



1 Department of Neonatology, The Children's Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, 310052 Hangzhou, Zhejiang, China
2 Department of Psychology, The Children's Hospital, Zhejiang University School of Medicine, National Clinical Research Center for Child Health, 310052 Hangzhou, Zhejiang, China
Academic Editor: Qiu-Lan Ma
Abstract
Bilirubin neurotoxicity is a serious consequence of hyperbilirubinemia, which is the most common disease of the neonatal period. Clinically, bilirubin neurotoxicity can result in motor deficit, auditory dysfunction, cerebral palsy, seizure and neurodevelopmental diseases, amongst others. Bilirubin neurotoxicity is one of the major worldwide causes of neonatal brain injury, especially in poorer developing countries. However, the mechanisms of bilirubin neurotoxicity are still unclear. After the failure of attempts targeting neurons in many neurodegenerative disorders, neuroinflammation has become a significant target of research. Here, recent advances concerning neuroinflammation in bilirubin neurotoxicity are reported with a focus on the clinical characteristics of bilirubin neurotoxicity, including age-dependency, region-specificity and its yin-yang properties. Effects of neuroinflammation on blood brain interfaces and treatments targeting neuroinflammation in bilirubin neurotoxicity are also reviewed, which may promote the precision of future treatment of bilirubin neurotoxicity.
Keywords
- neonate
- hyperbilirubinemia
- neurotoxicity
- neuroinflammation
- microglia
- astrocyte
Bilirubin neurotoxicity is a serious consequence of neonatal hyperbilirubinemia, the most common disease in the neonatal period [1]. Clinically, bilirubin neurotoxicity may manifest as acute bilirubin encephalopathy [2], chronic bilirubin encephalopathy (kernicterus) [3] and neurodevelopmental disabilities [4]. Bilirubin neurotoxicity is one of the major causes of neonatal brain injury worldwide, especially in poorer developing countries [5, 6, 7, 8].
The common molecular events of bilirubin neurotoxicity may include membrane disturbance, endoplasmic reticulum stress, mitochondrial injury, destructive enzyme induction, abnormal neurotransmitter/cell metabolism, excitotoxicity, oxidative stress and neuroinflammation [9, 10]. However, the initiator or core mechanism of bilirubin neurotoxicity is unclear. While attempts to target neurons in many neurodegenerative disorders have failed, neuroinflammation has gained growing attention. Evidence indicates the potential roles that bilirubin may play via neuroinflammation in neurodegenerative, neuropsychiatric and other neurological diseases [11]. Neuroinflammation has been considered as the core mechanism of several neurodegenerative diseases such as Alzheimer’s disease [12, 13] and schizophrenia [14, 15]. Animal models of bilirubin neurotoxicity show pathological changes very similar to those of Alzheimer’s disease [16] and schizophrenia [17]. Analysis of cerebrospinal fluid (CSF) reveals that inflammation associated cellular processes and signaling pathways are dominantly activated in infants with acute bilirubin encephalopathy [18]. Such evidence suggests neuroinflammation plays a key role in the pathogenesis of bilirubin neurotoxicity.
Neuroinflammation is defined as an immune response toward harmful insults through the release of inflammatory mediators (such as cytokines, chemokines and reactive oxygen species) primarily produced by microglia and astrocytes in the central nervous system (CNS) [19]. As an initial immune response, neuroinflammation is mainly beneficial and protective; however, when prolonged or excessive it plays a pathogenic role in several neurodegenerative and psychiatric disorders [20, 21, 22]. Free bilirubin can pass through the blood brain barrier and trigger a neuroinflammatory response in neonatal hyperbilirubinemia. Activation of neuroinflammation in the early life will affect fetus/infant neurodevelopment [23]. Excessive/prolonged neuroinflammation activated by bilirubin will interfere with development of the immature brain and lead to neurological sequelae. The role of glial cells and inflammation in bilirubin neurotoxicity has been reviewed, primarily on the basis of in vitro studies, by Brites [24]. Here, recent advances in understanding neuroinflammation in bilirubin neurotoxicity are reported, with a focus on the clinical characteristics of the latter, including age-dependence, region-specificity and its yin-yang properties. Effects of neuroinflammation on blood brain interfaces (BBIs) and treatments targeting neuroinflammation in bilirubin neurotoxicity are also reviewed.
The susceptibility of neonates to bilirubin neurotoxicity varies with the postnatal day [25, 26]. The main casualties of bilirubin neurotoxicity are early newborns and premature babies [27]. Alternatively, bilirubin has protective effects against metabolic syndrome and diabetic complications in the elderly [28]. The mechanisms of the age-dependent bilirubin neurotoxicity remain unclear. However, here the contributions of neuroinflammation to this age-dependent pattern are described.
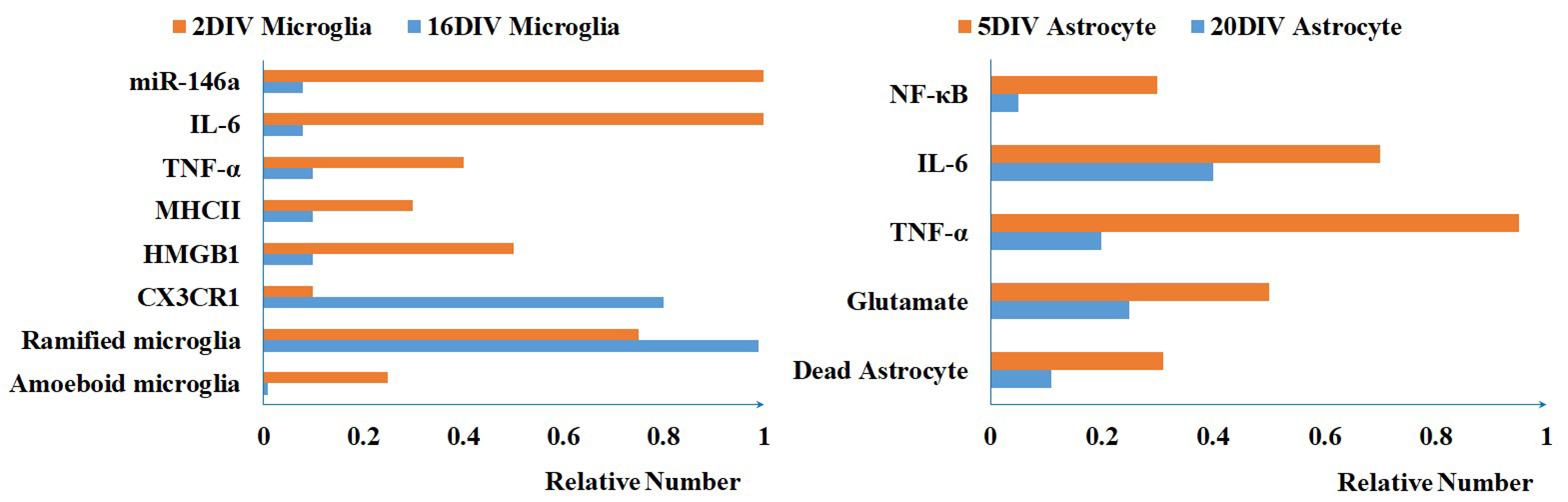
Microglia, in which age-related changes are found [29], have the greatest
heterogeneity during the early postnatal days [30, 31]. When treated with free
bilirubin, microglia at two days in vitro (2DIV) show early apoptosis
and amoeboid shape, lower levels of CX3C chemokine receptor 1 (CX3CR1) and microRNA (miRNA)-124, higher
expression of high mobility group box protein 1 (HMGB1), nitric oxide, tumor necrosis factor (TNF)-
Bilirubin selectively affects brain cells in specific regions, such as basal ganglia, brainstem nucleus, hippocampus and cerebellum [37]. This “affection” is where the name “kernicterus” originates [38]. Therefore, it is important to identify underlying mechanisms of this region-specific bilirubin neurotoxicity. It may be due to differences in bilirubin uptake, clearance, or cell sensitivity. However, more investigation is still required. Here, findings to date of region-specific neuroinflammation in this field are now described.
Single-cell analysis reveals region-dependent heterogeneity of microglia and
astrocytes [39, 40, 41]. The activation of microglia and astrocytes induced by
lipopolysaccharide depends on the brain region [42, 43]. Activation of microglia
and up-regulation of cytokines are observed in bilirubin affected brain regions
in vitro [44]. The activation of microglia and astrocytes is selectively
located in zones more affected by bilirubin in kernicterus mice [45, 46]. Whole genome gene expression analysis reveals that the NF-
Bilirubin has a dual role as toxic and therapeutics, called yin-yang properties [52]. It is generally considered a neurotoxin in newborns as a high incidence of severe hyperbilirubinemia ultimately leads to neurological dysfunction. Alternatively, bilirubin nanoparticles have a therapeutic potential against multiple diseases, such as autoimmune encephalomyelitis and ischemia-reperfusion injury [53]. The most prevalent theory postulates that a moderate bilirubin level may be beneficial while excessive bilirubin is toxic. However, the underlying mechanisms of the contrary effects remain unknown. The two sides of neuroinflammation in the pathogenesis of bilirubin neurotoxicity are now discussed.
Microglia are motile cells that investigate the brain environment. Upon brain invasion or insult, microglia quickly develop into different functionally overlapping phenotypes to exert neuroprotective and neurotoxic effects [54]. Phagocytic microglia are the first cells activated upon bilirubin insult [17, 46, 55], in an attempt to protect neurons from injury by suppressing the inflammatory response, clearing harmful substances and removing cellular debris. This type of activation is transient and decreases over time. Inversely, pro-inflammatory microglia increase at the late stage of bilirubin toxicity, inducing neuron death by secreting inflammatory cytokines, chemokines, proteases and other toxic substances [46, 55, 56, 57, 58]. The two-type microglia are not categorically “bad” or “good”. Pro-inflammatory microglia can prevent infection by producing inflammatory cytokines and decompose dying neurons to promote phagocytosis. On the other hand, excessive phagocytic microglia may lead to abnormal tissue repair. Here, it is speculated that a balance of microglia subtypes may be one of the mechanisms by which bilirubin exerts its neuroprotective effects.
Astrocytes can be activated to exhibit various phenotypes depending on the
properties of the microenvironment; exerting either neurotoxic or neuroprotective
effects [59]. Reactive astrocytes can upregulate the secretion of
pro-inflammatory cytokines such as TNF-
Reactive oxygen species (ROS) [69] act as central regulators of inflammatory signaling [70]. ROS can be either protective or destructive, depending upon its concentration. Moderate ROS serves as a physiological signal in cells, while excessive ROS is destructive, leading to cell death. Bilirubin can have either an anti-oxidant or pro-oxidant effect in a concentration-dependent manner [71]. The mechanisms of bilirubin scavenging ROS may involve combining with oxygen radicals, inhibiting nicotinamide adenine dinucleotide phosphate oxidase 4 (NOX4) activity and activating the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway [72]. Bilirubin nanomedicines have been developed for the treatment of ROS-mediated diseases [73]. Alternatively, many studies have reported the pro-oxidant properties of bilirubin. Bilirubin enhances cellular oxidative stress induced by oxygen-glucose deprivation in organotypic hippocampal slice [74]. The oxidative stress induced by free bilirubin appears to cause DNA damage in vitro [75]. Clinical studies in infants reveal contradictory outcomes. Some studies demonstrate that elevated bilirubin increases oxidative stress [76], while other studies report that increased bilirubin has either a negative or no effect on oxidative stress [77].
In summary, the double roles of microglia, astrocytes and ROS may contribute to the bilirubin yin-yang properties.
BBIs refer to the structural and functional barriers separating the brain and CSF from the blood, including the blood brain barrier (BBB) and the blood CSF barrier [78, 79]. BBIs serve as the first line of defense preventing bilirubin entering into the brain. Here the contributions of neuroinflammation to BBIs damage in bilirubin neurotoxicity it is reviewed.
The effect of bilirubin on endothelial cells is concentration- and time-dependent [80]. Sustained exposure to high levels of bilirubin leads to endothelial cell death, secretion of matrix metalloprotein (MMP)-9 and MMP-2 and increased cell permeability [80, 81]. These changes are even worse in endothelial cells co-cultured with astrocytes [81]. Pro-inflammatory cytokines can induce cerebral endothelial dysfunction [82]. These reports collectively indicate that neuroinflammation induced by bilirubin accelerates the disruption of endothelial cells.
Astrocytes cover 99% of the neurovasculature and provide a vital role in regulation of BBB functions [83, 84]. The response of astrocytes to bilirubin have been reviewed in previous sections. The effect of bilirubin on aquaporin 4 [85], which is highly expressed in astrocytes, has not yet been investigated.
Pericytes are embedded in the basal lamina and display a variety of functions in the neurovascular unit [86]. Bilirubin induces the secretion of endothelial nitric oxide synthase, IL-6 and vascular endothelial growth factor by pericytes and leads to loss of cell viability and apoptosis of pericytes in vitro [87].
Transporters [88, 89, 90] and enzymes [91] associated with BBIs play an active role in the efflux and/or elimination of bilirubin from the brain. The transport and metabolism of bilirubin at BBIs are reviewed by Gazzin et al. [92]. P-glycoprotein is upregulated in microvessels and MRP-1 is downregulated in the choroid plexus of the jaundiced Gunn rat [93]. P-glycoprotein and MRP-1 are reported to be upregulated in the hippocampus of a kernicterus infant at autopsy [94]. Collectively, these reports suggest that bilirubin affects the expression of transporters on the BBIs. Pro-inflammatory cytokines upregulate the expression of ABC transporters, P-glycoprotein and MRP-1, while they down-regulate cytochrome P450 oxygenases (CYPs) in the brain [95, 96, 97, 98, 99].
Taken together, over-activated neuroinflammation may damage structural cells, upregulate transporters and down-regulate CYPs on BBIs in bilirubin toxicity (Fig. 2). The disruption of BBIs’ structural cells may facilitate the entry of bilirubin into the brain, while the upregulation of transporters on the BBIs might promote its efflux. However, when the damage of BBIs reaches a certain extent, the benefit of the upregulation of transporters becomes negligible.
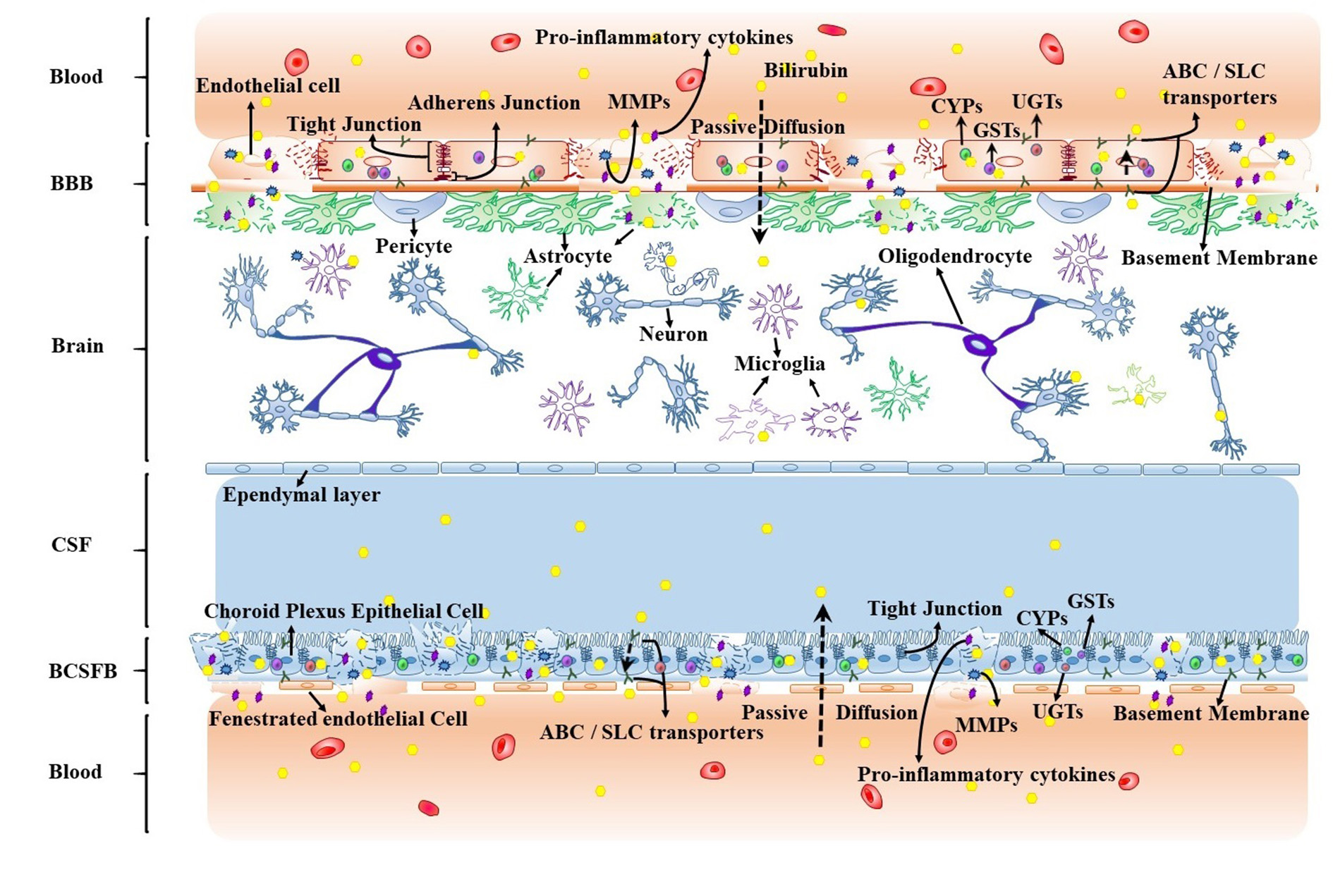 Fig. 2.
Fig. 2.Effect of over-activated neuroinflammation on transport and metabolism of bilirubin at BBIs. BBIs, Blood-brain interfaces; CSF, Cerebrospinal fluid; BCSFB, Blood-CSF barrier; BBB, Blood-brain barrier; ABC, ATP-binding cassette; SLC, Solute carrier; CYP, Cytochrome P450 oxygenase; GST, Glutathione-S-transferase; UGT, Uridine diphosphate glucuronosyltransferase.
Therapeutic drugs targeting neuroinflammation are being tested in bilirubin neurotoxicity. The main targets of these therapies in the attenuation of bilirubin toxicity (Table 1) [47, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110] are pro-inflammatory cytokines, signaling pathways, toll-like receptors and activated microglia. Anti-inflammatory therapies have achieved partial neural recovery in cell culture and animal models, but to date no drug has been moved into any phase of clinical trial.
| Drugs | Mechanism | Cells | Animal Model |
| Glycoursodeoxycholic acid [104] | Attenuate suppression of interleukin-6 secretion | Astrocyte | / |
| Reduce TNF- | |||
| Inhibit TNF- | |||
| Caffeine [105] | Increase activity of catalase, glutathione peroxidase, superoxide dismutase, glutathione, decrease malondialdehyde | Astrocyte | / |
| Decrease TNF- | |||
| Dexamethasone [106] | Reduce release of IL-8 and MCP-1 | NT2-N neuron | / |
| Baicalin [107] | Inhibit p38 MAPK pathway | Neuron | |
| Thalidomide derivatives [47] | Destabilize TNF- |
/ | CBA/Ca mouse |
| BAY 11708 [47] | Inhibit IκB kinase | / | CBA/Ca mouse |
| Git27 [47] | Antagonize TLR4 | / | CBA/Ca mouse |
| Minocycline [100, 101, 103] | Inhibit expression of TNF- |
Co-culture of glial cells and neurons | / |
| Inhibit p38 MAPK phosphorylation | / | Gunn rat | |
| Inhibit activation of microglia | |||
| Inhibit activation of astrocytes and microglia | / | Ugt1 | |
| Reduce expression of NFK | |||
| Decrease expression of TLR2 | |||
| Down-regulate MMP2 and MMP9 | |||
| Reduce oxidative and ER stress | |||
| Melatonin [108] | Inhibit expression of TNF- |
/ | PHZ treated SD rat |
| Increase expression of IL-10 | |||
| Decrease malondialdehyde, glutathione | |||
| Increase myeloperoxidase | |||
| Erythropoietin [109] | Inhibit expression of TNF- |
/ | PHZ treated SD rat |
| Increase expression of IL-10 | |||
| Decrease malondialdehyde, myeloperoxidase | |||
| Increase glutathione | |||
| Curcumin [110] | Prevent induction of IL-1 |
/ | Gunn rat |
| Prevent increase of glutamate | |||
| Prevent increase of heme oxygenase 1 |
Minocycline is a second-generation tetracycline with anti-inflammatory
properties in the CNS. Minocycline inhibits the p38MAPK and NFK
Nevertheless, neuroinflammation is not always harmful and can have an important defensive role in bilirubin-induced pathology and trigger reparative mechanisms, which may (partially) explain the inefficacy of anti-inflammatory therapies in neurodegenerative diseases. Thus, it is speculated here that immunomodulatory therapy to maintain a balanced neuroinflammation system may achieve a better outcome in the treatment of bilirubin encephalopathy.
Neuroinflammatory responses contribute to the clinical characteristics of bilirubin neurotoxicity, including age-dependence, region-specificity and its yin-yang properties. Over-activated neuroinflammation may accelerate the damage of BBIs and thus facilitate bilirubin influx into the brain. Strategies targeting balancing neuroinflammation, rather than anti-inflammatory therapies alone, may be of benefit for the precision treatment of bilirubin neurotoxicity.
Several questions have not been answered. What is the spatio-temporal heterogeneity of microglia and astrocytes in the pathogenesis of bilirubin neurotoxicity? Would a bilirubin induced neuroinflammatory response develop into a chronic or a sustained response? How should neuroinflammation be managed to achieve positive outcomes in bilirubin neurotoxicity?
FZ—substantial contributions to conception and design; drafting the article; LC—revising the article and final approval of the version to be published; KJ—substantial contributions to conception and design, revising the article and final approval of the version to be published.
Not applicable.
This work was supported by the National Natural Science Foundation of China (81871012, 81571263, and 81300975), the Zhejiang Provincial Technology Plan (2015C37105), the Key Laboratory of Reproductive Genetics (Zhejiang University), Ministry of Education, and the Key Laboratory for Diagnosis and Therapy of Neonatal Diseases of Zhejiang Province. These organizations had no part in the study design, data collection and analysis, publication decisions, or preparation of the manuscript.
The authors declare no conflict of interest.