations were identified as the first genetic factor associated with ALS and are observed in 15–30% of families and 1.2–1.5% of sporadic ALS cases. Under normal circumstances, SOD1 can protect cells; however, when a gene mutation is thought to make SOD1 toxic, this form of toxic protein may be directly related to the genetic form of ALS pathogenesis. Abnormal mutations in SOD1 only in neuronal cell death was found in the spinal cord area, which means that the abnormal protein may be related to cell death [27]. TAR DNA binding protein 43 (TDP-43) and fusion sarcoma are related to ALS and frontotemporal dementia, indicating a new pathological mechanism associated with RNA metabolism [28, 29, 30]. Gene-TE pairs have been found to be associated with several cell biological processes that might contribute directly to ALS. Of these, mutations in the TDP-43 can cause transposable elements (TEs) and DNA sequences capable of transposing within the genome to become dysregulated and transcribed. Genes in the extracellular matrix and RNA processing are closely related to TEs. Thus, all the regulatory pathways can be affected in ALS [31].
In summary, GWAS analysis of evidence-based medicine has clearly shown that the expression of neurodegenerative diseases is regulated by similar functional genes, including oxidative stress, immune regulation, mitochondrial abnormalities, and metal ion damage (Fig. 1).
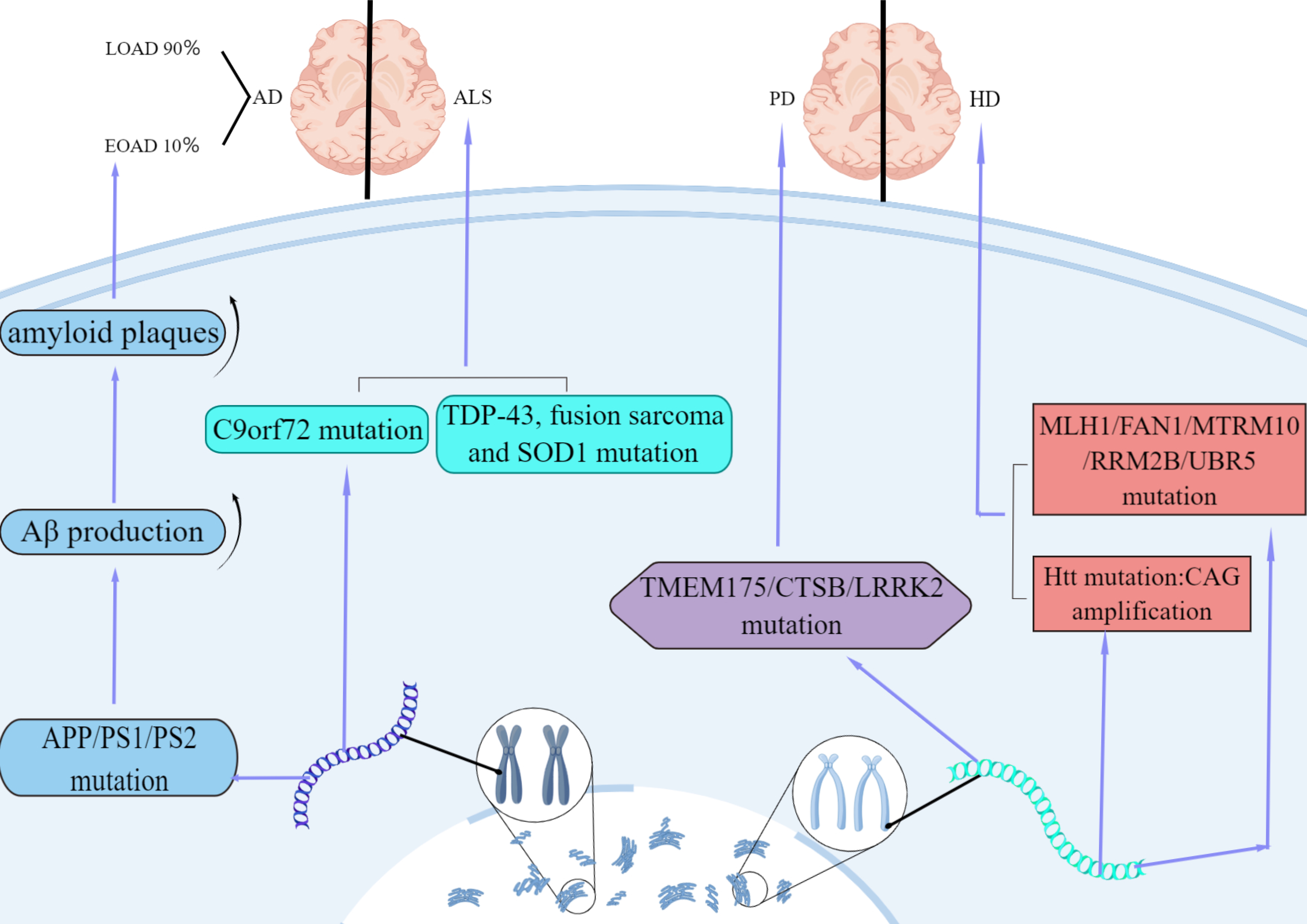 Fig. 1.
Fig. 1.Features of AD, ALS, HD and PD in genetic factor. AD is divided into early-onset (EOAD) and late-onset (LOAD). 10% of AD cases are
early-onset caused by autosomal dominant mutations in APP, PS1, or
PS2. The mutations increase A
Oxidative stress is mostly a reactive process in neurodegenerative diseases. Oxidative stress is caused by the accumulation of free radicals because of environmental changes, including chronic inflammation and mitochondrial dysfunction [32]. A crucial pathogenesis of neurodegenerative diseases is the dysfunction of specific regions of nerve cells due to oxidative stress, the main clinical manifestation of which is the dysfunction of memory and learning [33]. It has been demonstrated that oxidative damage to neuronal DNA is closely related to cognitive deficits and occurs early in the pathological changes of various diseases [34]. Reactive oxygen species (ROS) are fundamental free radicals that can aggravate tissue dysfunction and exacerbate oxidative stress. Under normal circumstances, the generation and scavenging of ROS presents a dynamic balance in the body. Appropriate ROS levels are considered to activate certain signaling pathways (such as AMPK/Ras pathway, epidermal growth factor receptor (EGFR) pathway and protein kinase C (PKC) pathway), adjust cell metabolism, and can also stimulate cell proliferation. However, when ROS scavenging is inferior to production capacity, ROS can accumulate and affect cells, thus resulting in DNA [nuclear and mitochondrial DNA (mtDNA)] damage, chromosomal instability, and protein misfolding [32].
Oxidative stress is an important factor in the pathogenesis of AD. The
pathophysiology of AD is primarily related to the formation of extracellular
amyloid beta (A
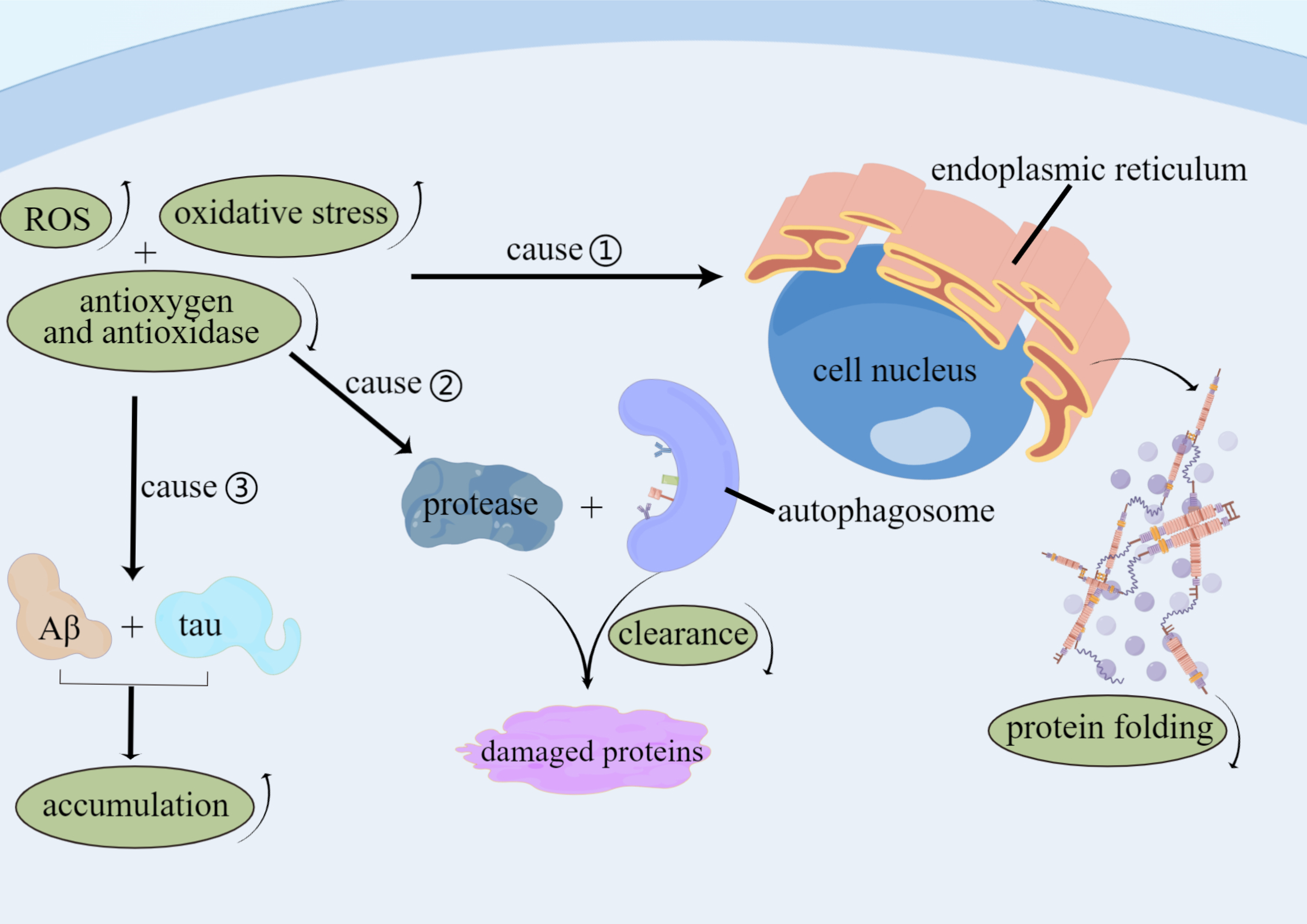
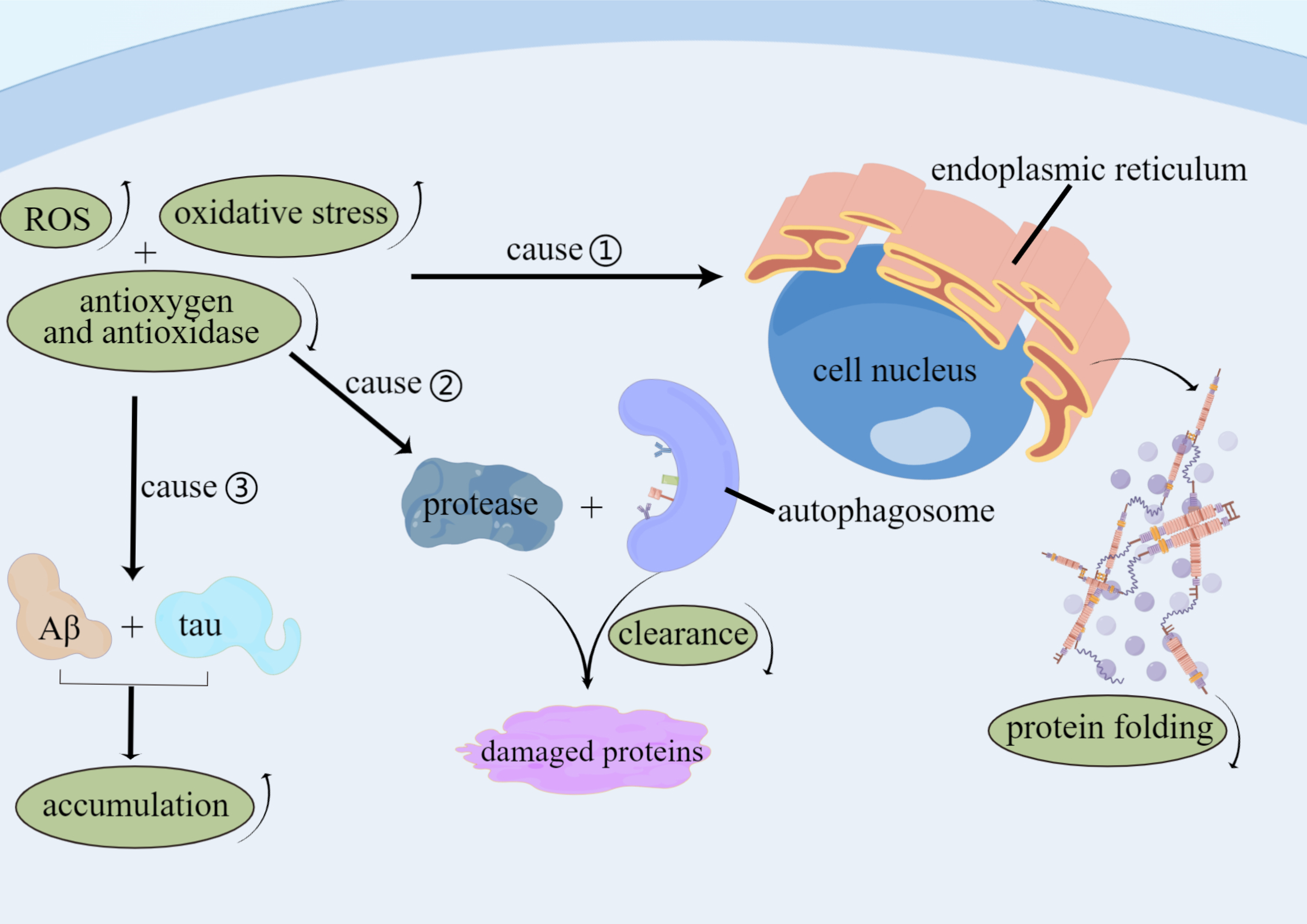 Fig. 2.
Fig. 2.Factors of Oxidative stress involve in AD. Levels of ROS and
oxidative stress are improved, and levels of antioxygen and anti-oxidase are
decreased in AD patients that lead to function of impaired protein folding in the
endoplasmic reticulum, reduced autophagy-mediated clearance and protease of
damaged proteins, and increased accumulation of tau proteins and A
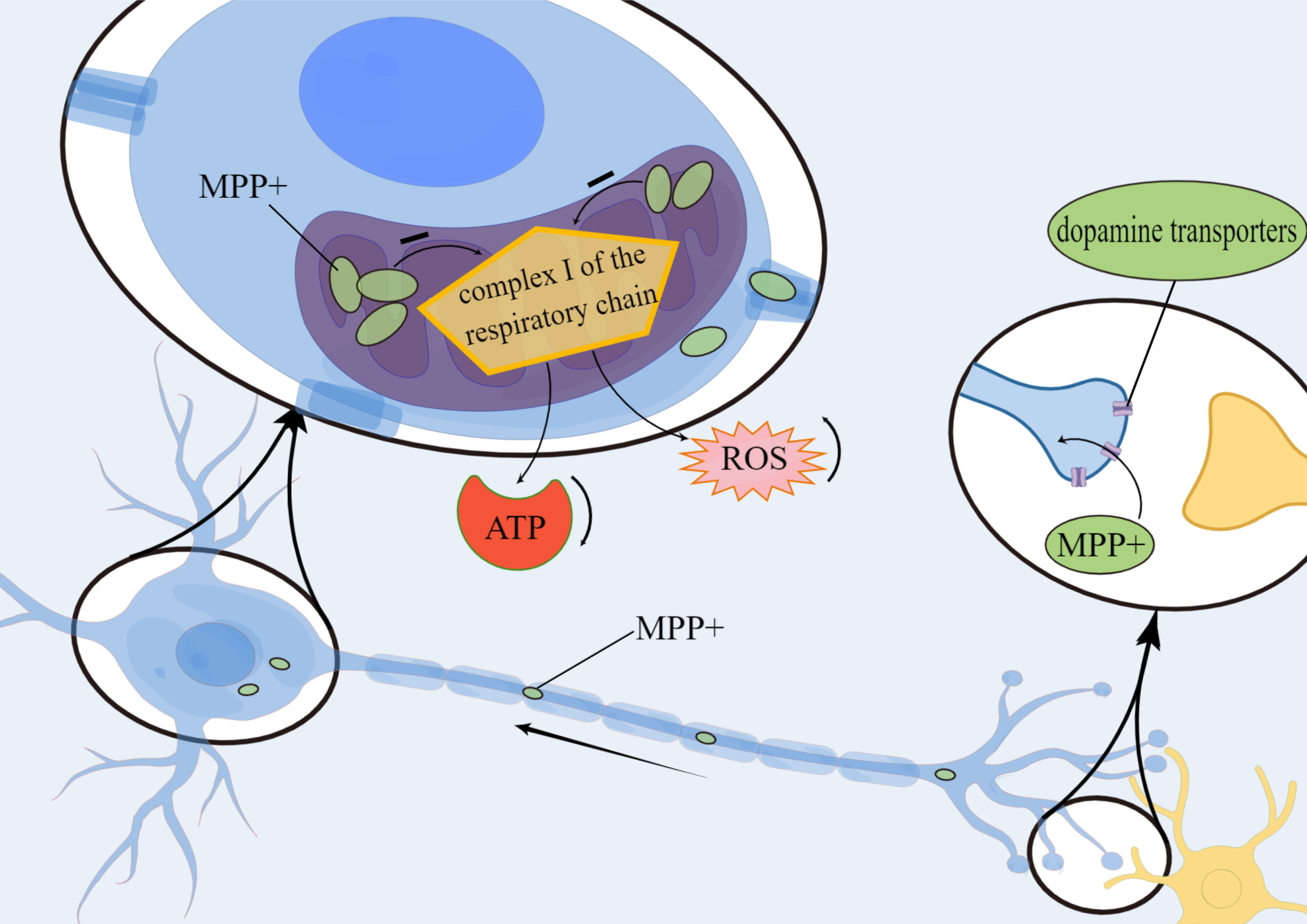
Several lines of evidence suggest that oxidative stress and ROS may be one of the major factors responsible for the progression of neurodegeneration. The neurotoxin metabolite 1-methyl-4-phenylpyridine ion is transported via dopamine transporters to neuronal mitochondria where they accumulate and inhibit complex I activity in the mitochondrial respiratory chain. This leads to a reduction in ATP production and an increase in ROS release, thus resulting in the degeneration of nigrostriatal and striatal dopaminergic neurons (Fig. 3).
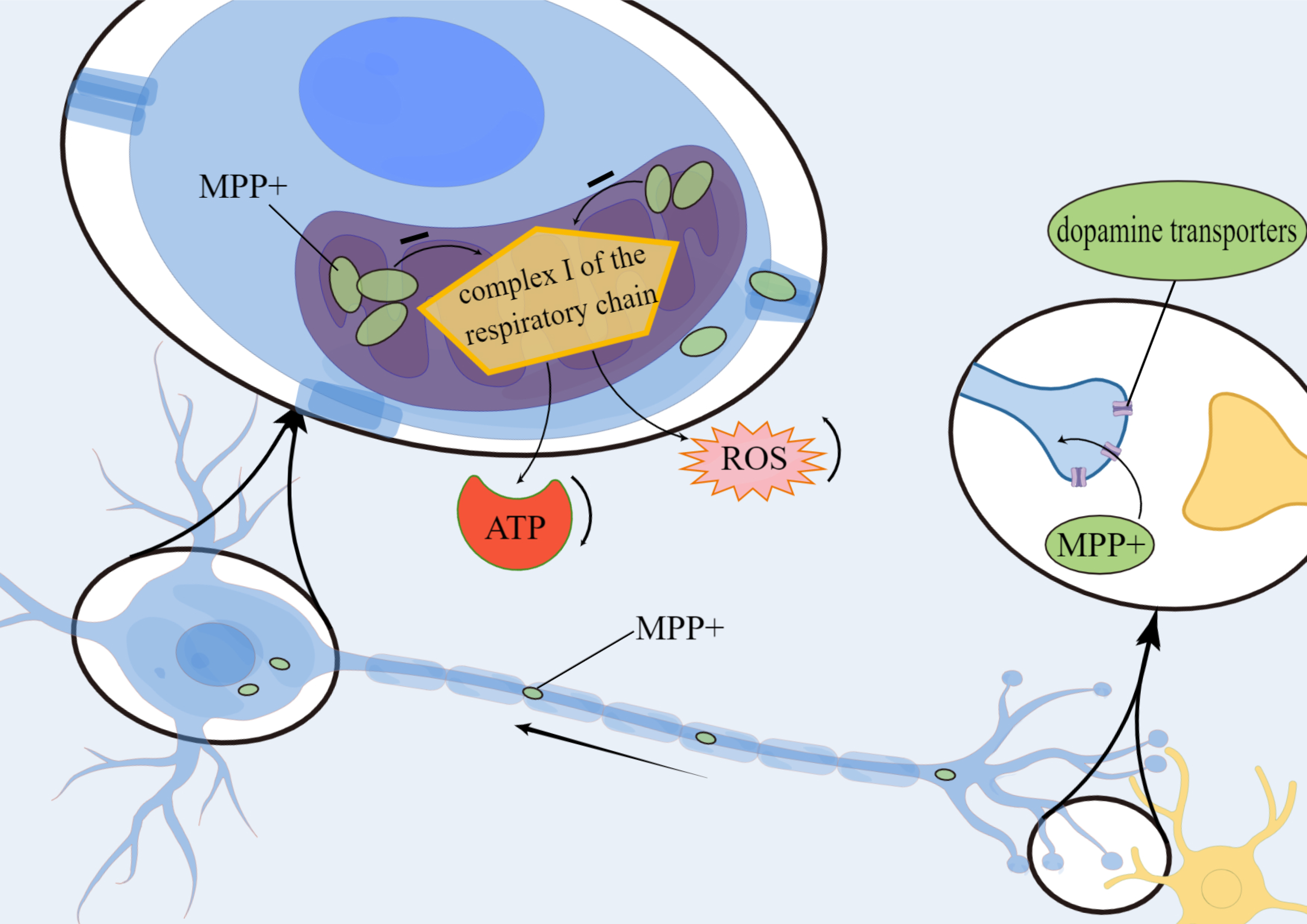 Fig. 3.
Fig. 3.Mechanism of metabolite 1-methyl-4-phenylpyridine ion
(MPP
This leads to degenerative necrosis of dopaminergic neurons in the striatum and
substantia nigra, and ultimately to the development of PD [38]. In addition,
metal-mediated toxicity in PD patients also plays a key role in the neuronal
damage, the enhancement of oxidative stress in the cellular environment, and
elevation of ROS [39]. Like other neurodegenerative diseases, oxidative stress
and ROS play a substantial part in the progression of HD. ROS generated during
DNA damage, lipid peroxidation and especially protein carbonylation are
particularly active in HD. Free radicals cause the peroxidation of intracellular
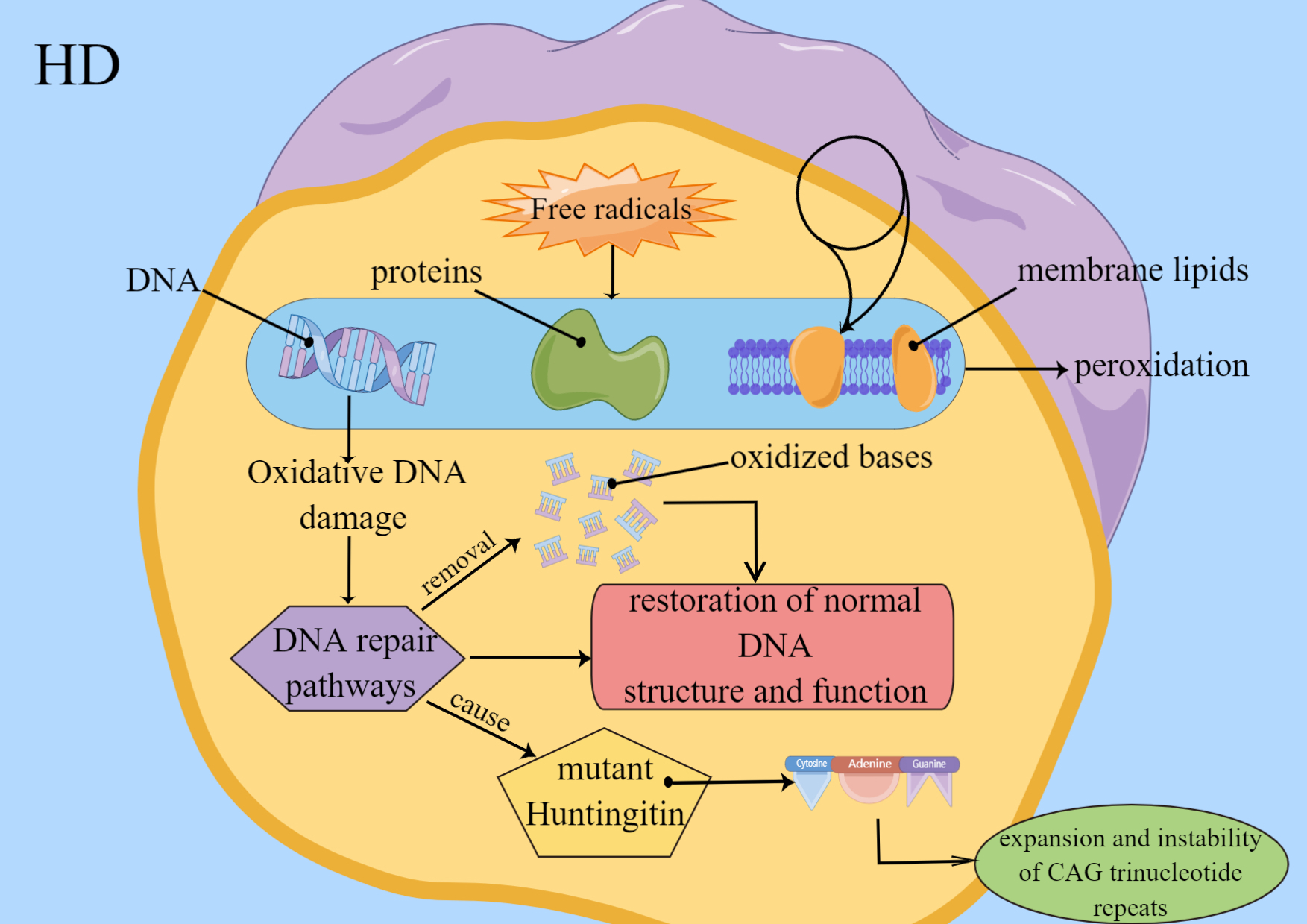
DNA, proteins and membrane lipids. Oxidative DNA damage induces DNA repair
pathways, and the behavior leads to restoration of normal DNA function and
structure and removal of oxidized bases. Repairment of damaged DNA may cause
instability and expansion of CAG trinucleotide repeats in Huntington mutants
(Fig. 4) [40]. Furthermore, the levels of iron increase; this can cause an
increase in the production of very harmful hydroxyl radicals, thus facilitating
interactions with the ferrous iron with H
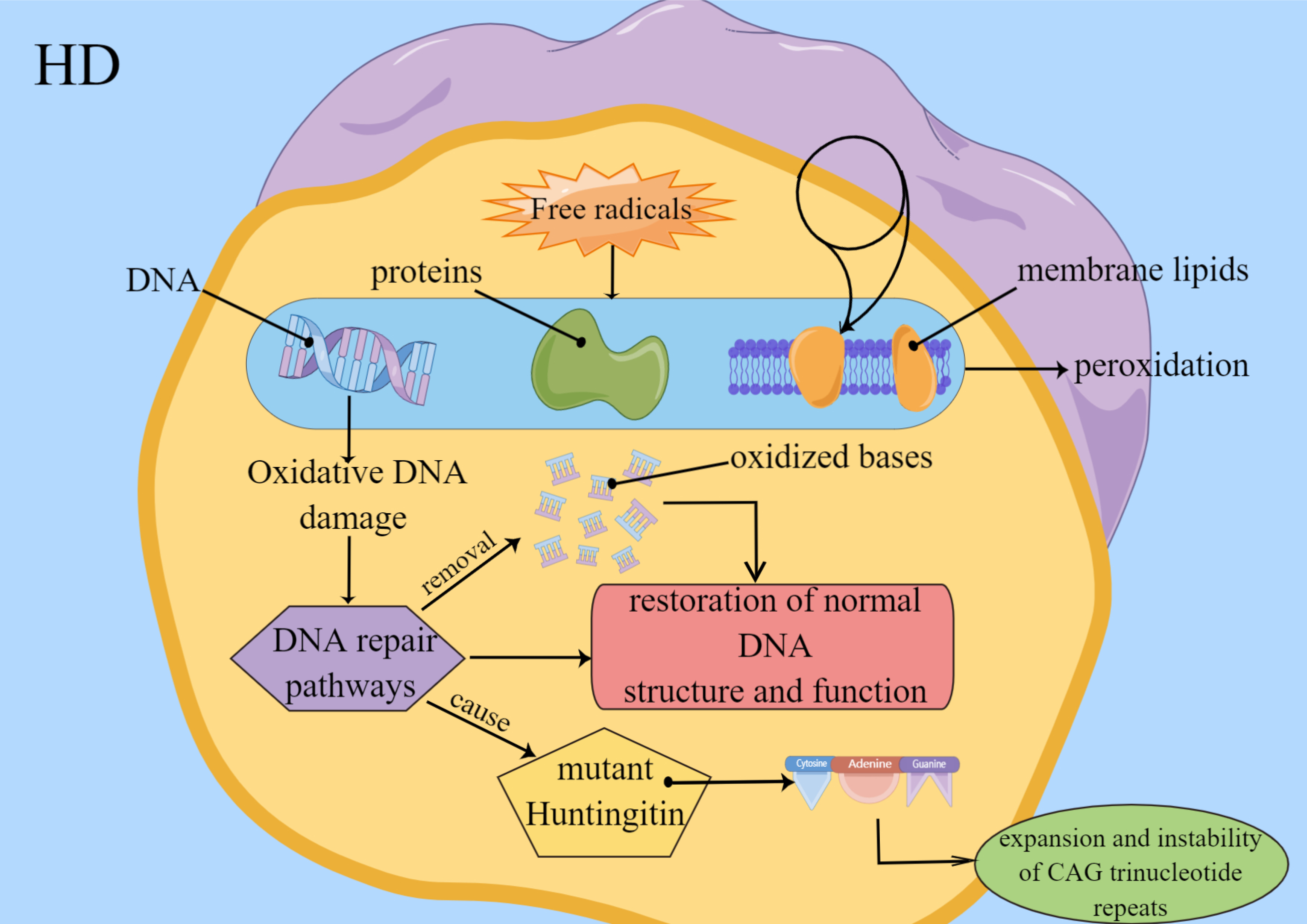 Fig. 4.
Fig. 4.Factors of Oxidative stress involve in HD. Free radicals lead to peroxidation of DNA, proteins and membrane lipids. Oxidative DNA damage induces DNA repair pathways, and the behavior leads to restoration of normal DNA function and structure. And oxidized bases are removed. Repairment of damaged DNA may cause instability and expansion of CAG trinucleotide repeats in Huntington mutant.
The key biomarker for ALS is oxidative stress; it is widely considered that oxidative stress may contribute to neuronal death and the progression of ALS. SOD1 is a member of the superoxide dismutase family and is the main enzyme for scavenging reactive oxygen species. The second most commonly known genetic cause of ALS is mutations in the SOD1 gene; these account for approximately 12% of familial cases and 1% of sporadic cases [41]. However, these mutations result in increased misfolding or toxic function of the SOD1 protein. In a previous study, Garg et al. [42] found that the direct interaction between mutant misfolded SOD1 and TRAF6 could lead to increased SOD1 functional toxicity.
Proteins are responsible to the most of organismal and cellular functions. The synthesis of protein involves multiple procedures; information held by a gene is transcribed into mRNA which is then translated by the ribosomes into a protein. Most proteins need to fold appropriately into a specific three-dimensional structure in order to function. Errors may occur at any time during this process. When errors occur, we can use chaperones and degradation machinery to help prevent their aggregation and folding [43]. Numerous studies have shown that protein misfolding and aggregation occur in many neurodegenerative diseases [44]. The misfolding of proteins can exert detrimental effects on their normal biological function and can generate harmful oligomers. These processes are associated with the binding of proteases and molecular chaperones during translation in cells [45]. The appearance of aggregated nuclei is the beginning of aggregation of protein [46]. Once nucleated, intracellular protein monomers with a tendency to aggregate undergo random folding to produce misfolded, non-functional protein oligomers, which further aggregate to form fibrils (also known as aggregates) [47]. Oligomers and multimers can be degraded by protein molecular chaperones, either by the ubiquitin-proteasome or by autophagy; damaged oligomers and multimers produce cytotoxicity, thus causing cell death and inflammation [47, 48].
AD, PD, and HD are closely related to the deposition and aggregation of proteins
that are misfolded, for example A
The link between neurodegeneration and immunity was established some time ago. However, little is known about neurodegeneration [50]. The immune cells in central nervous system are mainly microglia, oligodendrocyte and astrocytes [51]. These three types of cells undergo different physiological changes during neurodegeneration. Acute Neurodegeneration is associated with a relatively supernal rate of disease and a relatively low rate of immune-related development. However, chronic neurodegeneration is more persistent than immune-related development.
AD is characterized by progressive memory loss and cognitive impairment.
Astrocyte also secretes and ingests A
In the case of PD, there is evidence that Parkinson is associated with Specific
HLA variants (HLA-DRA and HLA-DRB1) [56], thus suggesting an association with the
autoimmune system. As a special case, idiopathic Parkinson’s syndrome is
diagnosed when the body detects the inflammation induced by T cells that target
Although astrocytes secrete and ingest
In a previous study, Ross et al. [64] reported that DNA damage was a common pathway responsible for neurodegenerative diseases. Furthermore, recent studies have shown that DNA damage can be divided into nuclear DNA damage and mitochondrial DNA damage [65]. Although relevant pathological data and the underlying reasons are open to question, Heather et al. [66] stated that dying or damaged cells released damage-related molecular proteins that can cause specific inflammatory cascades. Both have similar outcomes for neurodegenerative diseases.
There are now two main options, one is to increase free radical scavenging, such
as with superoxide dismutase 1 (SOD1), which protects neurons from
A
In a previous study, Cai et al. [68] found that mitochondrial DNA may be a key factor
associated with aging in neurodegenerative diseases. Tau tangles and
A
In addition to key proteins, related abnormalities have also been detected in exosomes. A recent study reported that extracellular vesicles contain misfolded SOD1 enzymes that are delivered to other normal cells, where they can spread to other normal cells [71]. Furthermore, exosomes have also been found in AD and PD. SOD1 has also been associated with mitochondrial DNA damage. Therefore, exosomes may represent a new mechanism to improve mitochondrial DNA damage.
The regulation of the body’s energy metabolism is essential to ensure the functioning of the organism. Mitochondria, as active controllers, play a central role in whole-body energy homeostasis [72]; neurons are one of the most energy-consuming cell types in the body and have a highly complex morphology. The normal functionality of neurons depends on mitochondrial morphology and functional integrity. Apart from the fact that mitochondria serve as primary energy providers, there are many other important functions in mediating some other activities of cells, such as calcium homeostasis, oxidative stress, amino and lipids acids metabolism and proliferation of cells [73]. In fact, mitochondrial dysfunction and mitochondrial abnormalities have been recognized as key and co-factors in the pathology of neurodegenerative diseases, which can disrupt the normal activity of cells and ultimately lead to neuronal loss [74].
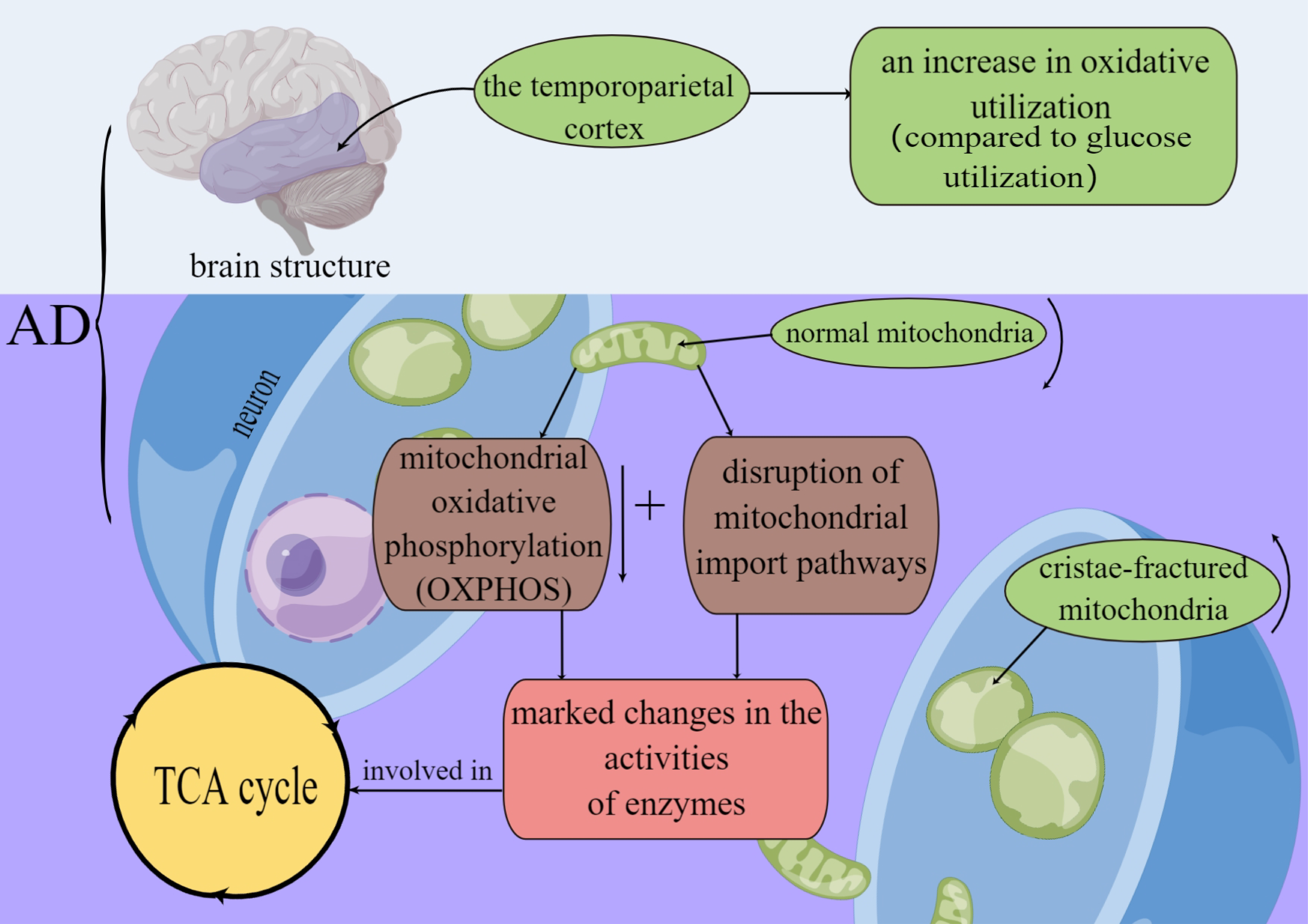
Compared with glucose utilization, by means of positron emission tomography, we detected a reduction in brain metabolism and an increase of oxidative utilization in the temporoparietal cortex of AD. The proportion of normal mitochondria was significantly lower, and the proportion of cristae-fractured mitochondria was significantly higher in AD neurons when compared with controls of the same age. In addition, mitochondrial size, number, and distribution, were significantly altered in AD-vulnerable neurons [75]. Furthermore, 15 out of 51 members of pathways related to glycolysis, oxidative phosphorylation, and the TCA cycle, were significantly downregulated in AD, as determined by microarray analysis and quantitative RT-PCR studies. Gene set enrichment analysis showed that disruption of mitochondrial import pathways and the downregulation of mitochondrial oxidative phosphorylation were prevalent in AD patients. These changes resulted in marked changes in the activities of enzymes that are involved in the TCA cycle in AD: dehydrogenases (including SDH and MDH) were increased, while dehydrogenases/decarboxylases (including PDHC, ICDH, and KGDHC) were decreased; these phenomena are associated with the clinical status of AD patients [76] (Fig. 5).
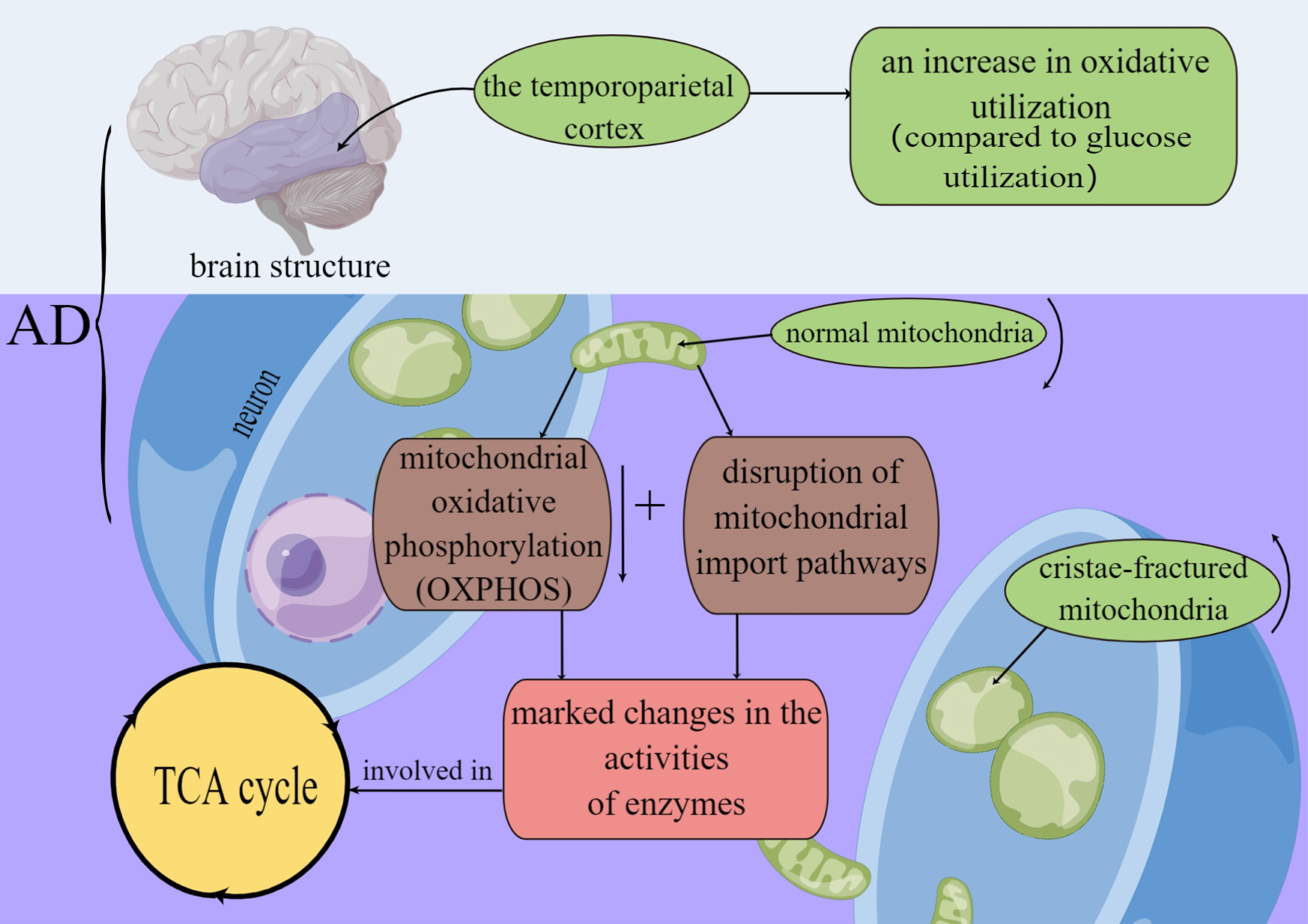 Fig. 5.
Fig. 5.Features of AD in Mitochondrial dysfunction factor. Disruption of mitochondrial import pathways and downregulation of oxidative phosphorylation (OXPHOS) are prevalent in AD. These changes resulted in marked changes in the activities of enzymes that are involved in the TCA cycle of AD.
Furthermore, other studies have suggested that damaged mitochondria played a key role in the pathophysiology of AD. Mitochondria regularly encounter endogenous stress such as DNA damage, oxidative toxicants and environmental stresses, thus generating structural or functional damage to these vital organelles. Neuronal activity is extremely energy-dependent and sensitive to disturbances in mitochondrial function [35].
Several studies have highlighted the crucial role of proteins and miRNAs in
regulating genes that are involved in mitochondrial integrity under different
pathological conditions of neurodegenerative diseases [77, 78, 79]. The mtDNA levels
in body tissues and fluids, and the ratio of mitochondrial genome to nuclear
genome, are usually the evaluative criteria for variations in mtDNA levels, these
represent biomarkers of mitochondrial aberrations [80]. Zhou et al. [81] previously
described the effects of microRNA-330, which targets the proto-oncogene vav
(VAV), on oxidative stress, mitochondrial dysfunction, and A
The maintenance of metal ion homeostasis is very important for the normal
biological functions of the brain, such as the synthesis and metabolism of
neurotransmitters and oxygen transport. Na
Imbalanced intracellular metal homeostasis and toxic metal exposure may play a contributing role in pathology and may represent a cause of neurodegenerative diseases. Karolina et al. [85] reported that an excessive level of trace metal ions could contribute to oxidative stress, endoplasmic reticulum stress, the activation of apoptosis, mitochondrial dysfunction, and the dysregulation of autophagy.
With regards to AD, there is evidence that it is associated with iron
homeostasis. And there are potential links with oxidative stress and glutathione
[86]; these associations may involve high levels of ACSL4 [87]. Researchers
previously demonstrated that the upregulation of xCT perturbed glutathione
metabolism and lipid peroxidation in ferroptosis in AD patients. Metal ions, such
as Zn
HD is another neurodegenerative disorder that is closely related to oxidative reactions catalyzed by transition metals. A growing number of studies have highlighted oxidative stress and the dysregulation of metals in the pathogenesis of HD. Studies of the effects of free radicals on HD have revealed a considerable increase in carbonylation in the striatum of the brain after HD. Similarly, changes in catalytic activity or the protein levels of antioxidant enzymes have been observed [91].
Neurodegenerative diseases are the most common diseases of the elderly. The mechanisms underlying such disease are both intricate and complex. Different theories have arisen that have their own advantages and disadvantages; however, precise mechanisms have yet to be elucidated. Furthermore, genetic factors, oxidative stress, and immunology all play roles in the pathogenesis process to a greater or lesser extent, and may complement each other. The current research focuses in neurodegenerative disease is mainly Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis. Due to recent in-depth studies of the enteric nervous system, we are now beginning to explore gut-related microbes, starting from the enteric neural network, to regulate disease and act as a target to improve regulation of disease as a target to improve disease. A current research hotspot relates to autophagy and mitochondria/lysosomes. These various theories may pave the way for a better understanding of the etiology or mechanisms of neurodegenerative diseases. With the advancement of global research, and increasing collaboration across different research topics, the mechanisms underlying neurodegenerative diseases will become increasingly clear and contribute to the development of clinical treatment plans.
All authors declared that they materially participated in the article preparation. LC, YH and JLong launched the viewpoint of manuscript. JLiu and WD wrote the manuscript and conceived the structure of Figures. JLiu finished the contents of Figures. QZ, CG and HC summarized the manuscript. RL, WA, YD and YQ revised the manuscript and collected the supplementary contents at the revision stage. YH polished the contents of manuscript at the revision stage. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank Fig.Draw (https://www.figdraw.com/) for editing figures.
This work was sponsored by National Key R&D Program of China (2022YFA1104900 & 2022YFA1104904), Natural Science Foundation of Guangdong Province (2021A1515010013), Department of Education of Guangdong Province (2021ZDZX2011), Traditional Chinese Medicine Bureau of Guangdong Province (20221275), Guangzhou Municipal Science and Technology Project (202201011760) and President Foundation of Integrated Hospital of Traditional Chinese Medicine of Southern Medical University (1202101003 & 1202103007).
Lukui Chen is serving as one of the Guest editors of this journal. We declare that Lukui Chen had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Gernot Riedel. The other authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.