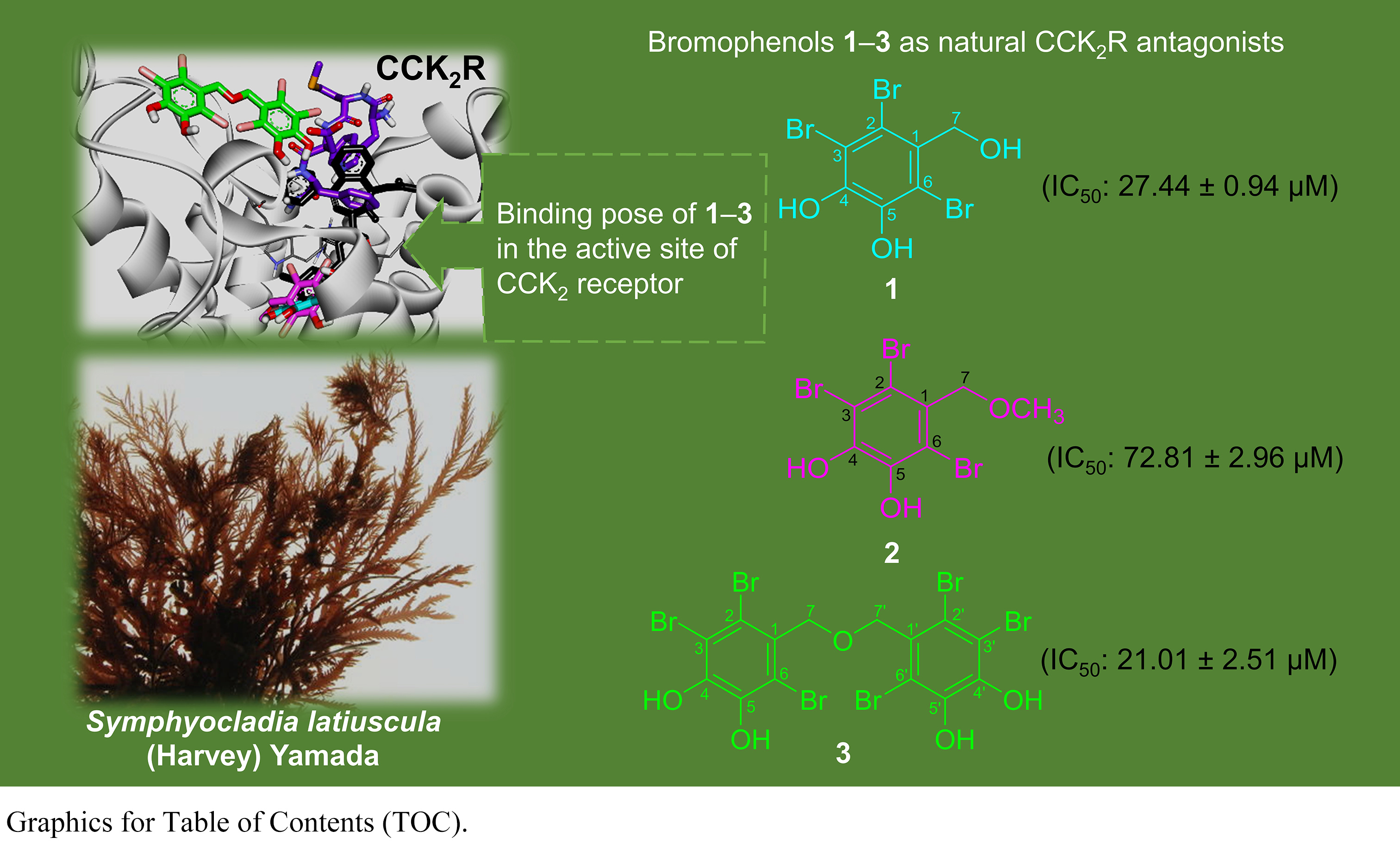
1. Introduction
l-Tryptophan (TRP) is one of the largest essential amino acids and an
essential component in the biosynthesis of proteins, muscle, and enzymes, an
essential substrate for the production of neurotransmitters and hormones. Acute
TRP deficiency results in increased pain sensitivity, auditory startle, increased
motor activity, and increased aggression, while chronic TRP deficiency causes
ataxia, cognitive impairment and dysphoria with skin hyperpigmentation [1].
Kynurenine (KYN) and serotonin (5-HT) are derived from the TRP metabolic pathway.
The kynurenine (KYN) pathway is involved in 95% of TRP degradation, resulting
in de novo synthesis of nicotinamide adenine dinucleotide (NAD) [2]. The
pathway begins with the conversion of TRP to KYN catalyzed by two enzymes,
tryptophan 2,3-dioxygenase and indolamine 2,3-dioxygenase. Furthermore, depending
upon the cell type, KYN is further metabolized through three distinct pathways.
(1) Synthesis of 3-hydroxykynurenine (3-HK) in microglia by kynurenine
3-monooxygenase (KMO) and its metabolites 3-hydroxyanthranilic acid (3-HAA) and
quinolinic acid (QUIN); (2) Synthesis of anthranilic acid in microglia by
kynureninase; and (3) Synthesis of kynurenic acid (KYNA) in astrocytes by
kynurenine aminotransferase (KAT) II [3]. QUIN is an endogenous glutamate
N-methyl-d-aspartate (NMDA) receptor agonist and KYNA is a
noncompetitive antagonist of alpha-7 nicotinic acetylcholine receptor
(7nAchR). Hence, abnormalities in the KYN pathway have been implicated
in the pathophysiology of depression.
Depression is a heterogeneous disorder, and the exact neuronal mechanisms
driving it have not yet been discovered. Evidence also suggests that
cholecystokinin (CCK) and its receptors play an important role in the
pathogenesis of anxiety-related behaviors and depression [4]. Furthermore, the
causal relationship between increased CCKergic neurotransmission in the socially
defeated cortex and depressive-like symptoms strongly suggests the relevance of
the CCKergic system as a novel target for antidepressant therapy [5, 6]. Anxiety
disorders are characterized by excessive fear, and Pavlovian threat conditioning
is an adaptive mechanism by which organisms learn to avoid potential threats,
thereby increasing their chances of survival [7]. The prefrontal cortex (PFC) is
a central brain region in the pathogenesis of depression and it has been
suggested that the ventromedial prefrontal cortex (vmPFC) plays an important role
in the acquisition of Pavlovian threat conditioning [7]. Amygdaloid CCK
receptors appear to be involved in the expression of fear-enhanced startle, as
fear-enhanced startle is blocked by systemic injections of CCK antagonists
[8]. Furthermore, injections of CCK-8 into the central nucleus of the amygdala
increases arousal and fear in rats [9]. Therefore, CCK and its receptors could be
one of the prime targets for antianxiety and antidepressant drugs.
Cholecystokinin (CCK) is a 33-amino-acid-long peptide hormone, first identified
in the gastrointestinal (GI) tract, that regulates gut motility [10], pancreatic
and gastric-acid secretion [11, 12], and gall bladder contractions [13]. It is a
secretagogue of insulin and pancreatic polypeptides in non-ruminants [14, 15].
Also, it is one of the most abundant peptides in the central nervous system (CNS)
that function as a neurotransmitter and has an inverse correlation of its level
with depression and anxiety [16, 17, 18, 19, 20]. CCK levels are elevated in depression [21],
and patients with high cerebrospinal fluid (CSF) CCK levels have more suicide
attempts than those with low CSF CCK levels [16]. The receptor exists as two
subtypes, CCK and CCK, previously classified as CCK and
CCK. CCK is predominant in the GI system and has roles in gallbladder
contraction and stimulation of pancreatic secretion [22, 23]. CCK is
predominant in the CNS and has roles in satiety, memory, anxiety, osmotic stress,
and neuropsychiatric disorders [24, 25]. Several studies have shown that CCK
peptides have anxiolytic effects in various experimental models of anxiety and
can induce panic attacks in humans [26, 27, 28]. In addition, activation of CCK2
receptors in the brain can cause anxiety. Therefore, selective CCK2 receptor
antagonists represent a unique class of anxiolytics [29]. Recently, much effort
has been made in developing potent and specific CCK receptor antagonists,
which were classified as:
(a) benzodiazepine group: L-365,260, L-36 718 (devazepide), CI-988, YM022,
Z-360, YF476 (netazepide), and YM022 [30, 31, 32, 33];
(b) tryptophan dipeptide derivatives: PD-134,308 [28, 34];
(c) ureidoacetamides: RP-73,870 [35];
(d) pyrazolidinones - LY-288,513 [36].
Despite the progress in developing several CCK receptor
agonists/antagonists [12, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46], none of them have reached the clinic, because
of unfavorable, insufficient, and various biological effects discovered in
clinical trials [39]. Only netazepide and Z-360 are currently under clinical
development for the management of gastric neuroendocrine tumors and pancreatic
cancer, respectively. Therefore, the discovery of selective CCK receptor
antagonists with suitable pharmacokinetic profiles is very important.
In drug discovery and development, halogenation plays a vital role in drug
optimization by modulating lipophilicity and cell membrane solubility, increasing
blood–brain barrier (BBB) permeability and the half-life, and improving membrane
binding and CNS delivery of pharmaceutical drugs [47, 48, 49, 50]. In recent years, the
halogen bond has received a great deal of attention for hit-to-lead-to-candidate
optimization to improve the drug-target binding affinity; as a result, 25% of
the marketed drugs are halogenated [49]. Marine sources are predominant in
bioactive, structurally diverse, and unique halogenated compounds, of which 45%
are Bromo-metabolites [51]. These active metabolites have become structural
models in developing synthetic derivatives with superior biological activity
[52].
The red alga Symphyocladia latiuscula (Harvey) Yamada is rich in
bromophenols with diverse biological activities, including anticancer [53],
antibacterial [54], antifungal [55], antiviral [56], free-radical scavenging
[57], aldose-reductase inhibitory [58], -glucosidase inhibitory [59],
and other properties [60, 61, 62]. Secondary metabolites from this alga contain a
2,3,6-tribromo-4,5-dihydroxybenzyl moiety with various substituents. Recently, we
have reported anti-diabetic [63], anti-Alzheimer’s disease [64], anti-tyrosinase
[65], and anti-Parkinson’s disease activity [66], of
2,3,6-tribromo-4,5-dihydroxybenzyl derivatives from S. latiuscula. The
anti-Parkinson’s disease activity of those derivatives were evaluated via their
effect on human monoamine oxidase and dopamine receptors [66]. Monoaminergic
neurotransmission is primarily based on G-protein coupled receptors (GPCRs)
signaling. The aminergic system is dysregulated in anxiety disorders and major
depressive disorder, and antidepressants act directly or indirectly through the
GPCRS. Therefore, GPCRs are potential therapeutic targets for intervention in
neurodegenerative diseases. However, apart from MAO and dopamine receptors, there
are no reports on how natural bromophenols affect other GPCRs receptors.
Therefore, our main objective in this study was to predict novel GPCRs targets
via proteocheminformatics modelling (PCM) and characterize the functional effect
of natural bromophenols on CCK receptors for the management of CCK-mediated
diseases, especially anxiety and depression.
2. Materials and Methods
2.1 Chemicals and Reagents
We isolated three bromophenols, namely, 2,3,6-tribromo-4,5-dihydroxybenzyl
alcohol (1), 2,3,6-tribromo-4,5-dihydroxybenzyl methyl ether
(2), and bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3)
(Fig. 1), from the leafy thalli of Symphyocladia latiuscula (Harvey)
Yamada and identified as described in our recent report [63]. We estimated the
purity of the test compounds to be 98% as evidenced by proton and carbon NMR
spectra. We obtained transfected Chinese hamster ovary (CHO) cells from Eurofins
Scientific (Le Bois I’Eveque, France). We obtained Hank’s balanced salt solution
(HBSS) buffer, Dulbecco’s modified Eagle medium (DMEM) buffer, and
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer from Invitrogen
(Carlsbad, CA, USA). We purchased reference drugs CCK-8s and YM022 from
Sigma-Aldrich (St. Louis, MO, USA).
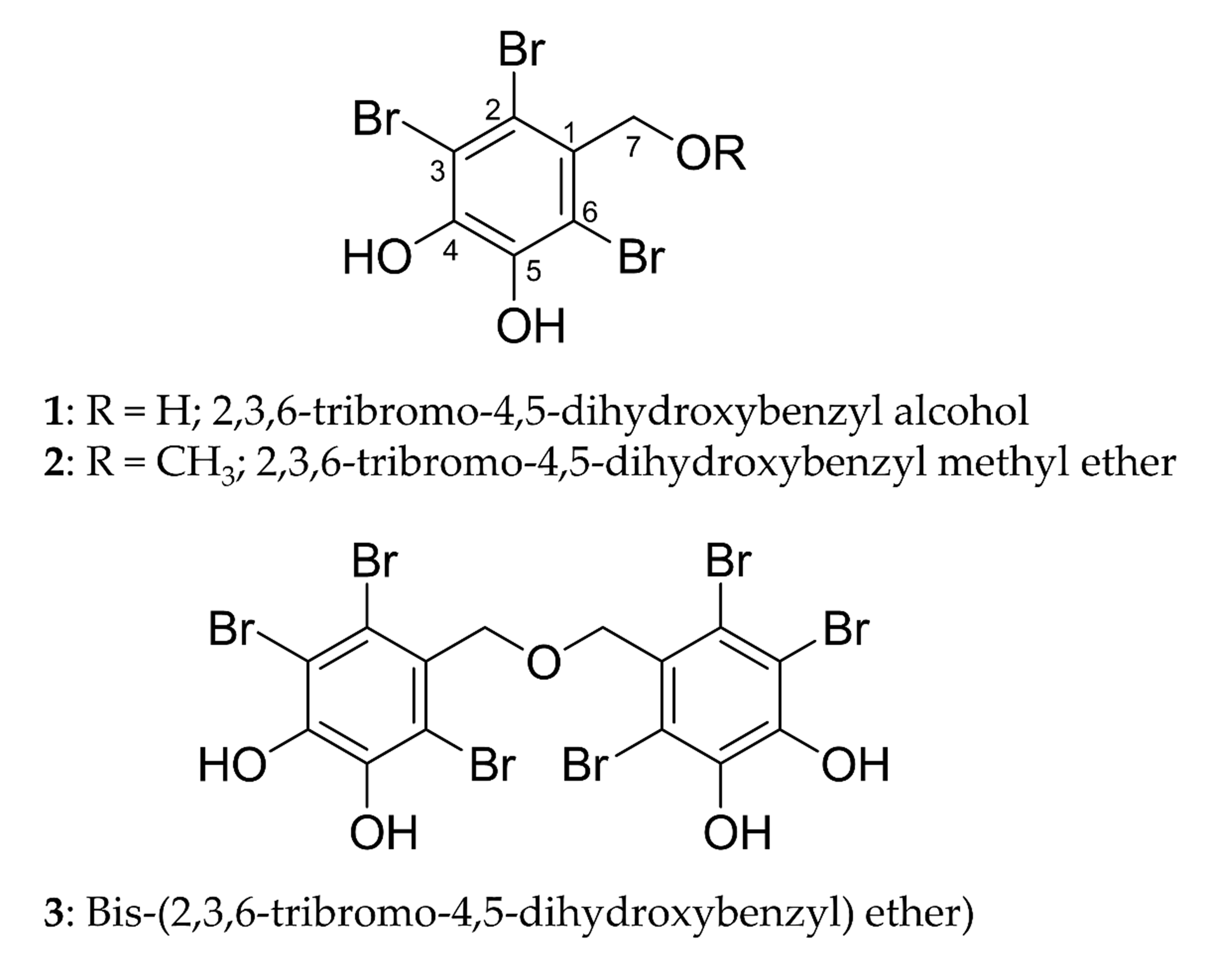 Fig. 1.
Fig. 1.
Structures of bromophenols isolated from S.
latiuscula.
2.2 In Silico Prediction of Targets
Proteochemometric modeling (PCM) is a biological activity modeling technique
based on the similarity of a set of ligands and a set of target proteins. It is
built based on chemical descriptors that describe datasets of compounds and
proteins of interest. Different interactions between a set of compounds and a set
of targets can be described when describing the specific interaction between each
compound and each target in the dataset. We did PCM as an in-silico way
to predict potential protein targets of test bromophenols. The model was directed
to chemically and biologically similar compounds (55,079 compounds active and
inactive) against 99 human proteins (11,537 active pairs vs 43,542 inactive
pairs). We selected 99 protein targets because these targets are considered
‘critical nodes’ in the biological network. Critical nodes need links that can
crosstalk with other pathways, should be member of related proteins i.e.,
isoforms where two or more of the proteins in the group have unique biological
roles and is highly regulated, either positively or negatively [67].
We evaluated the pattern that differentiates active and inactive complexes by
using Parzen Rosenblatt Windows (PRW) [68, 69] and predicted the activity of
novel compounds against the 99 protein targets. We calculated chemical
similarities using an Aitchison-Aitken kernel [70] and represented chemical
structures as an ECFP_4 fingerprint [71]. Protein sequences were subjected to
sequence alignment using MUSCLE [72], operated with the bio3d package [73],
before we calculated the similarities between two protein sequences. (For full
information on the model, see [74]).
2.3 Functional GPCRs Assay
We evaluated the functional effect of bromophenols 1–3 on
cholecystokinin 2 (CCK) receptor in transfected CHO cells expressing human
cloned CCK receptors by measuring their effects on cAMP production using
HTRF detection. In brief, we suspended stable transfectants (CHO-CCK) in
HBSS buffer (Invitrogen, Carlsbad, CA, USA), supplemented with 20 mM hydroxyethyl
piperazineethanesulfonic acid (HEPES) buffer and 500 M
3-isobutyl-1-methylxanthine (IBMX), then distributed them into microplates at a
density of 1 10 cells/well. Then, the plates were incubated for
30 min at 37 °C in the presence of HBSS (basal control), test
bromophenols 1–3 (12.5, 25, 50, and 100 M) or a
reference agonist (CCK-8s). We prepared separate cells with 100 nM CCK-8s for
stimulated control measurement. After incubation, cells were lysed, and a
fluorescence acceptor (D-labeled cAMP) and fluorescence donor (anti-cAMP
antibody with europium cryptate) were added. We measured a fluorescence transfer
at = 337 nm and = 620 and 665 nm, using a
microplate reader (EnVision, PerkinElmer, Waltham, MA, USA) after 60 min of
incubation at RT. The cAMP concentration was calculated as the ratio of the
signal at 665 nm to that of 620 nm (i.e., by dividing the signal measured at 665
nm by that measured at 620 nm).
The agonist effect was expressed as a percentage of the control response to 100
nM CCK-8s. Similarly, for the antagonist effect, we evaluated the percent
inhibition of the control response to CCK-8s 10 nM. We used CCK-8s as an agonist
and YM022 as antagonists as reference drugs. We tested the standard reference
drugs at several concentrations to generate a concentration-response curve from
which either EC or IC value was calculated.
2.4 Homology Modeling
We obtained the primary sequence of the human CCK receptor from UniProt
(ID: P32239, GASR_HUMAN). The human adenosine (A) receptor (PDB ID 3EML)
has a higher similarity to CCKR [75]. Hence, we built the model on the
template of the AR crystal structure from the RCSB protein data bank (PDB)
using ID 5UIG with SWISS-MODEL. We refined the model using the 3Drefine server
[76].
2.5 Statistics
The Kruskal-Wallis test and Dunn’s multiple comparison test was performed to
calculate statistical significance of the agonist/antagonist activity according
to the treatment of bromophenols 1‒3 using GraphPad Prism
version 5.03 (GraphPad Prism, GraphPad Software Inc., San Diego, CA, USA). The
significance levels were denoted as *p 0.05. A level of p
0.05 was considered statistically significant. Data in the figure and table
are represented as mean SD and result from at least three independent
experiments.
3. Results
3.1 In Silico Target Prediction
From PCM, we predicted the highest-ranked 15 potential protein targets for the
three bromophenols. Table 1 lists the target proteins with the normalization
rate. As shown in Table 1, most of the predicted target proteins (cholecystokinin
receptor 1/2, adenosine receptor 2A/2B, 5HT1A receptor, and PI3K-
isoform) were the same for the three bromophenols. However, only bromophenol
2 had greater normalization values, and the ranking order of target
proteins differed for each compound. Cholecystokinin type B/Gastrin receptor was
predicted as a prime target for 2, with the highest normalization value
(0.959). The same CCK receptor was predicted in a rank order of 5 and 14
with normalization value 0.896 and 0.648 for compounds 3 and 1,
respectively. Since bromophenol 2 is a major component in S.
latiuscula, we then proceeded to validate the highest-ranked CCKprediction in GPCRs cell-based functional assays (Table 2).
Table 1.List of fifteen protein targets predicted from PCM modeling for
bromophenols 1‒3 with normalization values.
| 2,3,6-Tribromo-4,5-dihydrobenzyl alcohol (1) |
2,3,6-Tribromo-4,5-dihydrobenzyl methyl ether (2) |
Bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3) |
| Serine/threonine-protein kinase mTOR (0.725) |
Cholecystokinin 2 receptor (0.959) |
Cholecystokinin 1 receptor (0.924) |
| Adenosine receptor A (0.724) |
Cholecystokinin 1 receptor (0.932) |
Serine/threonine-protein kinase mTOR (0.920) |
| Beta-3 adrenergic receptor (0.708) |
Serine/threonine-protein kinase mTOR (0.929) |
PI3K- isoform (0.901) |
| PI3K- isoform (0.704) |
Adenosine receptor A (0.922) |
Adenosine receptor A (0.898) |
| Beta-2 adrenergic receptor (0.692) |
PI3K- isoform (0.912) |
Cholecystokinin 2 receptor (0.896) |
| Adenosine receptor A (0.683) |
Adenosine receptor A (0.909) |
Adenosine receptor A (0.883) |
| Beta-1 adrenergic receptor (0.674) |
Substance-K receptor (0.908) |
5-hydroxytryptamine receptor 1A (0.875) |
| 5-hydroxytryptamine receptor 1A (0.664) |
Substance-P receptor (0.908) |
Muscarinic acetylcholine receptor M2 (0.874) |
| Mitogen-activated protein kinase 14 (0.662) |
Endothelin-1 receptor (0.906) |
Somatostatin receptor type 2 (0.873) |
| Dopamine D receptor (0.659) |
Vasopressin V receptor (0.905) |
Muscarinic acetylcholine receptor M3 (0.873) |
| Histamine H receptor (0.657) |
Somatostatin receptor type 2 (0.905) |
Endothelin-1 receptor (0.873) |
| Muscarinic acetylcholine receptor M3 (0.655) |
Vasopressin V receptor (0.905) |
5-hydroxytryptamine receptor 4 (0.872) |
| 5-hydroxytryptamine receptor 1B (0.650) |
Oxytocin receptor (0.904) |
Vasopressin V receptor (0.872) |
| Cholecystokinin 1 receptor (0.648) |
Cannabinoid receptor 1 (0.903) |
Vasopressin V receptor (0.872) |
| Cholecystokinin 2 (0.641) |
B1 bradykinin receptor (0.903) |
Cannabinoid receptor 1 (0.872) |
| Note: Values in the bracket represent normalization values. |
Table 2.Efficacy values (% stimulation and % inhibition) of
bromophenols 1‒3 and reference compounds at human cholecystokinin 2 receptor.
| Compounds |
Response at 100 µM % Stimulation /(% Inhibition ) |
EC (IC) |
| 2,3,6-Tribromo-4,5-dihydrobenzyl alcohol (1) |
–7.8 1.41 (101.80 0.42) |
ND (27.44 0.94) |
| 2,3,6-Tribromo-4,5-dihydrobenzyl methyl ether (2) |
–11.8 1.70 (81.65 9.40) |
ND (72.81 2.96) |
| Bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3) |
–8.05 1.34 (104.35 1.34) * |
ND (21.01 2.51) |
| CCK-8s |
ND |
1.6 |
| YM022 |
ND |
–0.49 |
| Kruskal-Wallis test |
p value |
Dunn’s multiple comparison test (p 0.05) |
| 1 vs 2 |
1 vs 3 |
2 vs 3 |
| % Stimulation at 100 µM |
0.0608 |
NS |
NS |
NS |
| % Inhibition at 100 µM |
0.0273 |
NS |
NS |
* |
ND: Not Determined.
NS: Not significant.
% Stimulation and % inhibition of control agonist response at 100
M of bromophenols, respectively.
EC; half maximal effective concentration values of test compounds
(M) and reference agonist CC-8s (nM).
IC; half maximal inhibitory concentration values of test compounds
(M) and reference antagonist YM-022 (nM).
* Represents significant difference at p 0.05. |
3.2 Bromophenols as CCKR Antagonists
Following cell-based functional assays in stable transfectant (CHO-CCK),
we characterized the functional effect of bromophenols 1‒3 in
modulating CCK receptor function by their potential to either stimulate or
inhibit receptor activity. We tested a reference agonist CCK-8s and antagonist
YM022 for comparison. The CCK agonist or antagonist effect of 100
M of 1‒3 is shown in Table 2. As shown,
1‒3 had a potent antagonist effect on CCKR with
inhibition of the control agonist response by 101.80 0.42, 81.65
9.40, and 104.35 1.34%, respectively. Statistically significant
difference (p 0.05) was found between the antagonist effect of
bromophenols 2 and 3. The agonist effect was negligible, as
shown by the negative percent of simulation of the control agonist response.
Fig. 2 represents a concentration-dependent antagonist response of
1‒3 and YM022 on CCKR along with their IC values.
As shown there, bromophenols 1‒3 inhibited a 50% agonist
response of CCK-8s in CCKR at 27.44 0.94, 72.81 2.96, and
21.01 2.51 M, respectively. The reference antagonist YM022
had an IC value of 0.49 0.01 nM.
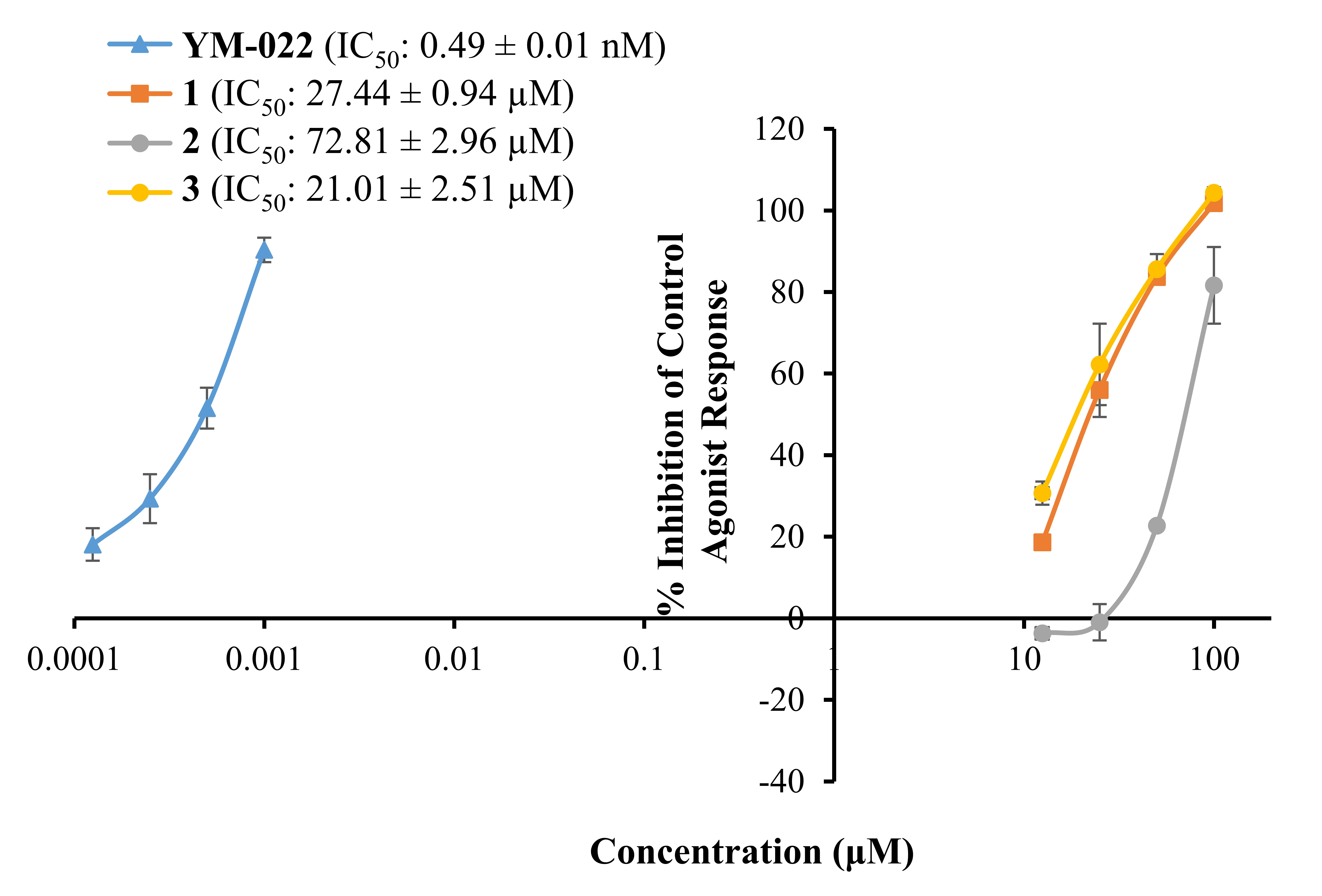 Fig. 2.
Fig. 2.
Concentration-dependent percentage inhibition of control agonist
response of bromophenols 1–3 and reference compound YM-022 on CCK
receptor. Bromophenols were tested at concentrations of 12.5, 25, 50, and 100
M. Reference antagonist YM-022 was tested at 0.125, 0.25, 0.5 and
1.0 nM concentrations, respectively. The experiment was carried out in
triplicates and inhibition values were expressed as the mean standard
deviation (SD) (n = 3).
3.3 Molecular Docking Simulation
To explore the binding environment where ligands 1‒3 interact
with human CCKR, ligands were docked against the 3D model of CCKR
using AutoDock 4.2 (Figs. 3,4); we analyzed the data based on interacting
amino-acid residues and binding score (Table 3). Furthermore, we compared the
docking results of test ligands with those of reference ligands. As shown in
Table 3, 1‒3 were predicted to bind with low binding scores
(–5.45 kcal/mol to –7.38 kcal/mol), and ligand 3 had the lowest
binding score (–7.38 kcal/mol) among the other test ligands. Ligands 1
and 2 formed two common H-bond interactions (Asn353 and Tyr380), as
shown by the green broken lines in Fig. 4. Also, the hydrophobic interacting
residues—Trp209 (- stacked, -alkyl), Val138 (alkyl),
Val349 (alkyl), Met134 (alkyl), His376 (-alkyl)—observed for the binding
of ligand 2 were in common with the ligand 1 binding. The only
difference is that ligand 1 interacted with an additional residue,
Phe227 (-alkyl). However, 3 showed five H-bond
interactions—Val194 with the C5 -OH group, Arg356 with the C4’-OH group,
Pro210 with the C4’ and C5’-OH groups, and Arg208 with C2 bromine atom—which
were not observed for ligands 1 and 2. The reference ligand
YM022 had the lowest binding score (–9.74 kcal/mol) and displayed two H-bond
interactions with His376 and Asn353.
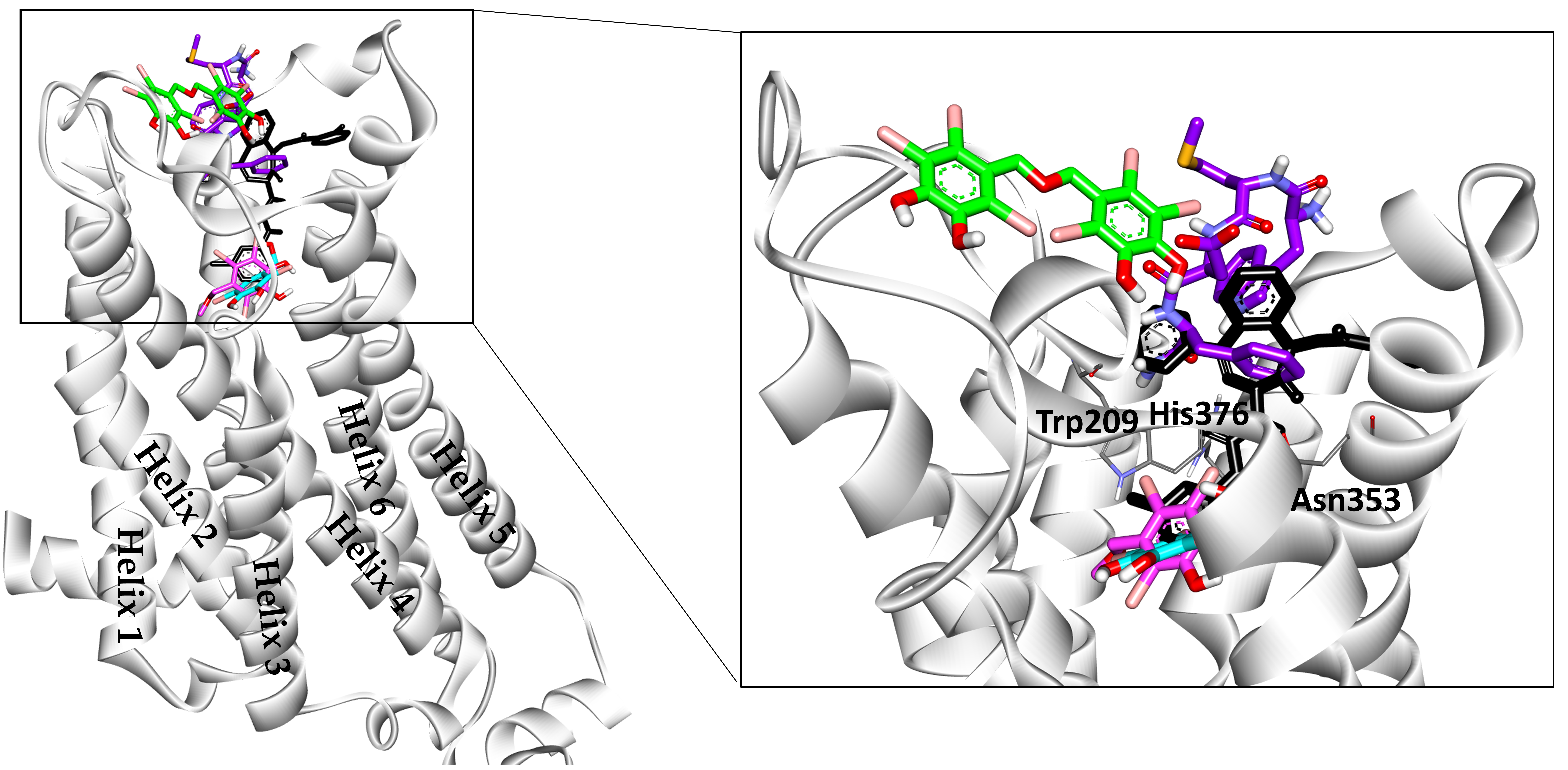 Fig. 3.
Fig. 3.
Binding pose of test and reference ligands in the active site of
CCK receptor. Each ligand is depicted in different color. For instance,
2,3,6-tribromo-4,5-dihydrobenzyl alcohol (blue), 2,3,6-tribromo-4,5-dihydrobenzyl
methyl ether (pink), bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (green),
YM-022 (black), and CCK-4 (purple).
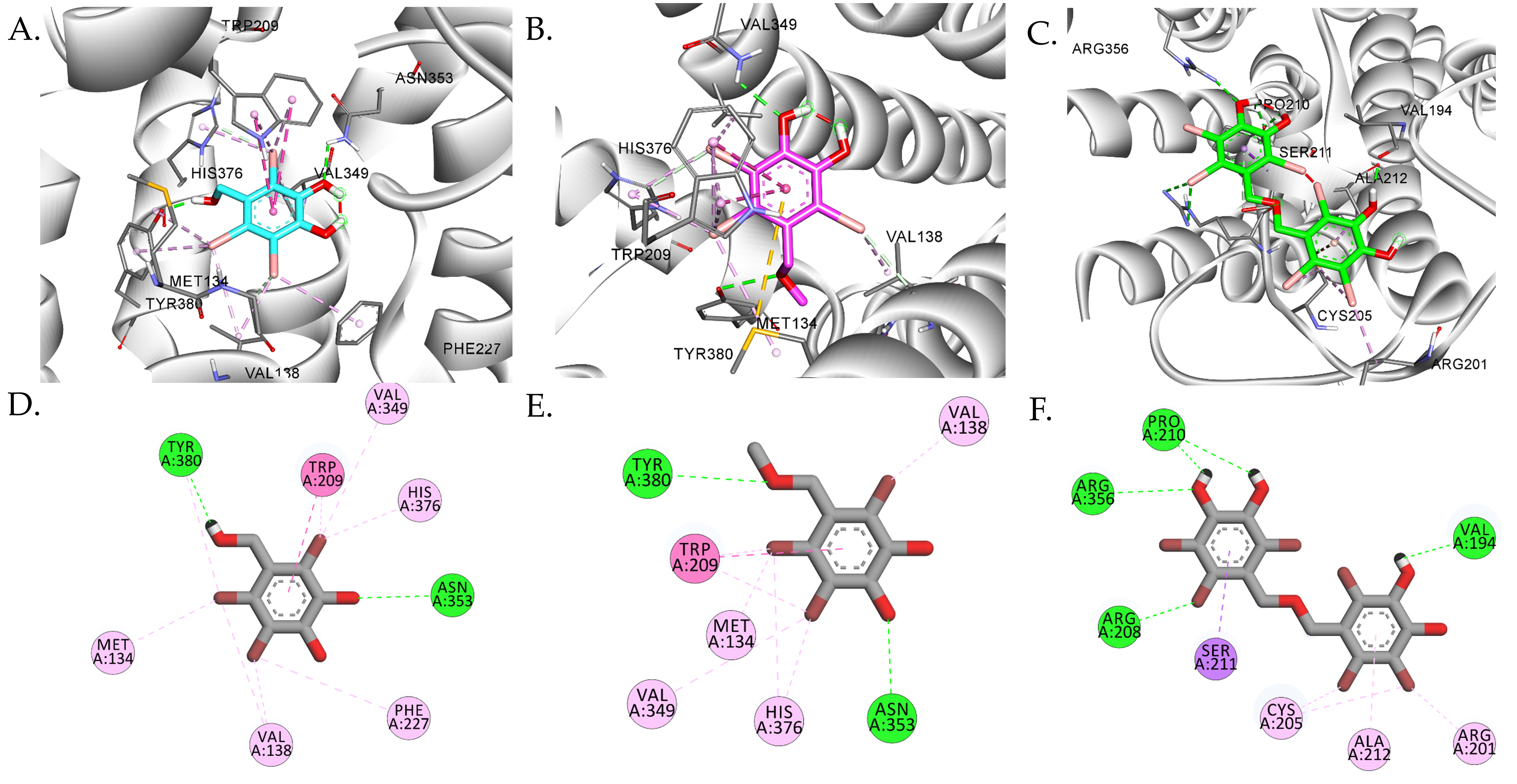 Fig. 4.
Fig. 4.
2Dbinding pose of test ligands in the active
site of CCK receptor. (A–C) Binding pose and (D–F) 2D-binding diagram of
2,3,6-tribromo-4,5-dihydrobenzyl alcohol, 2,3,6-tribromo-4,5-dihydrobenzyl methyl
ether, and bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether, respectively. Dotted
lines with different colors represent different types of interactions. For
instance, H-bond interactions are represented with green dotted lines and
hydrophobic interactions with light-purple dotted lines.
Table 3.Binding affinity of compounds against cholecystokinin B
receptor and reference ligands using AutoDock4.2.
| Ligand |
Binding Energy (kcal/mol) |
Interacting residues |
| H-bond |
Hydrophobic |
| 2,3,6-Tribromo-4,5-dihydrobenzyl alcohol (1) |
‒5.45 |
Asn353, Tyr380 |
Trp209(π-π stacked, π-alkyl), Val349(alkyl), Val138(alkyl), Met134(alkyl), Phe227(π-alkyl), His376(π-alkyl), Tyr380(π-alkyl) |
| 2,3,6-Tribromo-4,5-dihydrobenzyl methyl ether (2) |
‒5.54 |
Asn353, Tyr380 |
Trp209(π-π stacked, π-alkyl), Val138(alkyl), Val349(alkyl), Met134(alkyl), His376(π-alkyl) |
| Bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3) |
‒7.38 |
Arg208, Arg356, Val194, Pro210 |
Ser211(Pi-Sigma), Arg201(Alkyl), Arg205(Alkyl), Ala212(Pi-Alkyl) |
| YM022 (antagonist) |
‒9.74 |
His376, Asn353 |
Ala352(π-σ), Trp209(π-π stacked, π-alkyl), Met134(alkyl), Arg356(alkyl), Trp355(π-alkyl), His376(π-alkyl), Val349(π-alkyl), Pro210(π-alkyl), Ile372(π-alkyl) |
| CCK-4 (agonist) |
‒6.76 |
Arg208, His376, Ala366, Asn115 |
Ile372(π-σ, π-alkyl), Pro201(π-alkyl), Ala352(π-alkyl) |
1-(3-methylphenyl)-3-[(3R)-1-[2-(2-methylphenyl)-2-oxoethyl]-2-oxo-5-phenyl-3H-1,4-benzodiazepin-3-yl]urea.
Tetragastrin; (3S)-3-[[(2S)-2-[[(2S)-2-amino-3-(1H-indol-3-yl)propanoyl]amino]-4-methylsulfanylbutanoyl]amino]-4-[[(2S)-1-amino-1-oxo-3-phenylpropan-2-yl]amino]-4-oxobutanoic acid. |
4. Discussion
The aim of this study was to predict novel GPCRs targets of three natural
bromophenols from Symphyocladia latiuscula via proteochemometric
modeling (PCM), characterize the functional effect on CCK receptors and
predict the interactions between a compound and the binding site of the target
protein for the management of CCK-mediated diseases, especially anxiety and
depression. The pathophysiology of major depressive disorder and anxiety is
complicated and not fully understood. Selective serotonin reuptake inhibitors
(SSRIs), selective norepinephrine reuptake inhibitors, tricyclic antidepressants,
and monoamine oxidase inhibitors are classic antidepressants used to treat
depression. However, these drugs require a minimum of 2–4 weeks of continuous
treatment to elicit therapeutic relief in depressed patients which leads to
prolonged suffering and disability and increases suicide risk [6]. Due to the
incomplete effectiveness of current treatment and the burdening neuroscience of
fear-related behavior, however, compel the search for novel, more effective
therapies [77]. In this regard, we gave a continuous effort to discover novel
antidepressants from natural products.
Here, we focused on natural brominated compounds 1‒3 from a
red alga S. latiuscula to explore their functional effect on
cholecystokinin 2 receptor based on our PCM prediction. We predicted a total of
15 target proteins for compounds 1‒3, and most of the predicted
proteins were the same for the three compounds, with different rank orders
following the normalization value (Table 1). The CCK receptor was a prime
protein target predicted for compound 2; however, the rank order and
normalization values were different for 1 and 3. We attribute
this difference to their structural differences. So, to verify the PCM prediction
and get molecular insight into the test ligands‒CCK receptor interaction,
we proceeded with the functional assay followed by molecular docking simulation.
The results of a functional assay conducted in the stable transfectant
(CHO-CCK) indicated that bromophenols 1‒3 are potent
CCK receptor antagonists. Bromophenol 1 and 3 are full
CCK antagonist that showed 100% antagonist effect at a 100 M
concentration, whereas the antagonist effect was approx. 82% for 2 at
that concentration. All the tested bromophenols had no agonist effect, as
indicated by the negative percentage agonist effect. The activity fashion of
1‒3 was similar to our recent studies, where 1 and
3 showed better antidiabetic property by inhibiting protein tyrosine
phosphate 1B and -glucosidase enzyme activity, and by increasing
insulin sensitivity and glucose uptake in a human liver-cancer cell line [63],
anti-Alzheimer’s disease activity via cholinesterase, self-induced
A aggregation inhibition [64], anti-Parkinson’s disease
activity via the agonist effect on dopamine D/D receptors [66], and
anti-browning effect by inhibiting melanin content and intracellular tyrosinase
levels in -melanocyte-stimulating hormone-induced B16F10 melanoma cells
[65].
All test compounds have a 2,3,6-tribromo-4,5-dihydroxybenzyl moiety in
common, with different substituents at C-1. Bromophenol 1 has a
hydroxymethyl group at C-1, 2 has a methyl ether, and 3 is a
dimeric form, in which another 2,3,6-tribromo-4,5-dihydroxybenzyl moiety is
attached at C-1 via a methyl ether linkage. Compared to the CCKR antagonist
effect of 2, bromophenols 1 and 3 had a better effect.
However, the exact structure-activity relationship is uncertain, because we had
only a few test bromophenols.
To get molecular insight on the promising antagonist effect of 1 and
3 over 2, we did a molecular docking simulation. The
CCK-binding score for 3 was the lowest (‒7.38 kcal/mol) among the
test bromophenols. Bromophenols 1 and 2 bound to the active
site of CCK receptor with a similar binding score (approx. ‒5.5 kcal/mol).
Also, the interacting residues were almost common for 1 and 2
(Asn353, Tyr380 Trp209, Val349, Val138, Met134, His376, and Tyr380). The only
difference is that the Br atom at C2 of 1 had an additional
-alkyl interaction with Phe227. Likewise, the most potent of them,
3, had unique H-bond interactions with Arg208, Val194, Arg356,
and Pro210 that were absent for the binding of 1 and 2. The -OH
group at C5, C4’ and C5’ and bromine atom at C2 of 3 had H-bond
interactions with Val194, Arg356, Pro210, and Arg208, respectively. The reference
antagonist YM022 and 3 had interacting residues Arg356 and Pro210 in
common, whereas His376, Met134, and Trp20 are common for YM022, 1, and
2. Residues Arg356 (TM6) and Asn353 (TM6) are active site residues
involved in CCKR antagonism for non-peptide antagonists [78], and
bromophenols formed H-bond interactions with these residues. Two CCK receptor
subtypes—CCK and CCK receptors—are 48% identical to each other
and differ in terms of molecular structure, distribution, and affinity for the
natural ligand (CCK and gastrin). Both subtypes share the same COOH-terminal
pentapeptide amide sequence for receptor binding [79]. A study model conducted to
discover 1,4-benzodiazepine allosteric ligands targeting CCK receptors suggested
six prime amino-acid residues for the selectivity of subtype CCK: Asn98
(TM2), Thr117, Thr118 (TM3), Ile329, Phe330 (TM6), and Leu356 (TM7); those for
CCK were Thr111 (TM2), Val130, Ser131 (TM3), Val349, Tyr350 (TM6), and
His376 (TM7). Asparagine residue is a conserved amino-acid residue in the TM6 of
CCK receptor, and Asn353 is responsible for the H-bond interaction with the N4 of
the benzodiazepine in CCKR [80]. Residues Thr111 (TM2) and His376 (TM7)
were most important for binding CCKR selective ligands, and Asn353 (TM6) is
an anchor residue with excellent enrichment and selectivity for CCKR [80].
Likewise, Val349 is a conserved amino-acid residue responsible for CCKR
binding affinity for nonpeptide antagonists [81]. Our test ligands showed
interactions with Asn353, Val349, and His376 and acted as CCKreceptor-specific antagonists.
Drugs modulate more than one target (six on average) to exert a therapeutic
effect [82]. PCM is one of the quantitative bioactivity prediction techniques
that predicts the potency or affinity for compound-target pairs [83], enables
simultaneous modeling of chemical and biological information and thus compound’s
affinity and selectivity across a panel of targets [84]. Nonetheless, the effects
of a compound at the cellular or the organism level are poorly understood because
PCM cannot account for the interactions of a compound with other unrelated
targets [85]. In the present study, though 2 was predicted with the
highest normalization value for the CCK receptor, it was the least active
among the tested bromophenols in a functional assay. The reason behind this
variation might be the ligand-receptor interaction and the ligand-receptor
complex stability. This needs to be confirmed via molecular dynamics.
Since halogenation of drug molecules is a common strategy to improve ADME,
membrane binding, permeation, and protein-ligand recognition [47, 86, 87, 88], and
addition of bromine and chlorine to peptide drugs improves CNS delivery by
increasing BBB permeation [48]; these bromophenols 1‒3 with
excellent PPB and BBB values [64] could be developed into novel CNS drugs. Marine
red algae are promising natural sources that contain around 90% of halogenated
compounds [89]. Because of a wide array of biological activities of halogenated
compounds and little halogen content in other natural products, structural
modification of natural products by halogenation is emerging [49].
The sulfated octapeptide (CCK-8s) is the most abundant molecular form of CCK in
the brain, with 400 pmol/g tissue content [90]. Earlier studies had reported an
increase in behavioral arousal and fear in rats after CCK-8 injection into the
central nucleus of the amygdala [91, 92]. CCK receptors have been strongly
implicated in anxiety [93]. The systemic or intracerebral injection of the
CCK-like peptides (CCK or CCK/CCK non-selective agonists)
exhibited an anxiogenic-like effect in various animal models [94, 95]. However,
co-treatment with CCK antagonist LY288513 blocked those effects; when
administered alone, it attenuated anxious behavior in animals. Also, other highly
selective CCK receptor antagonists PD134308 and PD135158 were as effective
as was diazepam to antagonize aversive behavior [96].
Food intake releases gastrin that binds to the CCK receptor on
enterochromaffin-like (ECL) cells and aids the synthesis and secretion of
histamine, which stimulates parietal cells for acid secretion by proton pump via
histamine H2-receptor and muscarinic M3-receptor [97]. Mental health is
well-linked with the gut function [98, 99, 100] because of microbiota-gut-brain axis
functions in a bidirectional manner in the regulation of depressive-like
behaviors [101, 102]. Therefore, these CCK antagonist bromophenols can
regulate depression-like behaviors.
5. Limitations and Future Directions
Our study highlights the in vitro CCK antagonist effect of only three
bromophenols in the red alga Symphyocladia latiuscula and predicts the
binding mode. In silico molecular dynamics studies predicting the stability of
ligand-receptor complexes are lacking. Penetration into the central nervous
system and stability of the ligand-receptor complex remain to be studied
in vivo. The small number of test compounds limited the
structure-activity relationship. The effects of the tested bromophenols at the
cellular or organismal level remain unknown because proteochemometric modeling
cannot account for interactions between compounds and other unrelated targets.
In the future, a more detailed understanding of CCK receptor signaling and
regulation, especially using in vivo models, will be critical to ensure
the activity of these natural bromophenols as anxiolytics and antidepressants.
6. Conclusions
Natural bromophenols 2,3,6-tribromo-4,5-dihydroxybenzyl alcohol (1),
2,3,6-tribromo-4,5-dihydroxybenzyl methyl ether (2), and
bis-(2,3,6-tribromo-4,5-dihydroxybenzyl) ether (3) from
Symphyocladia latiuscula are potent CCK antagonists with good
binding scores and interactions with prime residues at TM7 of the receptor in a
similar manner to a reference antagonist YM022. This study suggests that
bromophenols 1–3 are natural CCK antagonists that could
be novel therapeutic agents for CCK-related diseases, especially anxiety
and depression, and provides a foundation for future studies to elucidate the
molecular mechanism in animal models of anxiety and depression.
Author Contributions
PP participated in study design, isolation, treatment, and biochemical analysis,
and drafted the manuscript. SEP and SHS performed the molecular docking studies.
FMF performed PCM modeling. HAJ was involved in spectral analysis. JSC conceived
the study, coordinated the study, and interpreted the data. All authors read and
approved the final manuscript.
Ethics Approval and Consent to Participate
The research reported here was conducted at Eurofins Cerep, France according to
the Guide for the Care and Use of Laboratory Animals of the National Institutes
of Health and was authorized from the Research Ministry to manipulate GMO, with a
certificate number 28099.
Acknowledgment
The authors thank Prof. Dr. Hye Jin Park (Changshin University, Republic of
Korea) for providing the leafy thalli of Symphyocladia latiuscula (Harvey).
Funding
This research was supported by the National Research Foundation of Korea (NRF)
grant funded by Ministry of Science and ICT (No. 2020R1C1C1008331
[HNIBR202100303]).
Conflict of Interest
The authors declare no conflict of interest. Pradeep Paudel is serving as one of the Guest editors of this journal. We declare that Pradeep Paudel had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Gernot Riedel.





 , Se Eun Park 1,3, Su Hui Seong 1,4, Fazlin Mohd Fauzi 5, Hyun Ah Jung 6,*
, Se Eun Park 1,3, Su Hui Seong 1,4, Fazlin Mohd Fauzi 5, Hyun Ah Jung 6,*