
†These authors contributed equally.
Academic Editor: Changjong Moon
Background: One of the most serious complications of sepsis is sepsis-associated encephalopathy (SAE), which impairs the cognition ability of survivors. Environmental enrichment (EE) has been demonstrated to alleviate cognition deficits under many kinds of brain injury conditions. However, EE’s effects on SAE remain unknown. Therefore, this study aimed to determine EE’s effect on cognition disorders under SAE conditions and the underlying mechanism. Materials and Methods: Adult male rats, subject to SAE or not, were housed under a standard environment (SE) or EE for 30 days. Subsequently, the rats were subjected to cognitive tests, such as the novel object recognition (NOR) test, the Morris water maze (MWM) test, an Open Field (OF) test, the elevated plus maze (EPM) test, and a sensory neglect (SN) test. Neuroinflammation, apoptosis, and oxidative stress changes in the brain were also detected. Results: The results revealed that SAE impaired somatesthesia, recognition memory, spatial learning and memory, and exploratory activity, which were significantly improved by EE housing. EE also prevented SAE-induced anxiety-like behavior. In addition, EE housing capable induced a decrease in pro-inflammatory cytokines, and an increase in anti-inflammatory cytokines and antioxidant properties in the brain. Moreover, EE housing exerted an anti-apoptosis function by upregulating the level of B-cell lymphoma/leukemia-2 (Bcl-2) level and downregulating the level of p53 level in the hippocampus. Conclusions: The results of the present study indicated that EE exerts a neuroprotective function on cognitive ability in SAE rats. The effect is achieved by increasing antioxidants, and anti-inflammatory and antiapoptotic capacities. EE can effectively rescue SAE-induced cognitive deficits.
Systemic responses to infection commonly result in sepsis, the incidence of which is increasing gradually worldwide [1]. Sepsis is often accompanied by sepsis-associated encephalopathy (SAE) [2]. Over 50% of patients suffering from severe sepsis develop SAE, which is possibly one of the leading causes of mortality in intensive care units (ICUs) [3]. Acute SAE always manifests as a disturbance of consciousness and can ultimately lead to death [3, 4, 5]. In addition, survivors of SAE might carry a significant risk of long-term cognitive function disorder, such as changes in mental speed, learning and memory ability, spatial discrimination function, executive ability, and attention ability [6, 7, 8]. Long-term cognitive function disorder is a major cause of disability, leading to increased burdens on families and society, and poor life quality for patients. However, currently, we lack effective treatments for SAE-induced cognition disorder. Methods to improve SAE-induced cognition disorders have become a research hotspot.
Housing conditions comprising social interactions, novelty, and various forms of exercise are referred to as environmental enrichment (EE). Numerous studies have reported the positive effect of EE on cognitive function under various brain injuries [9]. For example, EE can improve the sensory and memory ability for traumatic brain injury (TBI) rats by changing the levels of proinflammatory cytokines and anti-inflammatory cytokines [10, 11, 12]. EE also inhibits strokeinduced cognition disorder by suppressing neuroinflammation and oxidative stress [13, 14]. Moreover, in models of preeclampsia and Parkinson’s disease, accumulating evidence has demonstrated the inhibitory effects of EE on cognition disorders by regulating inflammatory conditions and apoptosis levels [15, 16, 17]. These findings suggest that EE might be capable of rescuing cognitive ability impairment under SAE conditions.
Accordingly, in the present study, we investigated the effect of EE on SAE-induced cognitive impairment and the underlying mechanism. To this end, adult male rats were housed in EE or standard environment (SE) conditions for 30 days. The changes in cognitive function were then evaluated. Additionally, we detected the changes in neuroinflammation, apoptosis, and oxidative stress in the brain.
The Animal Center of the China-Japan Friendship Hospital, Beijing, China
provided the Sprague-Dawley (SD) rats (male; 60 days old; weighing 200–250 g).
The Guidelines for the use of animals in neuroscience research
(published in the Membership Directory of the Society, pp 27–28, 1992) were
followed when performing all the animal experiments, and the committee of Animal
Use for Research and Education of the China-Japan Friendship Hospital approved
this study. The rats were reared in a temperature-controlled room (24
SAE was induced by cecal ligation and perforation (CLP) according to previous published methods [18, 19]. In brief, the rats were subjected to sodium pentobarbital (50 mg/kg) intraperitoneal anesthesia. The cecum and adjoining intestine were exposed under aseptic conditions. A 3.0 silk suture was used to ligate the cecum at its base, beneath the ileocecal valve, followed by single perforation employing a 14-gauge needle (about 1 mm depth). A small amount of feces was extruded into the peritoneal cavity by gentle squeezing. The cecum was then returned to the peritoneal cavity, and 4.0 silk sutures were used to close the laparotomy incision. After surgery, the rats were given “basic support” (saline, 50 mL/kg) subcutaneously (s.c.) for recovery from anesthesia, immediately after and 12 h after CLP. All rats received antibiotic therapy by subcutaneous injection of ceftriaxone (30 mg/kg) and clindamycin (25 mg/kg) every 6 h for 3 days. Sham rats were given an identical cecum exposure but without ligation or puncture. At 7 days post-surgery, the model rats showed a 51% mortality rate, comparable to some previous studies [19, 20] and indicative of severe sepsis. For the survivals, the neurobehavioral tests, including auricular reflex, corneal reflex, righting reflex, tail flick reflex and escape reflex were performed 3 days after the CLP as previous study [18]. 0 refers to no reflexes, 1 point refers to weakened reflexes (no reflexes within 10 s), 2 points refers to normal reflexes, and the highest score refers to 10 points. According to preliminary experiment, the SAE was determined when the scores was below 8.
In the present study, SE and EE were used. For SE, the rats were reared in a
standard-sized cage (25
For EE, the rats were reared in a large cage (40
According to the results of preliminary experiments and previous studies [23], the sample size for each experiment was determined. The rats receiving CLP or sham operation were placed into four groups (n = 18 per group): Sham-SE group (sham injury + SE housing); Sham-EE group (sham injury + EE housing); SAESE group (CLP + SE housing); and SAE-EE group (CLP + EE housing).
After EE or SE housing, the mechanism of EE was investigated. We detected the level of inflammatory/anti-inflammatory mediators, oxidative/antioxidant activity, and apoptosis-related molecules and pathways. Thus, in each group, six rats were used to detect the level of apoptosisrelated molecules, six rats were used to detect the oxidative/antioxidant activity, and six rats were used to detect the levels of inflammatory/anti-inflammatory mediators.
The experimental procedure is shown in Fig. 1. The rats were subjected to behavioral tests naively and were manipulated only when routine cleaning was performed. The tests were performed between 08:00 and 17:00 hours. To eliminate the confounding effect of testing time, we counterbalanced the testing order of the rats.
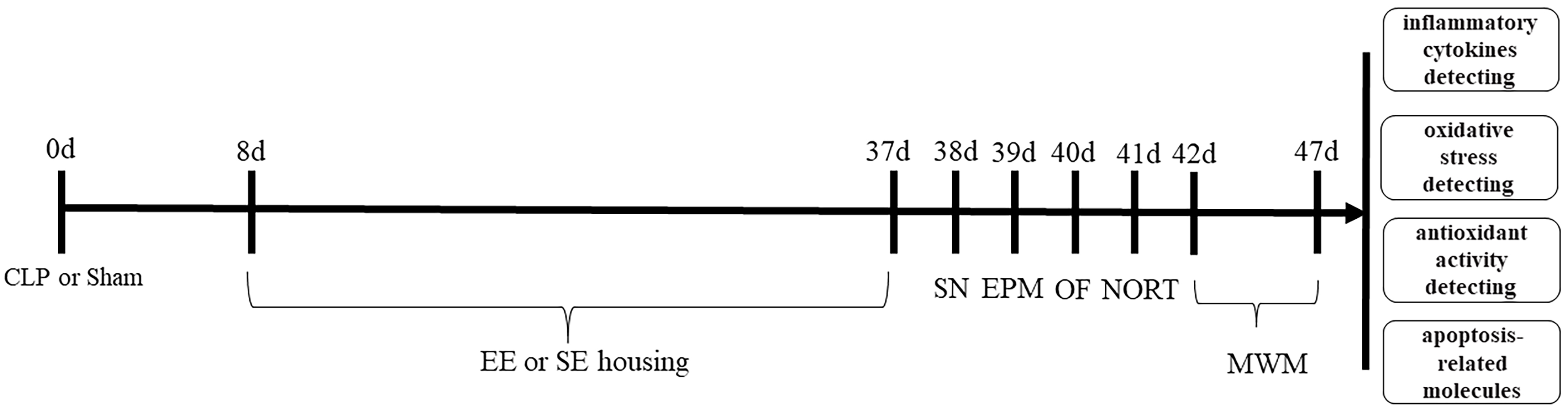 Fig. 1.
Fig. 1.Flow chart of the experimental process.
Somatesthesia was assessed using a sensory neglect (SN) test [24]. The SN test was performed 1 day after 30 days of SE or EE. In this test, we placed a 2 cm diameter sticker on the distal-radial area of both forelimbs. The rats were put into an empty cage and then we measured the time taken to remove the stickers (latency). Each rat was subjected to three trials at 5 min intervals and 2 min maximum trial time.
The elevated-plus maze (EPM) test was used to assess anxiety-like behavior. The
EPM comprises two opposing open arms (10.2
Locomotor and exploratory ability were evaluated using an open field (OF) test,
as previously described [26]. The OF was 60
Spatial learning and reference/working memory was assessed using a Morris water maze (MWM) [27, 28, 29]. The maze consisted of a dark circular tank with a diameter of 178 cm, which was filled with water to 37 cm deep and was separated into four quadrants (A, B, C, D). A plexiglass platform (diameter = 10.2 cm diameter) was placed at approximately 28 cm from the wall of the pool in quadrant C (the southeast quadrant) and was submerged at 2 cm below the waterline. Each animal was tested for 6 days. In this test, a 5-day testing block (four trials each day) was performed to detect the rat’s spatial learning ability. In each trial, the rats were put into a random quadrant, facing the wall. In 120 s of given time, the latency to the reach the platform was recorded. If the rats could not reach the platform, we physically guided them to it. The rats were kept on the platform for 30 s after reaching it, followed by a 5-minute break before the next trial. To determine the role of non-spatial factors, on day 6, we raised the platform to 2 cm above the waterline such that was visible to the rats. On day 6, we also performed a single probe trial to determine memory retention. We removed the platform and the rats were allowed to explore the maze for 30 s. The detection indices were: proportion of time spent in the target quadrant (%) and the number times they crossed the platform location.
The Novel Object Recognition (NOR) test was carried out to evaluate recognition
memory (non-spatial working memory) [30, 31]. The NOR test was performed according
to a previously published method [32]. This test had two phases. In phase 1
(familiarization phase), the rats were allowed to freely explore two identical
objects (placed in opposite corners of the field) for 5 min. Then, the rats were
removed to their cages and rested for 1 h. During phase 2 (test phase), a novel
object replaced one of the previous objects and the rats were allowed to explore
them for 3 min. To reduce emotional instability-induced variation, rats were
excluded from analysis when they exhibited excessive freezing behavior for more
than 60% of the test phase (1 rat in Sham-SE group, 1 rat in Sham-EE group, 2
rats in SAE-SE group and 1 rat in SAE-EE group). When the nose of rats pointed at
the objects and explored it at a distance less than 1 cm, the time taken to
explore the object was measured. If the rats used the objects as props, sat on
the objects or contacted but did not face the objects, these behaviors were not
regarded as object exploration [33]. The total exploration and
discrimination index (DI) was calculated as described in a
previous study [34]. The total exploration = the sum of exploration time of the
novel object (tn) + the sum of exploration time of the familiar object (tf). DI =
(tn–tf)/(tn+tf)
After behavioral testing, rats were killed by decapitation. The brain was removed immediately and washed with ice-cold isotonic saline solution and placed in liquid nitrogen and stored at –80 °C.
Inflammatory mediators and apoptosis-related molecules in the brain or blood
were measured using ELISAs. The brain tissues were obtained according to our past
study [35]. Briefly, the isolated brain was homogenized in normal saline,
followed by centrifugation for 10 min at 4 °C and 2000 rpm. Interleukin
(IL)-6, IL-10, IL-1
The extent of lipid peroxidation was measured by determining the level of
thiobarbituric acidreactive substances (TBARS), as previously described [35, 36].
Isolated brain tissue was sonicated in 10% (w/v) radioimmunoprecipitation assay
(RIPA) buffer added with a protease inhibitor cocktail (Sigma, St. Louis, MO,
USA). Following incubation for 30 min on ice, the homogenates were centrifuged
for 10 min at 4 °C and 600
To directly determine the amounts of oxidized proteins in biological samples,
AOPPS were measured. Tissue was subjected sonication for homogenization by
sonication in cold 50 mM NaH
In vivo, NO’s final and stable end products are nitrates and nitrites, whose sum (NOx) represents total NO production. A colorimetric assay (Cayman Chemical Company, Ann Arbor, MI, USA) was used to determine NOx [35, 38]. In sample homogenates, nitrates were converted to nitrites using nitrate reductase and NADPH. The Griess reaction was then used to monitor total nitrites (nmol/mg protein) at 540 nm.
Reduced glutathione (GSH) and glutathione disulfide (GSSG; oxidized glutathione)
concentrations were measured in brain extracts. Brain samples were subjected to
homogenization in a 1:1 mixture of cold 10% metaphosphoric acid, 5 mM EDTA (pH
6.8) and 0.1 M potassium phosphate, followed by a 30 min incubation of ice. The
homogenate was then centrifuged for 30 min at 10,000
Sonication was used to homogenize brain tissue as described in ‘Lipid
peroxidation’. The homogenates were subjected to centrifugation for 10 min at 4
°C and 600
All data are shown as the mean
First, we investigated somatosensory changes. The results of the two-way ANOVA
showed an obvious interaction effect (SAE factor: F (1, 68) = 35.08, p
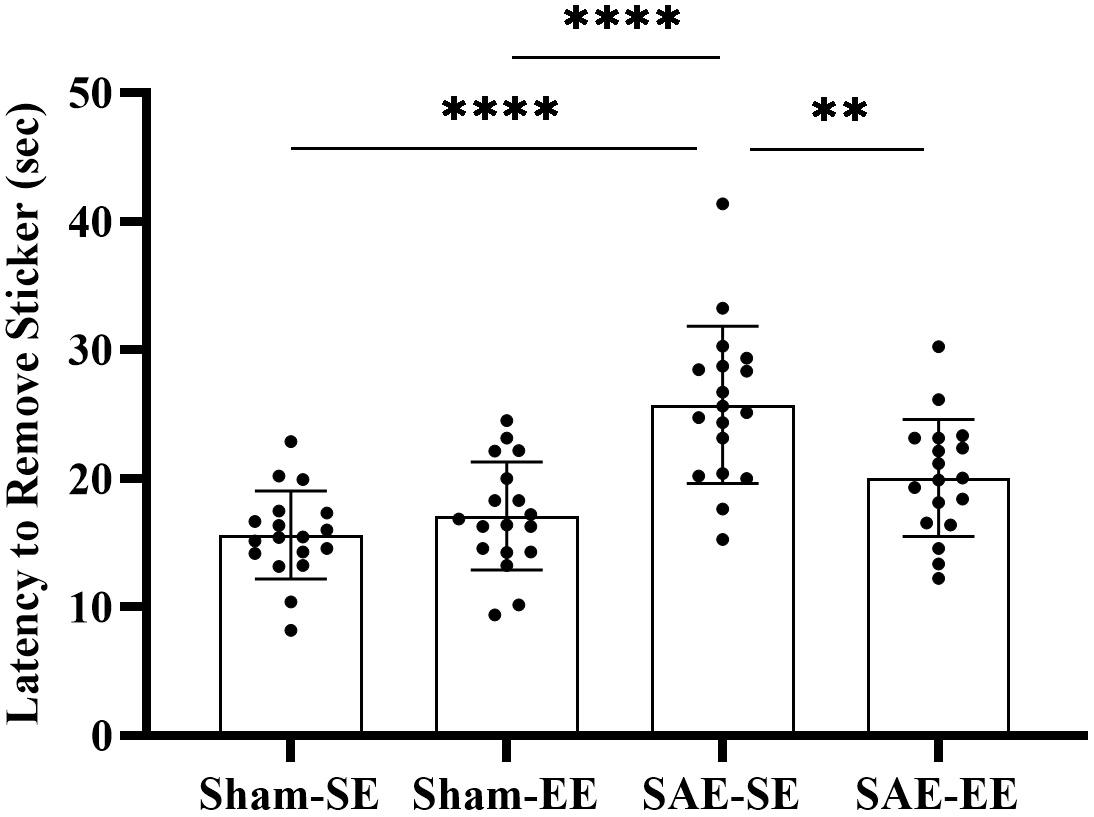 Fig. 2.
Fig. 2.The rats in the SAE-SE group had longer latency to remove
stickers than the rats in the Sham-SE or Sham-EE group. However, the rats in the
SAE-EE group had shorted latency to remove stickers than the rats in the SAE-SE
group. **p
The EPM test was used to detect changes in anxiety-related behaviors. The
anxiety behavior indices included number of entries into the
open/closed arms, latency of entry into an open arm, and time occupying
open/closed arms, head dipping, rearing, and grooming. Results
showed that SAE factor, environmental factor or interaction had no effect on
the time spent in head dipping, rearing, and grooming among the
four groups (head dipping: SAE factor: F (1,
68) = 0.9969, p = 0.3216; environmental factor: F (1, 68) = 0.1180,
p = 0.7323; interaction F (1, 68) = 0.2388, p = 0.6266;
rearing: SAE factor: F (1, 68) = 0.9700, p = 0.3282; environmental
factor: F (1, 68) = 1.598, p = 0.2105; interaction F (1, 68) = 0.4592,
p = 0.5003; grooming: SAE factor: F (1, 68) = 0.2117, p =
0.6469; environmental factor: F (1, 68) = 0.4602, p = 0.4998;
interaction F (1, 68) = 0.0035, p = 0.9529; Fig. 3A–C). However,
significant effects were found for open arm time (SAE factor: F (1, 68) = 15.72,
p = 0.0002; environmental factor: F (1, 68) = 6.229, p =
0.0150; interaction F (1, 68) = 2.993, p = 0.0881; Fig. 3G),
latency of entry into an open arm (SAE factor:
F (1, 68) = 64.47, p
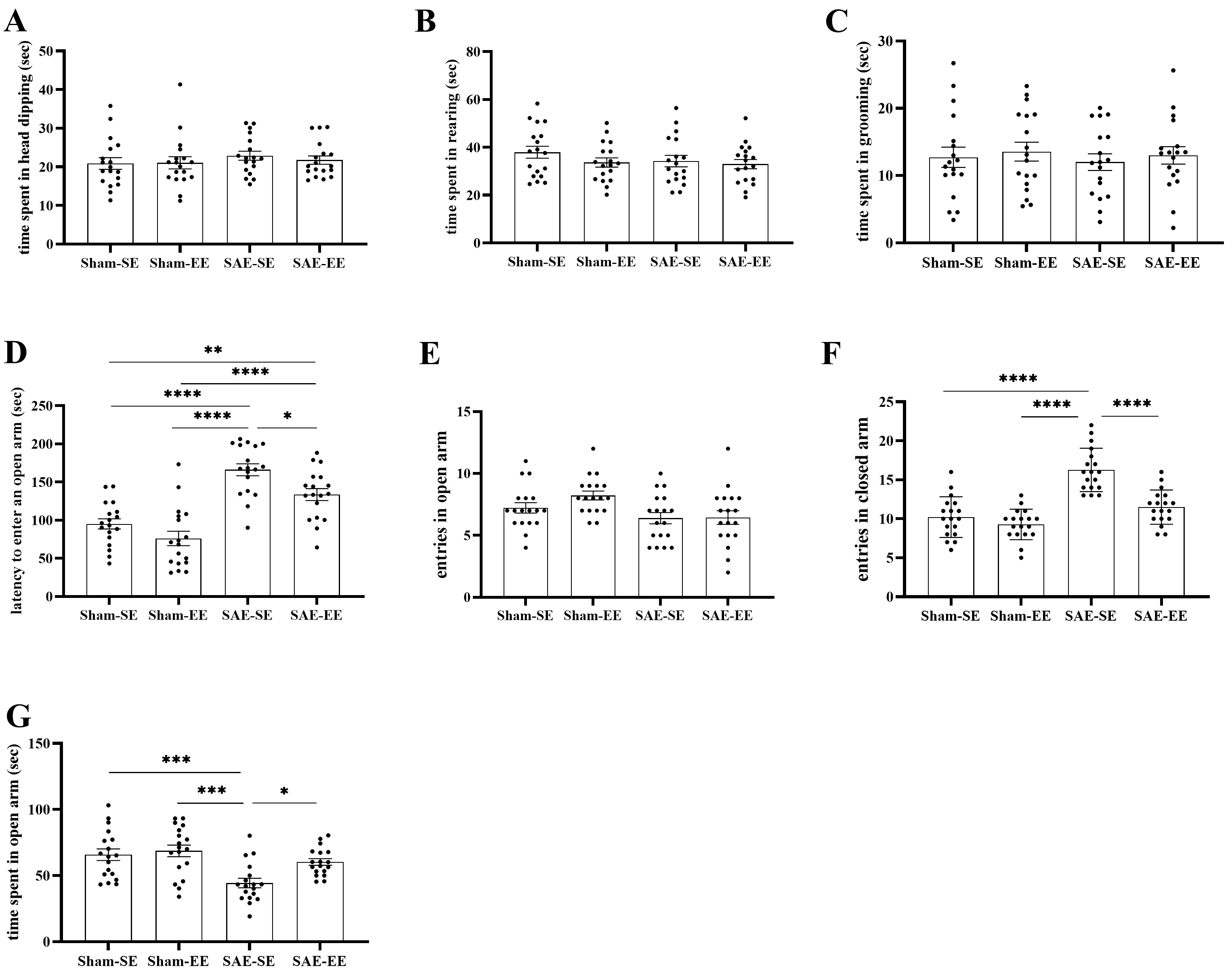 Fig. 3.
Fig. 3.EE attenuated the SAE-induced anxiety-related behaviors. (A–C)
No statistical differences were found in dipping, rearing or grooming. (D) The
rats in the SAE-SE or SAE-EE group spent more time before entering an open arm.
In addition, the rats in the SAE-EE group spent less time before entering an open
arm compared with the rats in the SAE-SE group. (E,F) There were no differences
in entries in the open arms among four groups. The rats in the SAE-SE group had
the increased entries in closed arms than the rats in the SAE-EE, Sham-SE or
Sham-EE group. (G) The lower open arm time was found in the SAE-SE rats when
compared with Sham-SE, Sham-EE or SAE-EE rats. *p
Exploratory activity was evaluated using the OF test. There were no effects of SAE factor, environmental factor or interaction in the number of line crossings, rearing, and total distance traveled among the four groups (number of line crossings: SAE factor: F (1, 68) = 3.956, p = 0.0507; environmental factor: F (1, 68) = 2.593, p = 0.1120; interaction F (1, 68) = 0.2714, p = 0.6041; number of line rears: SAE factor: F (1, 68) = 0.04711, p = 0.8288; environmental factor: F (1, 68) = 0.2944, p = 0.5892; interaction F (1, 68) = 0.2385, p = 0.6269; total distance traveled: SAE factor: F (1, 68) = 2.313, p = 0.1329; environmental factor: F (1, 68) = 0.03263, p = 0.8572; interaction F (1, 68) = 0.2541, p = 0.6159). These results indicated that locomotor activity was not affected by sepsis or environment factors.
However, the exploratory behavior was impaired in the SAE-SE rats: they spent
less time in the center square (SAE factor: F (1, 68) = 53.09, p
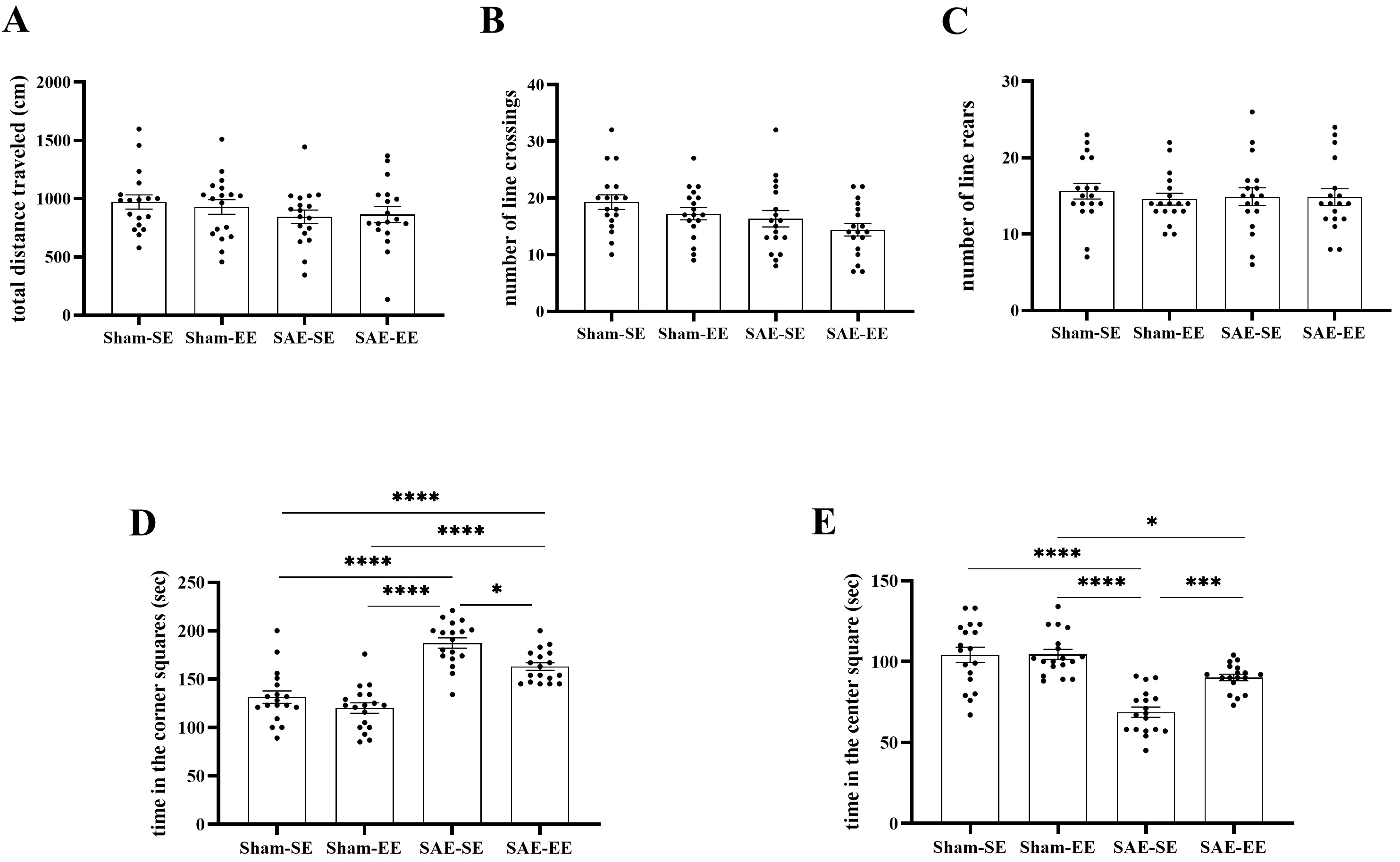 Fig. 4.
Fig. 4.EE inhibited SAE-induced exploratory activity
decrease. (A–C) No differences were found in total traveled distance and times
of line crossings and rears among the four groups. (D) The rats in the SAE-SE
group had more time in the corner squares when compared with the rats in the
Sham-SE, Sham-EE or SAE-EE group. (E) The rats in the SAE-SE group had less time
in the center square when compared with the rats in the Sham-SE, Sham-EE or
SAE-EE group. *p
The MWM was used to test the reference/working memory and spatial learning. The
results for spatial learning revealed that the SAE-SE rats took more time to
climb up the hidden platform compared with that of the Sham-EE or Sham-SE rats
from day 3 or day 4 onward, suggesting that SAE impaired their spatial learning
ability (SAE-SE vs. Sham-EE group; day 3: p
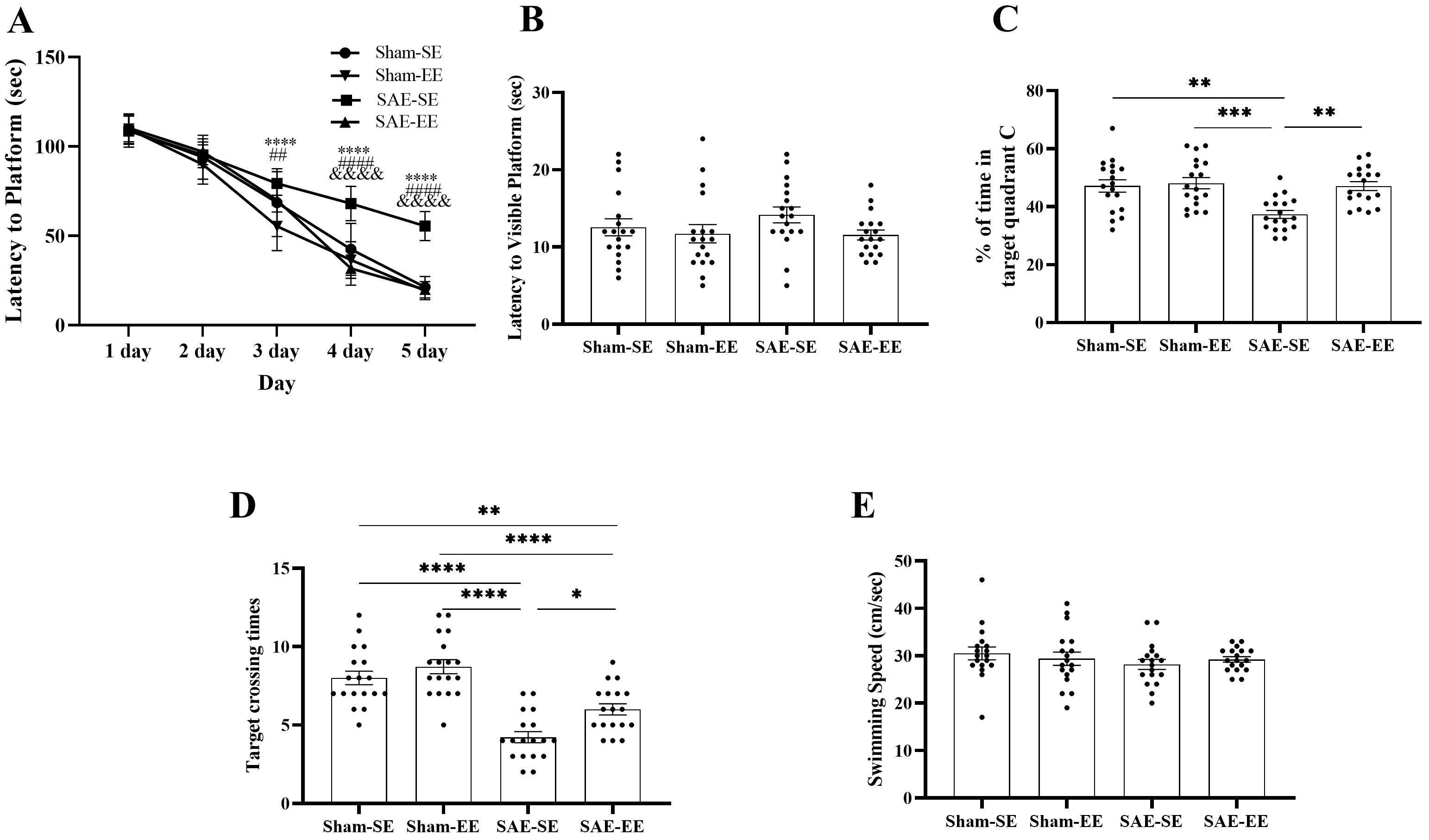 Fig. 5.
Fig. 5.EE inhibited the SAE-induced spatial learning and
memory deficits. (A) At 3 day, 4 day or 5 day, the rats in the SAE-EE group
exhibited shorter latency to platform when compared with the rats in the SAE-SE
group. In addition, the rats in the Sham-EE or Sham-SE rats had the shorter
latency to platform than the rats in the SAE-SE group from 3 day or 4 day on.
SAE-SE vs. Sham-EE: ****p
After removing the platform on day 6, the reference/working memory retention was
determined by the number of times the rat crossed the platform area and duration
of occupancy of quadrant C. The data showed that the Sham-SE, Sham-EE or SAE-EE
rats exhibited a duration of occupancy of quadrant C than the SAE-SE rats,
suggesting enhanced memory retention (SAE factor: F (1, 68) = 9.431, p =
0.0031; environmental factor: F (1, 68) = 9.238, p = 0.0034; interaction
F (1, 68) = 6.270, p = 0.0147; Post-hoc analysis: Sham-SE vs.
SAE-SE group: p = 0.0011; Sham-EE vs. SAE-SE group: p
= 0.0003; SAE-EE vs. SAE-SE group: p = 0.0012; Fig. 5C). The
number of times the rats crossed the platform area showed similar results. The
Sham-SE, Sham-EE, or SAE-EE rats crossed the platform more frequently than did
the rats in the SAE-SE group (SAE factor: F (1, 68) = 65.72, p
The NOR test was adopted to detect the changes in non-spatial working memory
(recognition memory). The results indicated that DI deteriorated after SAE (SAE
factor: F (1, 63) = 9.896, p = 0.0025; environmental factor: F (1, 63) =
14.93, p = 0.0003; interaction F (1, 63) = 10.16, p = 0.0022;
Post-hoc analysis: Sham-SE vs. SAE-SE group: p = 0.0002;
Sham-EE vs. SAE-SE group: p
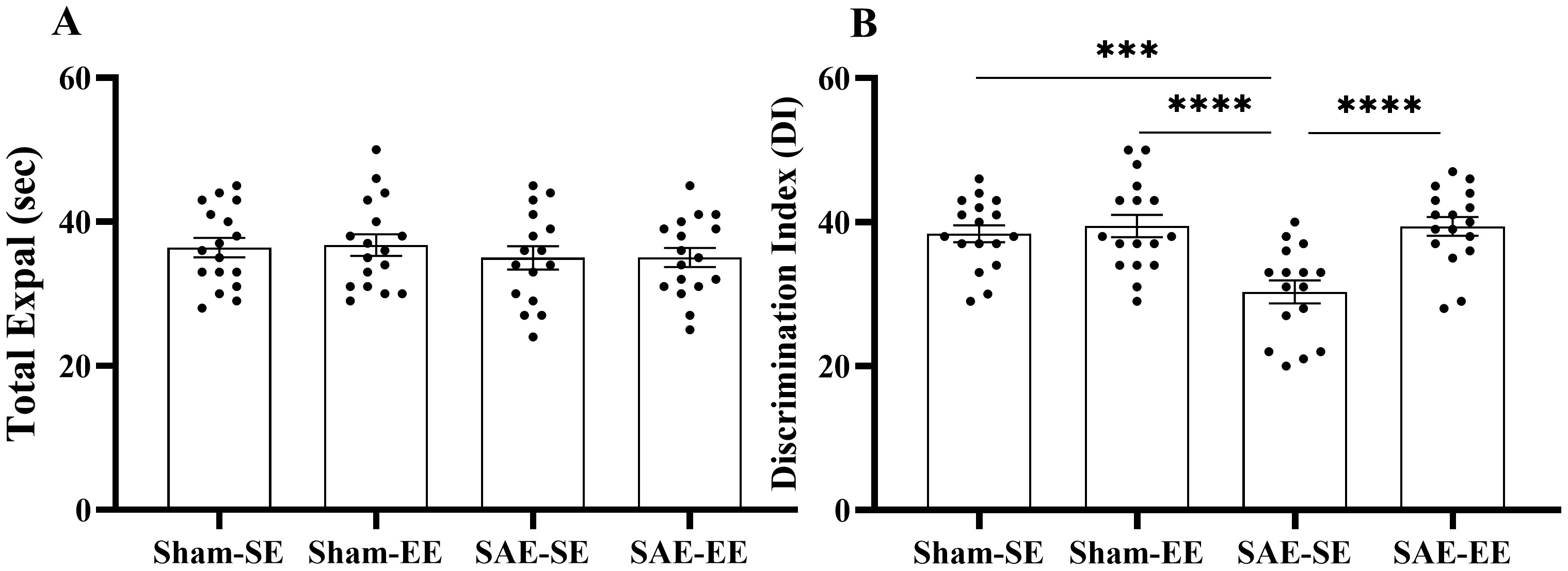 Fig. 6.
Fig. 6.EE ameliorated the recognition memory impairment induced by
SAE. (A) total exploration. (B) DI. The total exploration exhibited no
differences among four groups. The rats in the SAE-SE group had decreased DI when
compared with the rats in the Sham-SE, Sham-EE or SAE-EE group. ***p
Accumulating evidence demonstrates that inflammatory cytokines contribute to the impairment of cognitive function [39, 40, 41]. Therefore, we assessed the changes in brain inflammatory cytokine levels.
The results of the two-way ANOVA showed an obvious
interaction effect on the levels of inflammatory cytokines (IL-1
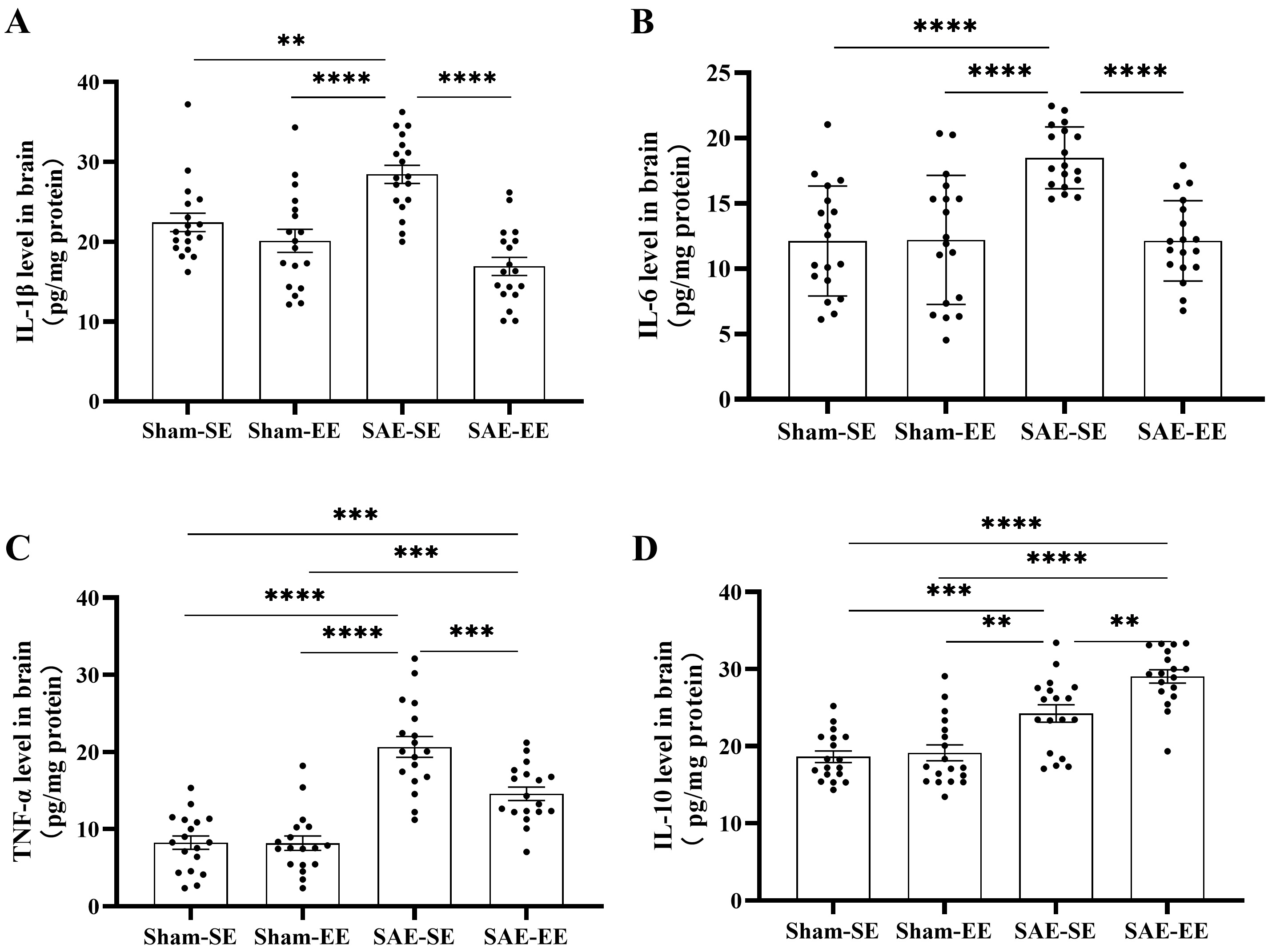 Fig. 7.
Fig. 7.EE inhibited inflammatory cytokines, but enhanced
anti-inflammatory cytokines in the brains of SAE rats. (A) The whole brain of
SAE-SE rats had higher IL-1
Additionally, the inflammatory or anti-inflammatory cytokines in serum exhibited
no differences among the four groups (IL-1
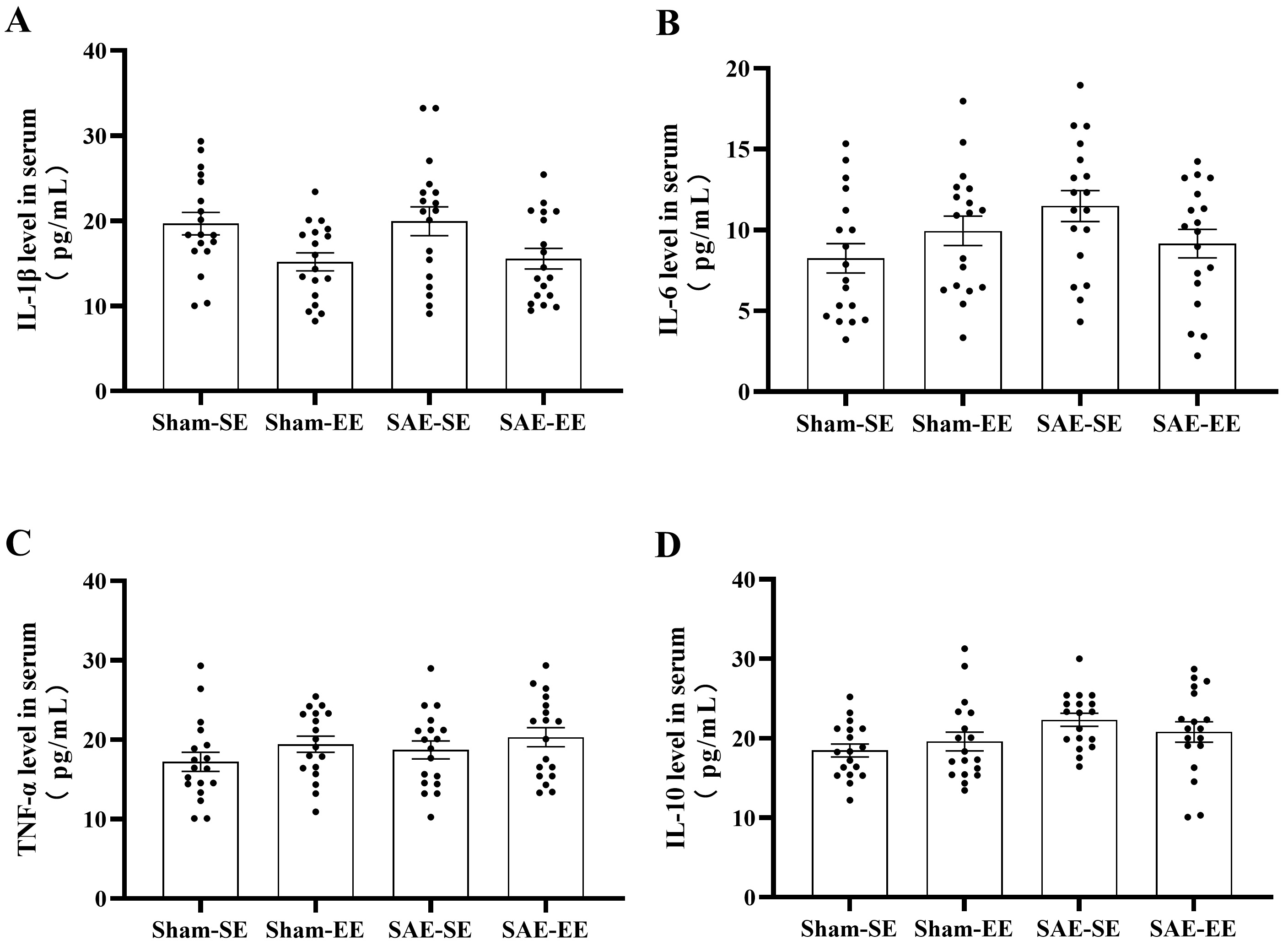 Fig. 8.
Fig. 8.EE had no effect on the levels of inflammatory
cytokines or anti-inflammatory cytokines in the serum. (A) No differences were
found in the IL-1
These results indicated that EE decreased pro-inflammatory cytokines and increased anti-inflammatory cytokines in the whole brain of the SAE rats.
SAE causes a significant increase in apoptosis, which also contributes to impaired cognitive function. p53 and Bcl-2 have been shown to regulate apoptosis in opposite manners [42, 43].
SAE factor, environmental factor or interaction contributed to the difference of
p53 or Bcl-2 level between four groups (p53: SAE factor: F (1, 68) = 56.97,
p
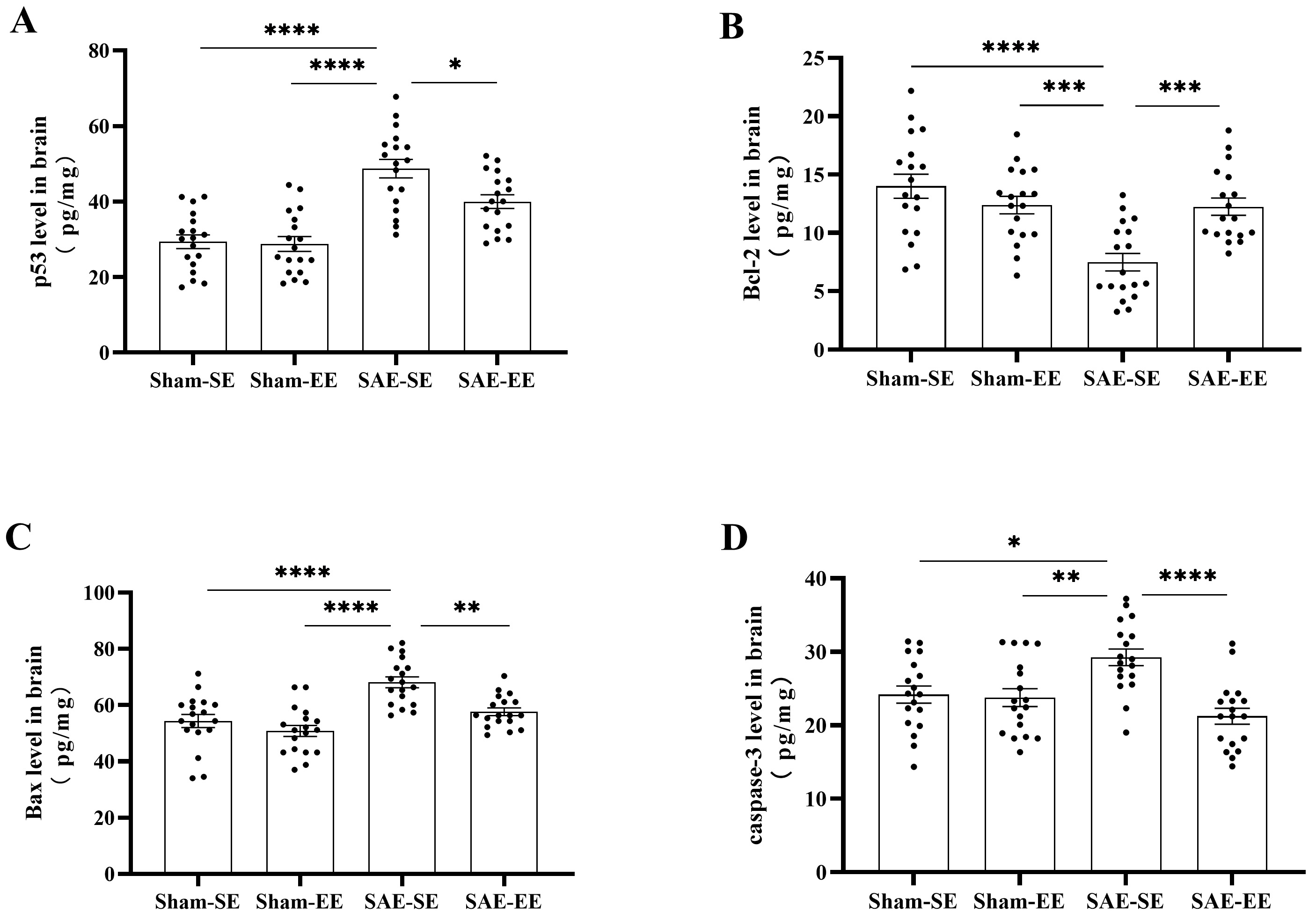 Fig. 9.
Fig. 9.EE regulated apoptosis-related molecules in the brain of SAE
rats. (A) The p53 protein level in the whole brain of SAE-SE rats was obviously
higher than that of the Sham-SE, Sham-EE or SAE-EE rats. (B) The Bcl-2 protein
level in the whole brain of SAE-SE rats was less than that of the Sham-SE,
Sham-EE or SAE-EE rats. (C) The Bax level in the whole brain of SAE-SE rats was
higher than that of the Sham-SE, Sham-EE or SAE-EE rats. (D) The caspase-3 level
in the whole brain of Sham-SE, Sham-EE or SAE-EE rats was higher than that of the
Sham-SE, Sham-EE or SAE-EE rats. *p
Previous studies have shown that Bcl-2 reduced apoptosis by influencing BAX and
caspase-3 [44]. Therefore, we also detected the changes in BAX and caspase-3
levels in the brain using ELISA. As expected, SAE increased the levels of BAX and
caspase-3 (BAX: Sham-SE vs. SAE-SE group: p
Thus, EE exhibited anti-apoptosis activity under SAE conditions by affecting the levels of p53 and the Bcl-2/BAX/caspase-3 signaling pathway.
It is widely reported that oxidative stress is responsible for the impaired cognitive function in various pathological conditions [45, 46]. Consequently, we detected the changes in oxidant activity in the whole brain.
As shown in Fig. 10, there was a significant increase in TBARS level in the
SAE-SE group, when compared with that in the Sham-SE or Sham-EE group
(SAE factor: F (1, 68) = 30.50, p
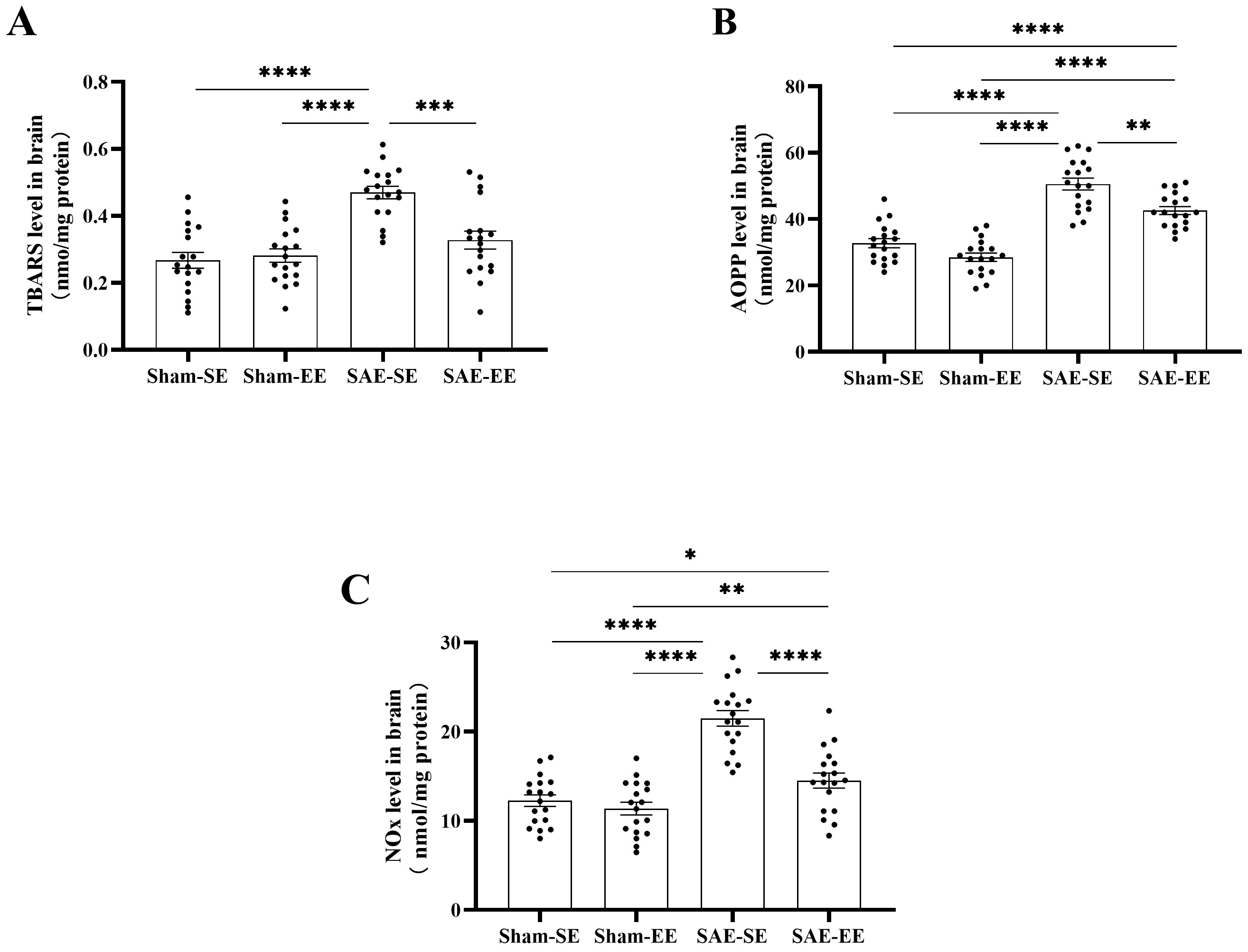 Fig. 10.
Fig. 10.EE attenuated oxidative stress in brain of SAE rats. (A) The
TBARS level in the whole brain of SAE-SE rats was higher than that of the
Sham-SE, Sham-EE or SAE-EE rats. (B) The AOPP level in the whole brain of SAE-SE
rats was higher than that of the Sham-SE, Sham-EE or SAE-EE rats. (C) The NOx
level in the whole brain of SAE-SE rats was higher than that of the Sham-SE,
Sham-EE or SAE-EE rats. *p
A similar response was found for AOPP (Fig. 10B). Under SAE conditions, the AOPP
level in whole brain of the SAE-SE rats was markedly increased compared with that
in the Sham-SE or Sham-EE groups (Sham-SE vs. SAE-SE group: p
We also detected the changes in NOx. As expected, SAE caused a significant
enhancement in NOx levels relative to those in the Sham-SE or Sham-EE group
(Sham-SE vs. SAE-SE group: p
Analysis of the markers of oxidative stress showed that SAE caused significant oxidative stress, which could be inhibited by EE housing.
An imbalance between oxidation and anti-oxidation leads to oxidative stress; therefore, the antioxidant activity in the brain was determined.
In the SAE-SE group, GSH was oxidized with a decreased level in whole brain
relative to that in the Sham-SE or Sham-EE group (SAE factor: F (1, 68) = 26.32,
p
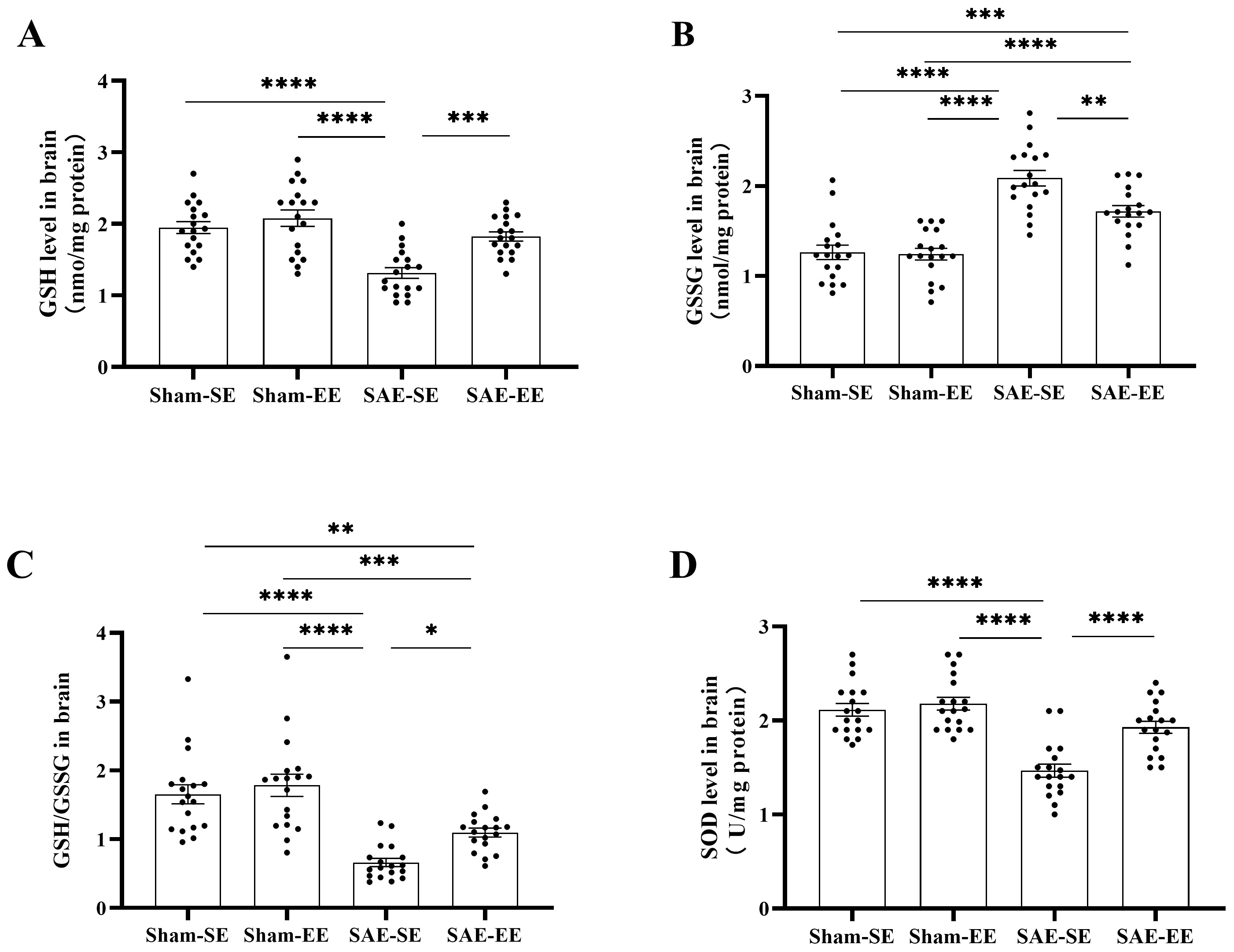 Fig. 11.
Fig. 11.EE enhanced the antioxidant activity in the brain of SAE rats.
(A) The GSH level in the whole brain of SAE-SE rats was less than that of the
Sham-SE, Sham-EE or SAE-EE rats. (B) The GSSG level in the whole brain of SAE-SE
rats was higher than that of the Sham-SE, Sham-EE or SAE-EE rats. (C) The ratio
of GSH/GSSG in the whole brain of SAE-SE rats was less than that of the Sham-SE,
Sham-EE or SAE-EE rats. (D) The SOD level in the whole brain of SAE-SE rats was
less than that of the Sham-SE, Sham-EE or SAE-EE rats. *p
SOD is another important antioxidant enzyme, whose activity decreased
significantly in the SAE-SE group (SAE factor: F (1, 68) = 44.25, p
Cognitive rehabilitation commonly uses EE as an effective and simple treatment. Our present results suggested that cognition impairment induced by SAE could be effectively alleviated using EE. Performances in behavioral tests (OF, EPM, MWM, and SN) indicated that after 30 days of enrichment, the negative effects of SAE on reference/working memory, spatial learning exploratory activity, anxiety-related behavior, and somatesthesia, were inhibited. Thus, rats with SAE reared under EE conditions had an improved outcome, which was similar to observations in other animal models of brain dysfunction [47, 48, 49, 50, 51, 52]. In addition, EE also provides positive benefits to humans. For example, an enriched lifestyle is effective in slowing down the process of cognitive aging [53], ameliorates TBI-induced cognitive deterioration [50], and delays the onset of dementia [54].
The SN test is an effective examination to detect somatesthesia and reveals somatosensory changes resulting from injury to the prefrontal cortex [24]. The SN test showed that EE-housed SAE rats showed a marked enhancement in their response to somatosensory stimuli relative to SE-housed SAE rats. The EPM test is a good indicator of anxiety behavior [55]. The EPM test revealed that EE-housed SAE rats showed less anxiety-related behaviors compared with SE-housed SAE rats. This was consistent with previous well documented observations of EE’s anxiolytic effect [25, 56, 57, 58]. It should be noted that SAE commonly induces prefrontal damage that leads to an increased risk-taking behavior [59]. A previous study showed that open arm time could be increased by risk-taking behavior via a mechanism not associated with anxiety [60]. Therefore, in this test, we could not ignore the possible contribution made by risk-taking behavior to the increase in open arms time. MWM is a commonly used approach to assess hippocampus-dependent spatial learning and memory [61]. Herein, the MWM test revealed that EE could decreased the SAE-induced increase in latency to the platform. The rats find the invisible platform dependent only on memory and spatial cues; thus, decreased latency suggests enhanced memory and spatial learning. We also showed that the performance in the MWM test was not affected by locomotor factors. In addition, land-based locomotor impairment is not related to swimming speed, which also accounts for the independence from locomotor effects of learning and memory performance in the MWM test. There was no randomization of the target quadrant in this test, which might have allowed the rats to find the platform more easily, which is a limitation of this study. Herein, recognition memory was assessed using the NOR test, the results of which demonstrated that recognition memory impairment by SAE was effectively ameliorated by EE, and the preference for novelty was also increased following exposure to EE. The NOR test is used widely to detect recognition memory and mainly depends on innate recognition behavior, without externally applied rules or reinforcement. In addition to revealing the existence of familiar object in the rats’ memory [62], the increased preference for novel objects also suggests a close relationship between EE and the desire to access novel stimuli [63]. In this study, we did not set a day of habituation to the testing arena prior to the familiarization phase as some previous studies [64] which may increase the novelty-induced anxiety and result in the excessive freezing behavior in the test phase.
The present study also showed that EE had no effect on Sham rats. In agree with this result, it has been shown that EE lacks effect on the cognitive function of control groups [65, 66]. However, some studies also indicated a positive impact on the cognitive function of control groups [67]. This difference might be due to the duration of EE housing, the age or gender of experimental animal.
Sepsis-induced central nervous system (CNS) neuroinflammation is believed to be
the mechanism of delayed cognitive dysfunction [68, 69]. During sepsis, an
impaired blood-brain barrier (BBB) allows brain
transmission of systemic inflammation [70, 71]. Increased levels of the
pro-inflammatory cytokine, IL-1
The results of the present study showed that EE exposure could inhibit the SAE-induced increase in p53 and decrease in Bcl-2. P53 mainly regulates cell-cycle arrest, senescence, and apoptosis, while Bcl-2 inhibits the apoptotic pathway [77, 78, 79]. Thus, our study demonstrated an inverse relationship between Bcl-2 and p53 protein levels in the whole brain under SAE conditions. This finding is in line with previous studies concerning relationship between Bcl-2 and p53 protein levels [80]. Increased p53 and decreased Bcl-2 ultimately results in increased apoptosis of the essential brain cells under SAE conditions. Neuronal apoptosis can result in brain atrophy (especially in the cortex and hippocampus) and ventricle enlargement [81], finally leading to cognitive impairment. Of course, activation of mitogen activated protein kinase (MAPK) [82, 83] and mechanistic target of rapamycin (mTOR) signaling pathways [84] produce apoptotic factors as a result of neuroinflammation.
Oxidative stress is also associated with long-term cognitive impairment [85, 86].
Herein, oxidative stress marker levels were enhanced after SAE and EE could
reduce these increases. Increased oxidative stress leads to mitochondrial damage,
energy failure, overproduction of toxins and inflammatory factors, BBB breakdown,
and ischemia, which all result in cognitive impairment. We also found that
antioxidant activity was suppressed by SAE, and EE could alleviate this
suppression, indicating that EE improved the antioxidant system. Increased
oxidative stress produces reactive oxygen species in the brain, which play a
positive role in modulating the production of pro-inflammatory mediators by
preventing MAPK and nuclear factor kappa B (NF-
This study had some limitations: Firstly, we assessed the cognition function, detected cytokine levels, oxidative stress, antioxidant activity and apoptosis-related molecules only at a single time point. In the future, the temporal effects of EE will be assessed by determining these indices at different exposure times. Secondly, we used young (60 days old), male rats; whereas, the majority of patients with SAE are middle-aged or older. EE positively affects rats of all ages; however, we consider that it might have more benefits in younger rats. Because female rats have estrus cycle that may disturb behavioral performance, we only used male rats in the present study. Thirdly, we believe that the SAE is a diffuse cerebral dysfunction and the SAE-induced cognition deficits attributes to the impairment of whole brain, not one brain region or several brain regions. Therefore, in the present study, we detected the inflammatory cytokines, apoptosis-related molecules, oxidative stress and antioxidant activity in the whole brain, not the discrete brain regions. Of course, hippocampus and perirhinal cortex were closely related with spatial orientation and object discrimination [88]. In the future, we will detect the changes of these discrete brain regions. Fourthly, the CLP rats showed a high mortality rate (51%) in the present study, which is similar with some previous studies [19, 20], However, this high mortality rate may bias the research outcome. Therefore, a model of SAE by lipopolysaccharide (LPS) administration may be preferable.
The results of the present study indicated that EE could effectively counteract the cognitive deficits induced by SAE via a process related to reduced levels of pro-inflammatory cytokines, oxidative stress, and apoptosis, and enhanced levels of anti-inflammatory cytokines and antioxidant activity. This study highlights the potential rehabilitation effect of EE to treat the consequences of SAE.
AOPP, Advanced oxidation protein products; BBB, blood-brain barrier; Bcl-2,
B-cell lymphoma/leukemia-2; CNS, central nervous system; DI, discrimination
index; EE, environmental enrichment; ELISA, Enzyme-linked immune-absorbent assay;
EPM, Elevated Plus Maze test; GPx, glutathione peroxidase; GR, glutathione
reductase; ICUs, intensive care units; MDA, malondialdehyde acid; MWM, Morris
water maze test; NF-
YJD and YFT—Collection of data and writing manuscript. SJ and JWG—Supervision and direction the project. BYG and SJ—Analysis and interpretation of data, and preparation of manuscript. RDG and XL—Collection of data. The authors read and approved the final manuscript.
The study was approved by the committee of Animal Use for Research and Education in China-Japan Friendship Hospital (approval number: zryhyy61-21-03-24).
We thank the reviewers for excellent criticism of the article.
This study was supported by the Beijing Major Science and Technology Projects (CN) (Grant No. D181100000318005), Natural Science Foundation of Beijing Municipality (CN) (Grant No. 7212102), National Natural Science Foundation of China (CN) (Grant No. 81902294) and Elite Medical Professionals project of China-Japan Friendship Hospital (Grant No. ZRJY2021-BJ04).
The authors declare no conflict of interest.