








 , Anna R. Carta 1
, Anna R. Carta 11 Department of Biomedical Sciences, University of Cagliari, 09042 Cagliari, Italy
Academic Editor: Robert Friedman
Abstract
Major depressive disorder is one of the primary causes of disability and disease worldwide. The therapy of depression is prevalently based on monoamine reuptake blockers; consequently, investigations aimed to clarify the aetiology of depression have mostly looked at brain areas innervated by monamines and brain circuitry involved in inputs and outputs of these areas. The recent approval of esketamine as a rapid-acting antidepressant drug in treatment-resistant depression, has definitively projected glutamatergic transmission as a key constituent in the use of new drugs in antidepressant therapy. In this review we have examined the role of several brain areas: namely, the hippocampus, the medial Prefrontal Cortex (mPFC), the nucleus accumbens (NAc), the Lateral Habenula (LHb), the amygdala and the Bed Nucleus of Stria Terminalis (BNST). The reason for undertaking an in-depth review is due to their significant role in animal models of depression, which highlight their inter-connections as well as their inputs and outputs. In particular, we examined the modification of the expression and release of the brain derived neurotrophic factor (BDNF) and associated changes in dendritic density induced by chronic stress in the above areas of animal models of depression (AnMD). We also examined the effectiveness of ketamine and standard antidepressants in reversing these alterations, with the aim of identifying a brain circuit where pathological alteration might trigger the appearance of depression symptoms. Based on the role that these brain areas play in the generation of the symptoms of depression, we assumed that the mPFC, the NAc/Ventral Tegmental Area (VTA) and the hippocampus form a primary circuit of depression, where regular performance can endure resilience to stress. We have also examined how this circuit is affected by environmental challenges and how the activation of one or more areas, including amygdala, LHb or BNST can produce local detrimental effects that spread over specific circuits and generate depression symptoms. Furthermore, we also examined how, through their outputs, these three areas can negatively influence the NAc/VTA-PFC circuit directly or through the BNST, to generate anhedonia, one of the most devastating symptoms of depression.
Keywords
- depression
- ketamine
- BDNF
- prefrontal cortex
- hippocampus
- nucleus accumbens
- lateral habenula
- amygdala
- bed nucleus of stria terminalis
Depression is a complex pathology, in terms both of its aetiology and symptomatology. Although the aetiology of depression is still undefined, it is widely accepted that it is likely to emerge from the interaction of genetic and epigenetic factors [1]. Symptoms of primary relevance include a marked diminished interest or pleasure in most or all activities, the feeling of worthlessness or inappropriate guilt, and recurrent suicidal ideation or attempts [2]. Although these symptoms are hardly reproducible in animal models of depression (AnMD), the evaluation of peculiar behavioural features in rodents have markedly contributed to the preclinical development of many antidepressants [3], but the limits of AnMD in terms of predictive, face, construct and translation validity [4, 5], have limited the progress in understanding the role of specific brain areas in the aetiology of depression. Despite this, the objective morphological and biochemical changes that have been observed in specific brain areas of patients and AnMD (i.e., hippocampal neurogenesis and Brain Derived Neurotrophic Factor (BDNF) expression and release) have allowed researchers to consider the function of these areas as crucial in depression onset and in therapeutic response. In addition, other brain areas are thought to be involved in depression because they support certain functions that are lost in depression, leading to the onset of relevant symptomatology. Moreover, future attempts and attempts made thus far to characterize the brain circuits that are involved in depression can benefit from the identification of the sites of binding and the mechanism of action of antidepressants.
It has been estimated that, even when they are sufficiently treated for Major Depressive Disorder (MDD), more than one-third of patients do not respond to first line Standard Antidepressant (StAD) treatment for reasons that have not yet been clarified [6, 7]. The search for effective and well-tolerated new antidepressants is clearly an urgent need. At the moment, the development of new antidepressants can be pursued predominantly through testing their effects in AnMD [8]; the next phase of development and an essential intermediate goal in this process will entail acquiring a better understanding of brain circuits underlying antidepressant behavioural response in rodents.
Among recently produced antidepressants, ketamine stands out for its effects, although its precise mechanism of action has not yet been clarified [9, 10]. Ketamine is an anaesthetic and analgesic drug [11, 12] and possesses dissociative properties that have prompted its use as a recreational drug of abuse, mostly among adolescents [13]. Many years of investigations into the involvement of glutamatergic transmission in depression [14] have led to the discovery of the rapid and sustained antidepressant properties of ketamine [15]. The relevance of this finding is that the antidepressant effects of ketamine do not require 2–3 weeks of treatment to emerge, as is the case with StAD; indeed, many investigations have shown that the reduction of depression symptoms can be observed within 48–72 hours after the administration of i.v. ketamine at a dose of 0.5 mg/kg for 40 min [16, 17]. In 2019, (S)-ketamine, under the name Esketamine, was approved by the FDA for the treatment of patients affected by Treatment-Resistant Depression (TRD) [18]; the repurposing of ketamine in depression therapy and related disorders can henceforth become a concrete alternative to StAD and may soon enable clinicians to define the ideal patient who can benefit mostly from ketamine or esketamine therapy [19, 20].
Understanding the mechanism of action of ketamine in the treatment of depression is undoubtedly complicated by the difficulty of comparing its mechanism of action with that of StAD. Indeed, ketamine differs from these drugs in its capacity to block the N-methyl-D-aspartate (NMDA) receptor and in its capability of manifesting a rapid antidepressant effect [15, 21]. Nonetheless, serotonin [22] and norepinephrine transmission [23, 24], which are the target of StAD, could be involved in producing the antidepressant effects of ketamine. By contrast, therapy with StAD is known to be based on blocking either the serotonin or the noradrenaline reuptake or both [25], while the dopamine reuptake, with the exception of bupropion, is not a target of these drugs. Long term therapy with antidepressants results in the enhancement of serotonergic and noradrenergic transmission through a complex mechanism that involves changes in adrenergic and serotonergic receptor density or sensitivity, changes in receptor G-protein coupling and cyclic nucleotide signalling. This therapy can also determine induction of neurotrophic factors, as well as increasing neurogenesis in the hippocampus [26].
Interestingly, although the dopamine reuptake system does not appear to be directly involved in the effects of StAD, norepinephrine reuptake blockers may increase dopamine synaptic concentrations in areas implicated in depression, such as the medial Prefrontal Cortex (mPFC) the Nucleus Accumbens (NAc) and the BNST. This is likely to be the case because dopamine is recaptured by the norepinephrine transporter [27]. Consequently, in the light of the important role that dopamine transmission has acquired in the treatment of depression, with regard specifically to anhedonia and motivated behaviour [28, 29], it does appear surprising that dopaminergic drugs do not play a relevant role in the therapy of depression [30, 31]. On the other hand, dopamine involvement in the antidepressant effects of ketamine is to be considered highly likely [32].
This review aims to examine which of the brain circuits that are involved in depression play a crucial role in the antidepressant mechanism of ketamine action and which circuits are also involved in the effects of StAD.
In order to better explain the role of specific brain areas in the mechanism of action of ketamine, it will be initially necessary to evaluate the pharmacological action of ketamine on those parameters, such as BDNF expression and release, or hippocampal neurogenesis, which are objectively altered in the specific brain areas of depressed patients as well as in AnMD [33]. Considering that chronic stress has been used as a powerful tool in reproducing rodent models of depression, and that chronic stress generates biochemical changes and receptor adjustments at the neuronal level in specific brain regions, we will also discuss the capacity of ketamine and StAD to reverse these changes in order to better clarify their mechanism of action. This evaluation will enable us to highlight some specific brain circuits that are involved in ketamine effects but could also play a pivotal role in understanding the therapeutic action of new potential antidepressant agents and in the aetiology of depression itself.
Ketamine is a non-competitive antagonist for the NMDA-type glutamate receptor site [34, 35, 36]. Ketamine binds to the phencyclidine site within the channel in the open ionic state, preventing the flux of sodium and calcium ions [37]. The involvement of NMDA-type glutamatergic transmission in depression is suggested by its adaptive response observed following treatment with antidepressants [38, 39]; although the antidepressant action of ketamine was initially reported as early as 2000 [10], the role of glutamatergic transmission in depression and in ketamine’s mechanism of action is far from being fully clarified [40, 41] (Fig. 1). One reason why there is an obstacle to understanding this role is undoubtedly because of the extensive distribution of glutamatergic transmission in the mammalian brain, and the different role of NMDA receptors in healthy versus diseased brains [42, 43, 44, 45] (Fig. 1).
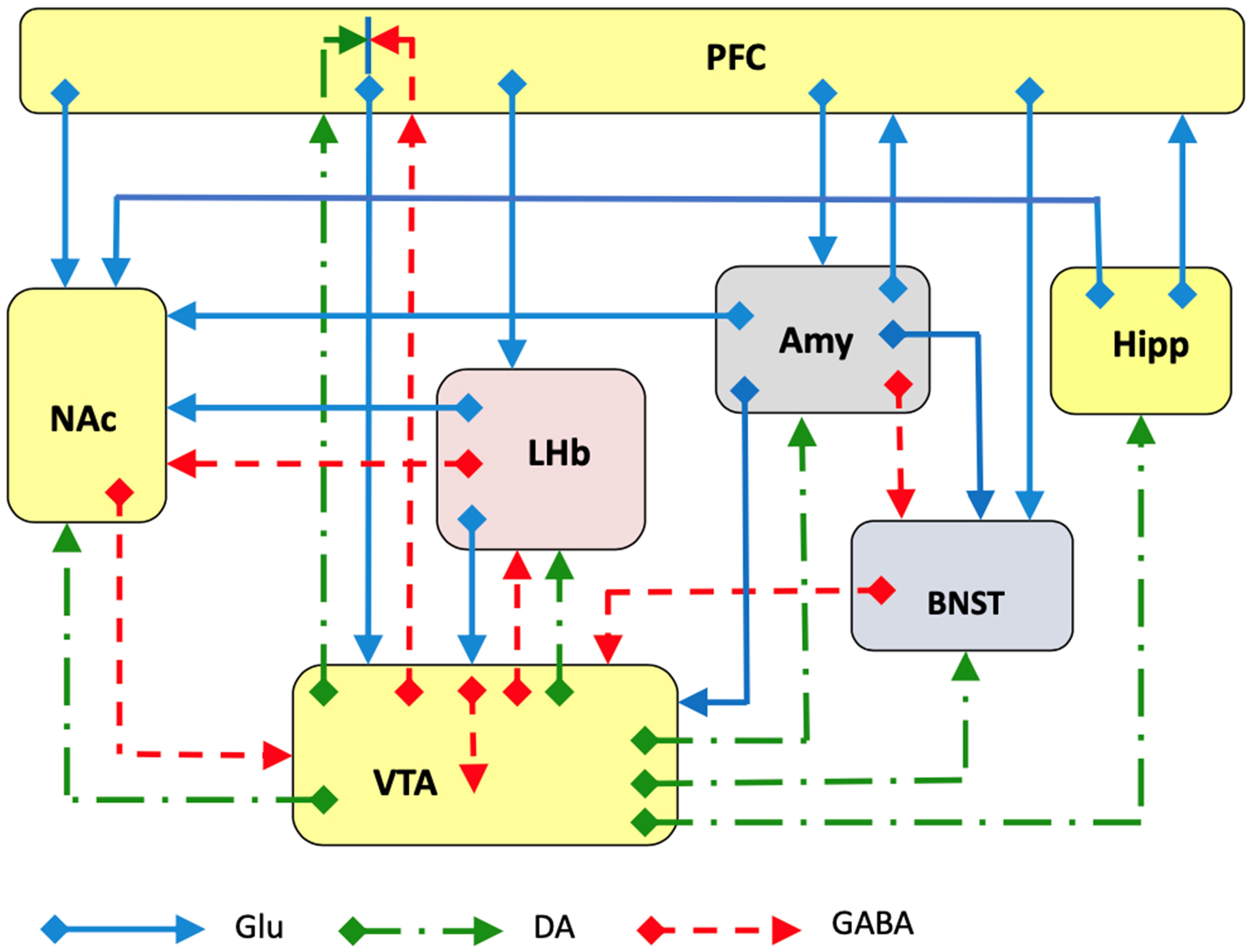 Fig. 1.
Fig. 1.Schematic representation of several major glutamatergic input pathways on mammalian brain nuclei where ketamine might interact with glutamate transmission by acting on NMDA and/or AMPA receptors to produce the antidepressant effects. Abbreviations: PFC, prefrontal cortex; Hipp, hippocampus; NAc, nucleus accumbens; LHb, lateral habenula; Amy, amygdala; VTA, ventral tegmental area; BNST, bed nucleus of stria terminalis. Glutamate (Glu), dopamine (DA) and GABA neurons and axons are represented.
Consequently, even though the antidepressant effects of ketamine were initially observed (as early as 1999) to overlap with those of imipramine in AnMD [46] and the antidepressant effects of ketamine have since been extensively reproduced using the raceme (R,S)-ketamine, at a dose of 10 mg/kg [47], a full understanding of the mechanisms underlying the rapid antidepressant action of ketamine has remained limited. Furthermore, if we were to accept that ketamine acts as an antagonist of NMDA glutamate transmission, how can we reconcile this observation with the fact that chronic stress reduces glutamate transmission in both the hippocampus [48, 49] and prefrontal cortex [50]? Indeed, ketamine is known to be able to curb the typical changes brought about by chronic stress-induced depression [41, 49]. To provide an answer to this question, it has been suggested that ketamine may act through the reduction of glutamatergic stimulatory transmission on a GABAergic inhibitory interneuron, which under physiological conditions inhibits a cortical pyramidal or hippocampal glutamatergic neuron [37, 51]. This explanation may corroborate the evidence that chronic stress in general reduces glutamate transmission and is in agreement with the observation that ketamine restores it in animal models of depression [49].
A second obstacle to understanding ketamine’s mechanism of action is the
possibility that both the S and R enantiomers and their related metabolites are
involved in its therapeutic effect [52]. Indeed, it is interesting to note that
the S-enantiomer of ketamine has a fourfold higher affinity for NMDA receptors
(Ki
A more in-depth discussion of the role of metabolites in the rapid and sustained antidepressant action of ketamine is not among the objectives of this review and can be examined in previous publications [19, 37, 57]. The above observations essentially suggest that the antidepressant effects of ketamine may manifest either through its antagonist action on the NMDA receptor or through potentiation of glutamate transmission through AMPA receptors. In addition, although these features enable us to explain how ketamine modifies the glutamate transmission in the hippocampus and in the mPFC, what remains to be clarified is the role of ketamine in the other brain areas that are involved in depression such as the NAc, the BNST, the amygdala and the LHb and how these areas interact with the mPFC and the hippocampus. Another important aspect of ketamine’s mechanism of action is its ability to induce a rapid and long-lasting antidepressant effect. It should be noted though, that this effect can last up to one week, during which time the plasma concentration of ketamine becomes irrelevant [15].
Although an in-depth pharmacokinetic and pharmacodynamic discussion of ketamine’s action is not among the objectives of this review, we feel it will be useful to clarify a few details as we refer to some recent publications for an argued evaluation [19, 58]. A first intriguing point serves to comprehend what the mechanism that activates the antidepressant response of ketamine may be; in particular, when evaluating the response to a single infusion of 0.5 mg/kg of (R,S)-ketamine, one observes the very rapid appearance of dissociative and psychotomimetic effects, which then gradually disappear in 60–120 min [59]. Nevertheless, given that this appearance is clearly generated by the achievement of an effective plasma concentration, it is hard to associate it with the activation of neuronal circuits, which allow the therapeutic antidepressant effect to take effect. Furthermore, there is a temporal correlation between the onset of behavioural effects, the occupation of NMDA receptors and the plasma or cerebral concentration of ketamine in different animal species [58]. A recent meta-analysis compared the efficacy of racemic ketamine with that of esketamine in bipolar and MDD subjects, concluding that intravenous ketamine appears to be more efficacious than intranasal esketamine [60]. In conclusion, ketamine and esketamine represent an innovative pharmacological opportunity for TRD, although concerns do remain about safety and tolerability of repeated and long-term treatment [20, 61].
To shed light on the role that brain circuits play in depression and in the mechanism of action of ketamine, we will look at BDNF expression and release in specific brain areas. BDNF is a 118 amino acid peptide and possesses wide neurotrophic effects implicated in neurogenesis, neuronal survival and differentiation, synaptic plasticity and Long-Term Potentiation (LTP) [62]. An altered function of BDNF has been implicated in the emergence of several neurological and psychiatric syndromes such as mood and anxiety disorders [63, 64] (Fig. 2).
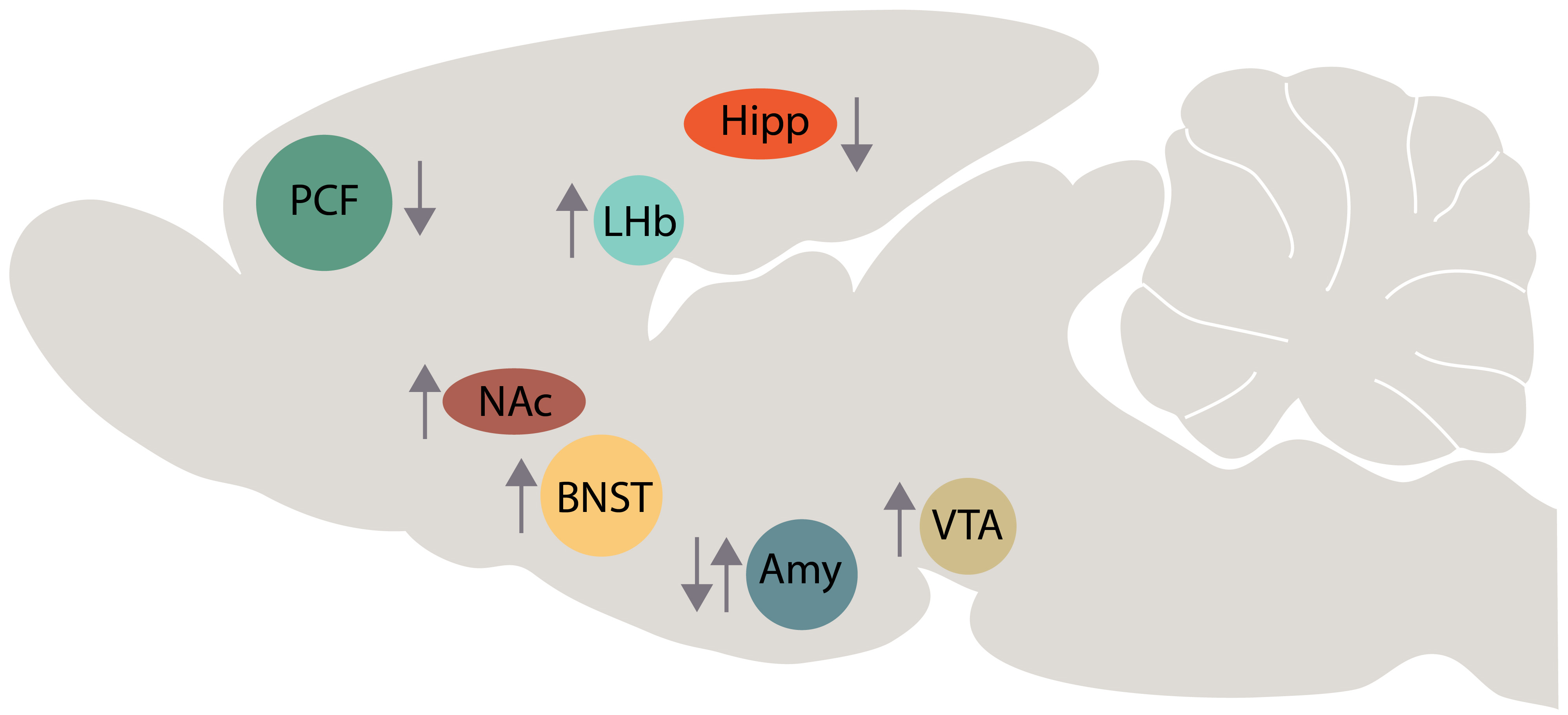 Fig. 2.
Fig. 2.Schematic representation of BDNF changes in several brain areas of animal model of depression (up arrow, BDNF increase; down arrow, BDNF decrease). Abbreviations: PFC, prefrontal cortex; Hipp, hippocampus; NAc, nucleus accumbens; LHb, lateral habenula; Amy, amygdala; VTA, ventral tegmental area; BNST, bed nucleus of stria terminalis.
BDNF can be released from presynaptic terminals or from dendrites, and act on TrkB (Tropomyosin receptor kinase B) receptors to activate cell growth and differentiation [65]. In particular, by instigating neuronal circuitry rewiring [9] BDNF is involved in the therapeutic action of StAD and ketamine. Depending on the brain area considered, ketamine will determine different BDNF responses. We will now examine its effects in the hippocampus, mPFC, NAc, amygdala, LHb and BNST.
BDNF has a unique role in the hippocampus, where it is involved in learning and LTP [66]. It is widely recognised that expression of the BDNF is reduced in the hippocampus of depressed patients and in the hippocampus of AnMD [33, 67], although the cause-effect relationship of this observation is not clear. Furthermore, chronic stress is known to cause a down-regulation of BDNF protein and mRNA in the hippocampus [68], an effect that is closely linked to depression [53]. Conversely, while treatment with StAD increased the expression of BDNF mRNA in the hippocampus [69, 70, 71], repeated electroconvulsive seizure treatment can also increase expression of the BDNF protein in the hippocampus [72].
The effect of ketamine on the hippocampus is also closely linked to that of the BDNF; indeed, the presynaptic modulation of vesicular glutamate release [73] and the postsynaptic modulation of glutamate receptor trafficking can be attributed to BDNF, as demonstrated by the increase of AMPA receptors in hippocampal slices and neuronal culture [74]. Bearing in mind that in the hippocampus, as well as in the mPFC, ketamine activates the cellular and molecular mechanisms that underlie synaptic strengthening and dendritic spine formation, it is quite likely that BDNF mediates these effects, and that ketamine can generate them through the release of BDNF [49, 51]. Direct evidence of this effect is provided by Autry et al. [75], who observed that ketamine (3 mg/kg i.p.) increased BDNF protein expression in the hippocampus after only 30 minutes’ treatment. Recently, it has been suggested that ketamine can increase BDNF release in the hippocampus through the blockade of postsynaptic NMDA receptors [37] followed by synthesis of eukaryotic Elongation Factor2 (eEF2), similar to that proposed by Autry et al. [75]. Moreover, these authors provided evidence for an increase of TrkB phosphorylation, an indicator of TrkB activation in the hippocampus, suggesting that BDNF-TrkB signalling in the hippocampus is involved in the antidepressant response to ketamine [74]. Complementary to this observation is the finding that a loss of hippocampal BDNF per se is not sufficient to mediate depression-like behaviour, but BDNF in the dentate girus might be essential in mediating the therapeutic effects of antidepressants [76].
Together with BDNF, hippocampal volume and hippocampal neurogenesis are parameters that can be assessed to test the response of ketamine or StAD in AnMD or in patients affected by MDD. Indeed, the volume of the hippocampus is reduced in MDD patients [77, 78] and although a few studies have reported contrasting results [79], most have confirmed this observation. Interestingly, the antidepressant effects of ketamine were observed in patients who have a relatively smaller hippocampus and were refractory to StAD [80]. Another crucial issue in the aetiology of depression is the hippocampal neurogenesis, a process by which 700 new-born hippocampal granule cells are normally added daily in adult humans [81]. This process should allow the distinguishing and storing of similar experiences as distinct memories; the weakening of this process is of relevance as a biomarker of MDD, although some limits should be considered [82]. Neuroimaging and post-mortem studies in MDD patients have shown that neuronal atrophy is likely to be associated with a reduction of neurogenesis [83, 84, 85, 86]. Similar to the effects on hippocampal BDNF, chronic stress decreases hippocampal neurogenesis, while StAD can reverse this decrease [87, 88, 89]. It is known that neurogenesis can be stimulated either by therapy with StAD, or by physical exercise by activating not only progenitors but also stem cells, which are normally not stimulated [90]. Interestingly, the delay in the onset of the StAD effect after chronic administration has often been attributed to the time needed for new neurons to be recruited into memory circuits.
Given this, it is even more challenging to try and understand how ketamine produces its rapid and long-lasting antidepressant effects. Deyama and Duman [51], who recently reviewed this issue, have suggested that neurogenesis is involved in the sustained but not in the fast actions of ketamine. Although it reconciles the effects of ketamine with those of StAD, this hypothesis raises a doubt about the indispensability of the reactivation of hippocampal neurogenesis for the therapeutic manifestation of the rapid effects of antidepressants. On the other hand, a further testimony to the crucial role of hippocampal neurogenesis has been provided by Yamada and Jinno [91], who reported that, just two days after the administration of a single dose (10 mg/kg i.p.), ketamine elevated the density of neuronal progenitors and new-born granule cells in the ventral but not in the dorsal hippocampus in adult mice. Notably, the densities of neuronal stem cells were not affected by ketamine in either area. The preferential action of ketamine on ventral hippocampus (vHipp) neurogenesis is consistent with the role of this area in emotional processes and memories, while the dorsal part is related to spatial memory [92]. These results not only confirm neurogenesis to be a complex phenomenon but also suggest that new therapeutic tools, to be used as a complementary therapy of StAD, could generate a partial and temporary reactivation of neurogenesis.
Furthermore, it has been suggested that ketamine activation of hippocampal neural activity may bring about protective action against stress-induced depressive-like behaviour, as suggested by the results of Mastrodonato et al. [93]. They showed that a single injection of ketamine (30 mg/kg, one week before), selectively increased DeltaFosB expression in the vHipp though only in mice exposed to social defeat stress [93]. Interestingly, the inactivation of the vHipp with lidocaine was able to prevent the sustained but not the acute antidepressant effect of ketamine, evaluated by forced swim test in Sprague-Dawley rats [94]. The authors therefore suggested that the activity in the vHipp-mPFC pathway is both necessary and sufficient for the antidepressant-like effect of ketamine to take place [94]. In short, we are suggesting that the antidepressant action of ketamine may occur as a result of a direct effect on the hippocampus, as supported by its effect on BDNF release and neurogenesis, although the connections that the hippocampus establishes with the mPFC and the NAc/Ventral Tegmental Area (VTA) suggest that it would be more appropriate to consider the global interaction of ketamine with a hypothetical primary circuit of depression (Fig. 3). This is also in consideration of the role of the mPFC in the cognitive aspects of depression, that of NAc/VTA in motivational behaviour and that of the hippocampus in its ability to control emotional memories.
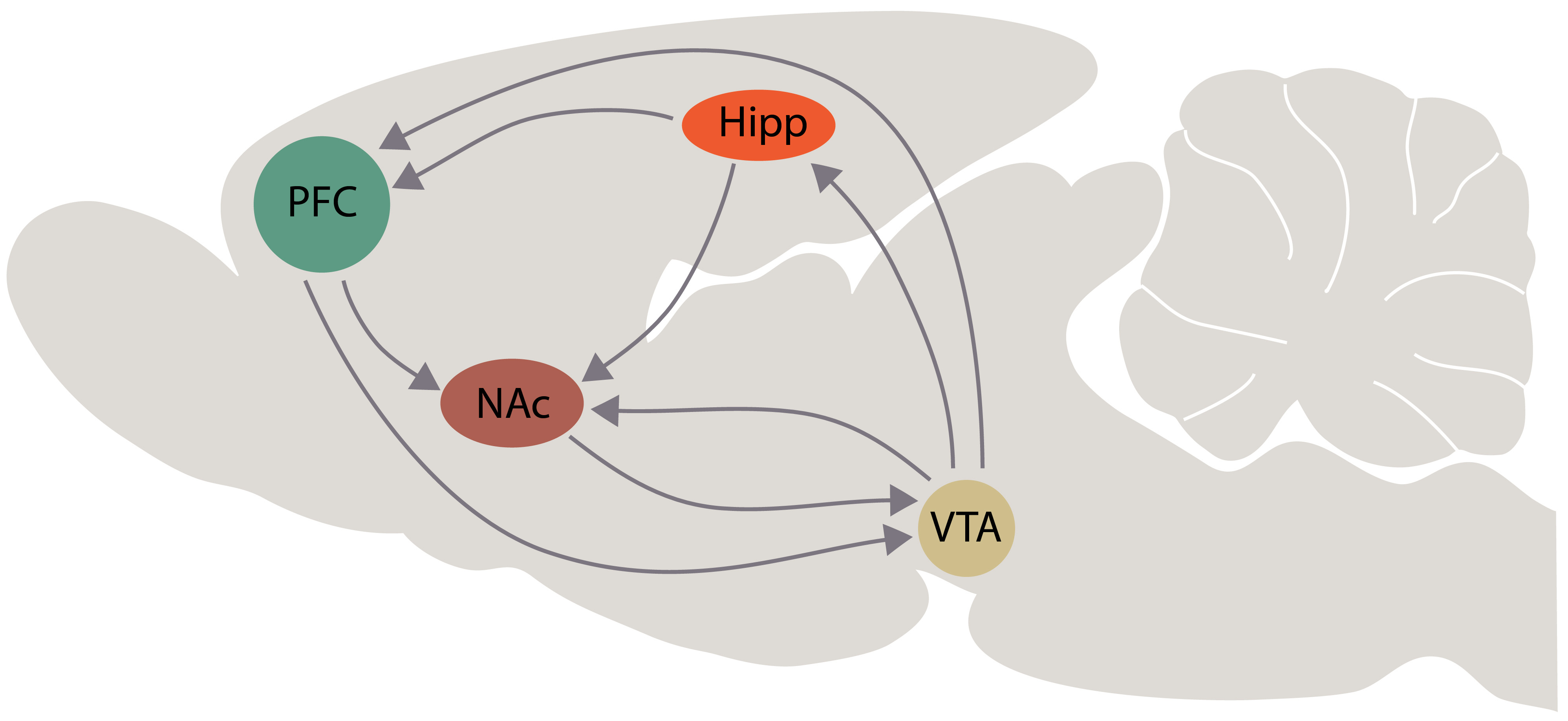 Fig. 3.
Fig. 3.Schematic representation of interactions among the brain areas that are involved in the “primary circuit of depression”, according with our hypothesis. Abbreviations: PFC, prefrontal cortex; Hipp, hippocampus; NAc, nucleus accumbens; VTA, ventral tegmental area.
Among the brain areas affected by depression, the mPFC has been widely investigated and many stimulating hypotheses about its role in depression have been advanced on the basis of several objective observations [95]. Among these, it is of some relevance that post-mortem studies have reported a reduced volume of several mPFC subregions in patients suffering from MDD and/or exhibiting suicidal thoughts and behaviours [96]. In particular, a reduction in size and density of neurons of dorsolateral mPFC has been observed [97, 98]. In addition, a decrease in synapse-related genes has been observed in AnMD [99]. An interesting report by Deyama and Kaneda [100] showed that a single injection of BDNF into the mPFC of mice produced long-lasting (8 days) antidepressant effects, in some way similar to the rapid and sustained antidepressant effect produced by ketamine [69]. Moreover, in the mPFC of subjects with MDD, the expression level of BDNF transcript variants and synaptic markers are selectively reduced [101].
Several studies have shown that the increased release of BDNF in mPFC may be the
means by which ketamine produces its antidepressant effects. Indeed, preclinical
studies have shown that both (S)-ketamine (10 or 50 mg/kg i.p.) and the ketamine
metabolite (2S,6S)-HNK (10 mg/kg i.p.) stimulate BDNF release in the mPFC of mice
[102, 103]. Furthermore, it was observed that the antidepressant effects of
ketamine (10 mg/kg i.p.) can be blocked by the administration of an antibody
against BDNF in the mPFC of rats [104]. Additionally, a direct effect of ketamine
(0.5
It is apparently incongruent that ketamine activates BDNF release because it reduces rather than stimulates glutamate transmission. One explanation for this apparent incongruity holds that NMDA receptor blocking activity occurs pre-synaptically on the terminals of GABA interneurons in the medial mPFC and thereby reduces the inhibitory activity of these neurons. Thus, the reduction of GABA inhibitory activity would allow an increased release of glutamate, which through NMDA receptors would in turn activate BDNF release; this is followed by rapid exposure of AMPA receptors located on pyramidal neurons projecting towards subcortical areas, or on other pyramidal cells [19, 37, 51, 105, 106]. Activation of AMPA receptors allows calcium entry via voltage-dependent calcium channels and the subsequent release of both BDNF and the vascular endothelial growth factor. These two factors induce transduction and synthesis of further factors that mediate activation of synaptogenesis and dendritic spine maturation, including GluA1 and postsynaptic density protein 95 through the TrkB/Flk-1—mammalian Target of Rapamycin (mTOR) complex1 signalling pathway [107]. Likewise, these investigators suggested that direct activation of postsynaptic AMPA receptors, resulting in increased extracellular glutamate and GABA levels in the mPFC, is involved in the antidepressant effects of the ketamine metabolite HNK [37].
The rapid effects of ketamine on neuronal plasticity in the mPFC has been observed in rodents [108]; these authors have shown that a single administration of a low dose of ketamine promptly activated the mTOR signalling, resulting in a long-lasting increase of spine and synapse-associated proteins in the rat mPFC. Notably, repeated stress induces an opposite effect, with dendritic spine loss in the rat mPFC [109]. Furthermore, Moda-Sava et al. [110], demonstrated that ketamine reversed the loss of postsynaptic dendritic spines induced by chronic stress or chronic corticosterone in a mouse model of depression. These authors also demonstrated that specific elimination of postsynaptic dendritic spines on mPFC projection neurons was temporarily associated with the manifestation of depressive behaviour and that antidepressant doses of ketamine, in addition to counteracting behavioural manifestations, resulted in regeneration of the spines; they further suggested that synaptogenesis might be a means by which the long-lasting antidepressant effects (but not the rapid ones) of ketamine are manifested [110]. This last hypothesis is not in agreement with the increased level of synaptic proteins (i.e., PSD-95, synapsin-1, and GluA1) that Li et al. [108] observed as early as 2 h after ketamine administration. Therefore, it can be attested that a local activity-dependent synaptogenesis in the mPFC pyramidal neurons is an essential feature of the antidepressant effects of ketamine [51].
The NAc receives excitatory glutamatergic innervation from different brain areas involved in depression such as the hippocampus, mPFC, basolateral amygdala and thalamus [111, 112, 113, 114] (Fig. 1), supporting the hypothesis of its key role in modulating individual susceptibility to depression and thus its potential to be a target area for the antidepressant actions of ketamine. The VTA provides the dopaminergic input to the NAc [115] but also to many other brain areas involved in depression such as the mPFC, hippocampus, BLA and BNST [116]. VTA neurons are in turn innervated by GABA projection from NAc and BNST, whereas glutamate neurons originating in the mPFC, LHb and amygdala also innervate VTA [19] (Figs. 1,4).
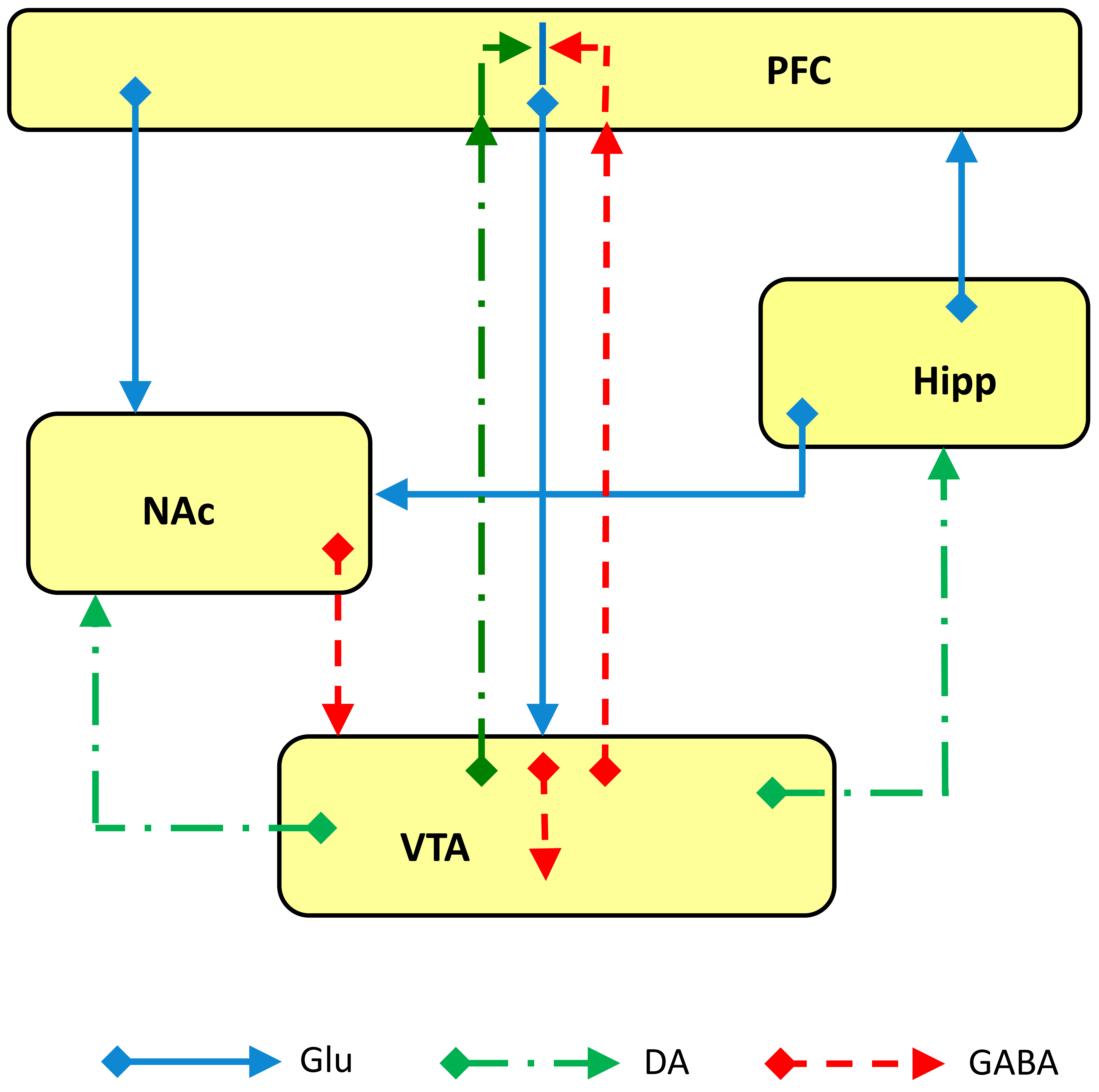 Fig. 4.
Fig. 4.Schematic representation of glutamate (Glu), GABA and dopamine (DA) inputs and outputs among the brain areas involved in the “primary circuit of depression”. Abbreviations: PFC, prefrontal cortex; Hipp, hippocampus; NAc, nucleus accumbens; VTA, ventral tegmental area.
Interestingly, inputs from the hippocampus and mPFC converge on the same NAc neuron, a feature that would suggest the existence of a hippocampal gating of other limbic inputs [117] and in particular that vHipp afferents to NAc regulate susceptibility to depression [118]. The NAc is a central component of the ventral striatum and integrates limbic and cortical signals to modulate goal-directed behaviour through the modulatory action of dopamine [119]. For its role in the reward circuit, in motivated behaviour, in hedonic appraisal, mood regulation and in anhedonia in general (decreased drive for activities because of the reduced capacity to feel pleasure and satisfaction for the associated outcomes), NAc has been implicated in the aetiology of depression [28, 120].
NAc mesolimbic pathway and the BDNF expression, has a fundamental role in mediating the effects of social defeat stress and those of antidepressants; in particular, after local deletion of BDNF in the VTA, the effects of social defeat stress on gene activation in the NAc was lost or reversed, as was the effect of chronic fluoxetine [121]. BDNF signalling is highly represented in the VTA-NAc circuit; in particular, one major source of BDNF in both regions is provided by a glutamatergic afferent from mPFC, hippocampus and amygdala, while release of BDNF is recognised by TrkB receptors expressed on VTA dopaminergic and NAc GABAergic neurons [28]. In general, the changes in BDNF release induced by chronic stress in the NAc further support its pivotal role in the generation of depression [28]. Additionally, the phasic stimulation of VTA neurons that project to NAc, but not those that project to the mPFC, induced susceptibility to social defeat stress in mice [122]. In particular, stress increases the release of BDNF in the NAc, an effect that was mediated by Corticotrophin Releasing Factor (CRF) [123]. It was subsequently suggested that BDNF–TrkB signalling but not dopamine signalling in the VTA-NAc circuit, was an essential component in the development of depressive-like behaviours induced by chronic stress [124]. Interestingly, it has also been shown that a BDNF increase (namely 90% elevation) in the NAc occurs only in susceptible mice [125]. Direct evidence of the NAc-BDNF role in promoting depression-like behaviour has been provided by the observation that direct infusion of BDNF into the NAc increases the immobility in forced swim tests (FST), while inhibition of TrkB signalling in the NAc decreases immobility [126].
Looking at the role of NAc in mediating the antidepressant effect of ketamine, it is of interest that ketamine and LY341495 (a potent and selective antagonist of group II metabotropic glutamate receptor [127]), but not the selective serotonin reuptake inhibitor (SSRI) citalopram, increased the number of spontaneously active dopamine neurons in the VTA, and the extracellular dopamine levels in the NAc as well as in the mPFC [128], an effect that was shown to be AMPA-dependent. Although the abuse potential of ketamine [129] would suggest there may be direct ketamine action on dopamine transmission through an interaction with dopamine reuptake, or with dopamine receptors, it has been demonstrated that such an occurrence is unlikely [130].
Stress is known to affect synaptic plasticity by inhibiting LTP and/or facilitating long term depression (LTD) in the hippocampus of rodents [131] and we also know that ketamine and its metabolite (2R,6R)-HNK restore LTP and long-term memory in the Wistar-Kioto rat model of depression that likely own an alteration of dopamine stores [132, 133]. As regards NAc, ketamine restores normal neuron population activity and restores LTP in the ventral subiculum-NAc pathway, thus suggesting that normal LTP in the NAc shell prevents the generation of helplessness [134]. On the other hand, a single low dose injection of ketamine (3–5 mg/kg) dose-dependently induced the impairing of LTP in the NAc at 24 h after administration; this effect was maintained for 7 days and was not associated with any alteration of basal synaptic transmission mediated by AMPA and NMDA receptors [135]. Similar results were previously shown by Réus et al. [136], who reported that both ketamine and imipramine impaired histone deacetylation in the NAc; this function was found to increase in the NAc but not in the mPFC, hippocampus, and amygdala in maternally deprived adult Wistar rats. This may well be relevant with regard to long-term changes related to stress and antidepressant treatment.
Further evidence of NAc involvement in ketamine’s effects was obtained by Abdallah et al. [137] in a MDD subgroup of patients displaying an enlarged bilateral NAc volume; these authors observed that ketamine treatment normalized NAc volume in patients who achieved remission. On the whole, the role of NAc in the appearance of peculiar symptoms of depression and in the antidepressant effects of ketamine supports its pivotal role in a primary circuit of depression; interestingly, the effects of stress, as well as the response to antidepressants (i.e., StAD and ketamine) on BDNF related actions in the VTA-NAc circuit appear to be opposite to those observed in the mPFC and hippocampus (Fig. 2).
The Lateral Habenula (LHb) has recently been implicated in the coding of negative emotions [138]. In particular, the functions of the LHb are associated with anxiety, stress, pain, avoidance learning, attention, human reward processing, psychosis and depression [139, 140, 141]. In consequence of the strong inhibition of dopamine neurons after a weak electrical stimulation of the LHb, it has been proposed that LHb provides a measure of control on VTA dopamine neurons, negatively influencing reward-related signals [138]. The capacity to reduce dopamine-mediated reward responsiveness validates the involvement of LHb in the occurrence of anhedonia, a typical feature in MDD patients [142] and possibly, in stress induced depression as reported by Shabel et al. [143]; these authors observed that acute stress can transform LHb reward responses into punishment-like neuronal signs. LHb is primarily a glutamatergic nucleus, and its output reaches the VTA and the dorsal raphe nuclei (DRN), where an inhibition of monoamine neurons can occur through the activation of GABAergic inhibitory neurons [144]. Glutamatergic LHb neurons are transiently activated by acute stress or aversive stimuli and likely mediate a wide range of responses, ranging from stress evasion to value-based decision-making [145]. The LHb receives projections from the mPFC, providing a link for conveying cognitive and affective information from prefrontal and insular regions to the midbrain monoaminergic centres [146] (Fig. 5).
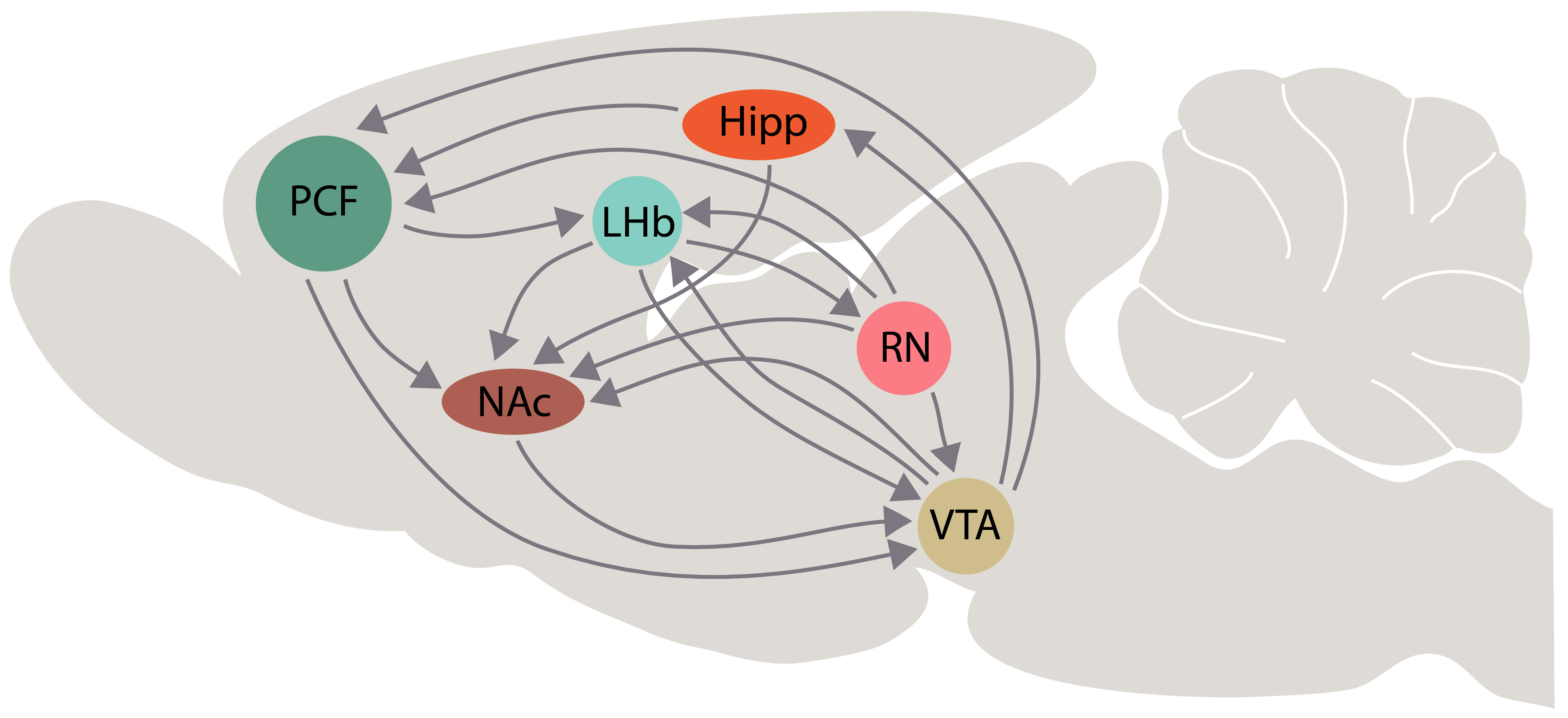 Fig. 5.
Fig. 5.Schematic representation of interactions among the lateral habenula (LHb) and the brain areas that are involved in the “primary circuit of depression”, according with our hypothesis. Some interactions of dorsal and median raphe with these brain areas are represented schematically. Other abbreviations: PFC, prefrontal cortex; Hipp, hippocampus; NAc, nucleus accumbens; VTA, ventral tegmental area; RN, raphe nuclei.
Based on this assumption, and considering the role of ketamine in the mPFC, it should not be surprising that ketamine may well produce its rapid antidepressant action through modulating the LHb function either directly or through the modulation of mPFC activity. Indeed, in a rat and mouse model of depression, in which burst-evoking photo-stimulation of the LHb can result in despair and anhedonia, it has been reported that because it blocks NMDA receptor-dependent burst firing in the LHb, ketamine can reduce stimulation of inhibitory neurons in the VTA, disinhibiting the NAc reward centre [142]. Similarly, ketamine can persistently (up to 72 h post injection) reverse maternal deprivation-induced intrinsic neuronal excitability in the LHb and decrease immobility in the forced swimming test in late adolescent male rats [147].
As far as the LHb BDNF role in AnMD is concerned, the few reports available have not enabled us to gain any clearer understanding of the mechanism involved. In addition, BDNF immunoreactivity in the habenula is mostly distributed in the medial part and is concentrated in fibers [148]. BDNF level expressions in the LHb were found to be higher than in controls of Chronic Restraint Stress (CRS) exposed mice, together with higher levels of GluN2B and c-Fos. Interestingly, the systemic administration of the specific GluN2B antagonist rislenemdaz, a drug used in phase II clinical trials as an antidepressant, reversed CRS effects while its infusion in the LHb decreased immobility time in CRS-exposed mice [149]. These authors have also showed that knocking down BDNF in the LHb prevented CRS-induced despair-like behaviour and inhibited neuronal activity in the LHb. Curiously, 6-hydroxydopamine lesions in the ventral region of the caudate putamen determined a reduction of BDNF and TRkB mRNA levels in the hippocampus, as well as in the BDNF mRNA in the habenula. By contrast, the lesions did not influence BDNF and TRkB expression in the NAc and in the VTA, whereas chronic imipramine reversed BDNF mRNA reduction in the dentate girus of the hippocampus [150].
A recent interesting study investigated the role of the LHb in depression-associated inflammation by using the Unpredictable Chronic Stress (UCS) animal model of depression [151]. These researchers observed an increase of inflammatory cytokines in the CNS, especially in the LHb; conversely, local administration of tumor necrosis factor-alpha in the LHb, can induce depressive-like behaviour in control rats. Lastly, they showed that lesioning LHb prevented the impairment of hippocampal neurogenesis and a reduction of the hippocampal function in UCS exposed rats [151].
Summing up, it appears that an inappropriate LHb firing activity could play a decisive role in the aetiology of depression, while its peculiar intrinsic activity and connections with the other brain areas involved in depression means that we can consider the LHb to be a further site through which the rapid action of ketamine occurs [152] (Figs. 5,6).
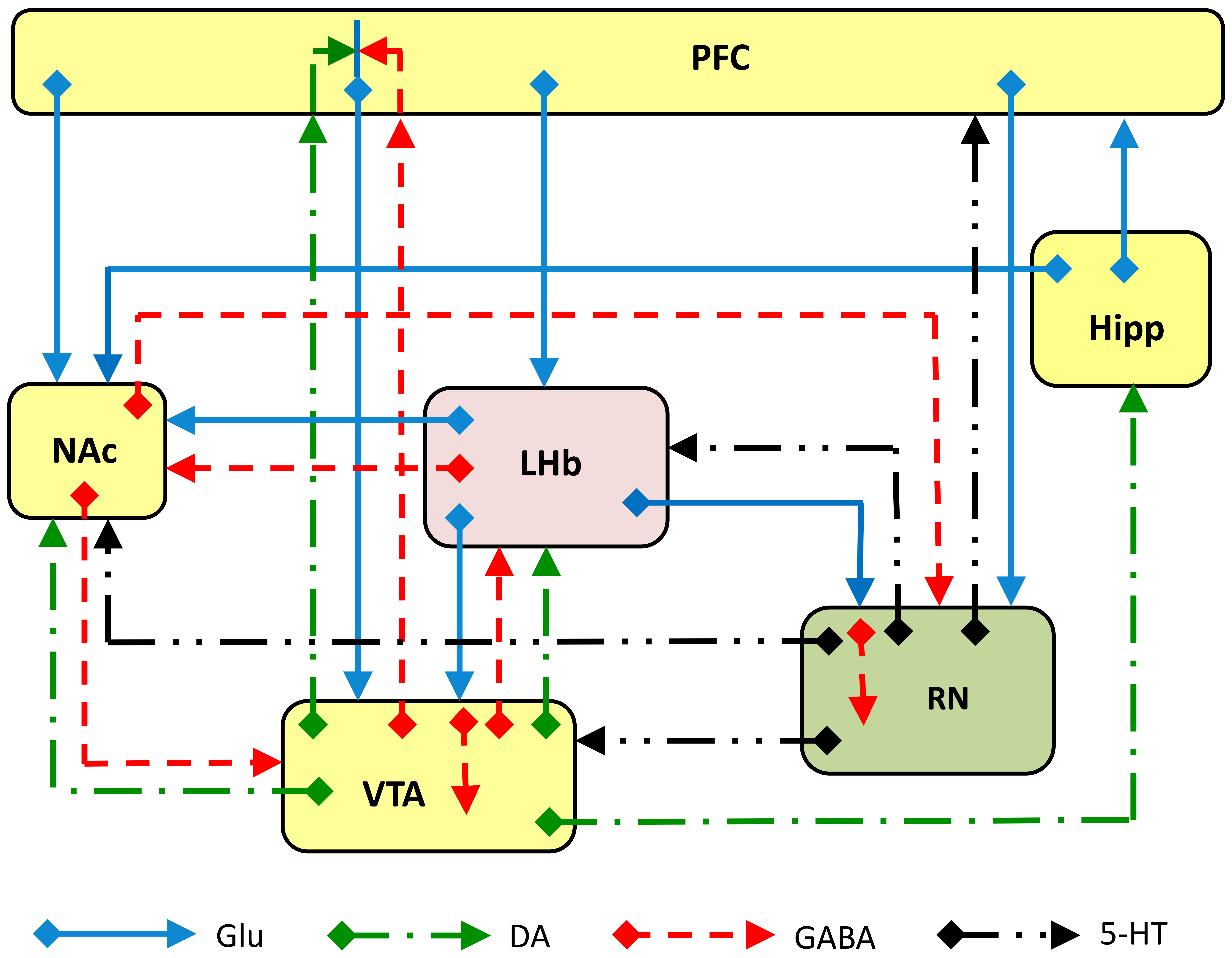 Fig. 6.
Fig. 6.Schematic representation of glutamate (Glu) GABA, dopamine (DA) and serotonin inputs and outputs among the lateral habenula (LHb), and the brain areas involved in the “primary circuit of depression”. Some inputs and outputs of dorsal and median raphe with these brain areas are represented schematically. Other abbreviations: PFC, prefrontal cortex; Hipp, hippocampus; NAc, nucleus accumbens; VTA, ventral tegmental area; RN, raphe nuclei.
The amygdala is composed of a series of heterogeneous subnuclei that differ both morphologically and functionally. Notably, the basolateral amygdala (BLA) is primarily glutamatergic [153], whereas the central nucleus of amygdala (CeA), which includes the centrolateral and the centromedial nuclei, consists of 95% GABAergic medium spiny neurons [154]. The BLA projects to the mPFC, hippocampus and NAc [155] thus interacting directly with the primary circuit of depression. CRS triggers dendritic growth and spinogenesis in the neurons of the BLA, regardless of whether or not they project to the mPFC [150]. CRS leads to increases of BDNF mRNA and BDNF protein and dendritic spine density in the BLA, which are strikingly different from those seen in the hippocampus and in the mPFC [156, 157, 158, 159].
Structural alteration and decreased function in the mPFC and hippocampus, but an increased function and an altered morphology in the amygdala were observed in MDD patients during clinical and post-mortem studies [160, 161]. The decreased connectivity and function in the mPFC may well contribute to the appearance of cognitive symptoms of depression while any augmented function of the amygdala may alter emotions and mood, increase fear and anxiety and hyper-activate the hypothalamic-pituitary-adrenal axis [159]. In this regard, it is of interest to note the suggestion that prelimbic mPFC excites amygdala output, while infralimbic mPFC inhibits it, providing a mechanism of bidirectional modulation of fear expression [162]. In a fear-generalized mouse model, a single administration of ketamine (30 mg/kg i.p.) reversed the reduction of BDNF levels induced by fear generalization in the BLA and in the infralimbic mPFC [163].
As for the evaluation of intracellular proteins (i.e., PSD-95) and NMDA glutamate receptor subunits (i.e., NR2A), it is worth noting that they are both increased in the amygdala of depressed humans, whereas they are decreased in the mPFC [164]. In addition, chronic variable stress reduces histone deacetylases-5 in the CeA of male rats [165], similar to that caused by chronic social defeat stress in mice [166]; by contrast, histone deacetylase does not seem to change in the mPFC after acute or chronic stress [167, 168]. In brief, it seems that the amygdala is definitely involved in the generation of depression, because of the biochemical and morphological changes induced by chronic stress, although there is scarce evidence of its involvement in the antidepressant effects of ketamine. Interestingly, many of the BDNF related or unrelated changes induced by chronic stress in the amygdala are opposite to those seen the hippocampus and in the mPFC.
The BNST is a complex limbic structure that plays a crucial role in the regulation of fear, anxiety and aversion [169, 170, 171], in the integration of stress response [172, 173, 174, 175] as well as in the acquisition of emotions and motivated behaviour [176] (Fig. 7). Consequently, since diminished motivation is an essential feature of depression [28], it is likely that it is extensively involved both in developing and sustaining depression as well as in the mechanism of action of StAD and ketamine [19, 24, 177]. The BNST’s role in anxiety and depression is also evidenced by a serotonergic innervation of the BNST [178, 179]; interestingly, the availability of serotonin reuptake sites in the BNST is positively correlated with individual differences in anxiety behaviour [180]. The BNST receives glutamatergic projections from the mPFC, from the hippocampus and the basomedial amygdale, whereas GABAergic inputs arise from central and medial amygdaloid nuclei [181] (Fig. 8). The noradrenergic innervation of the BNST arises from the A2 region of the nucleus of the solitary tract and the A1 region of the caudal brainstem, with a minor projection from the locus coeruleus [182, 183]. These inputs are involved in stress response [184] and in arousal [185]. In particular, the BNST may act as a node for signal integration and coordination for appropriate homeostatic physiological and behavioural responses, a function which is likely reinforced by more than a dozen neuropeptides co-expressed in BNST neurons [175]. Hence, the insufficient resilience capacity of the BNST in handling long-duration threats associated with chronic stress can be a crucial occurrence in the development of depression.
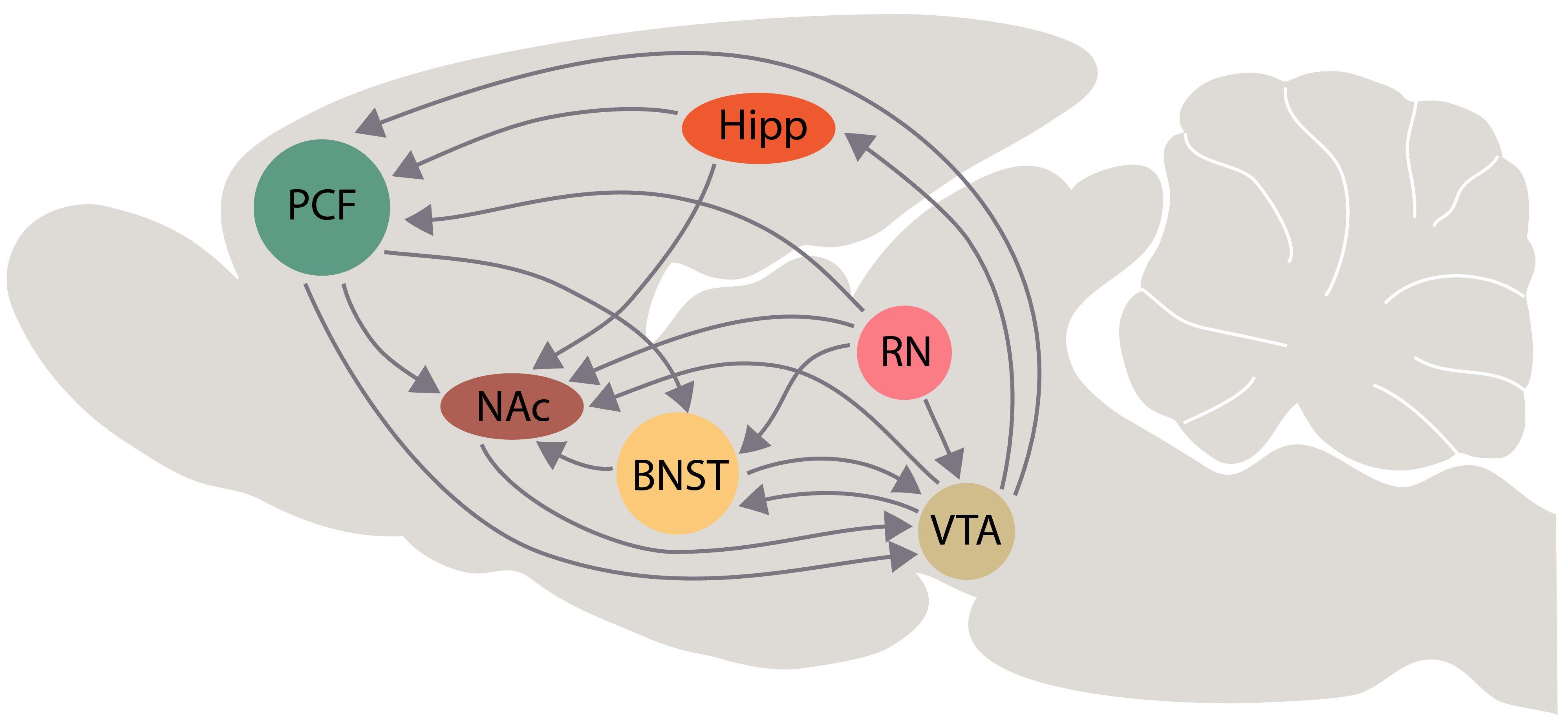 Fig. 7.
Fig. 7.Schematic representation of interactions among the bed nucleus of stria terminalis (BNST) and the brain areas that are involved in the “primary circuit of depression” according with our hypothesis. Some interactions of dorsal and median raphe with these brain areas are represented schematically. Other abbreviations: PFC, prefrontal cortex; Hipp, hippocampus; NAc, nucleus accumbens; VTA, ventral tegmental area; RN, raphe nuclei.
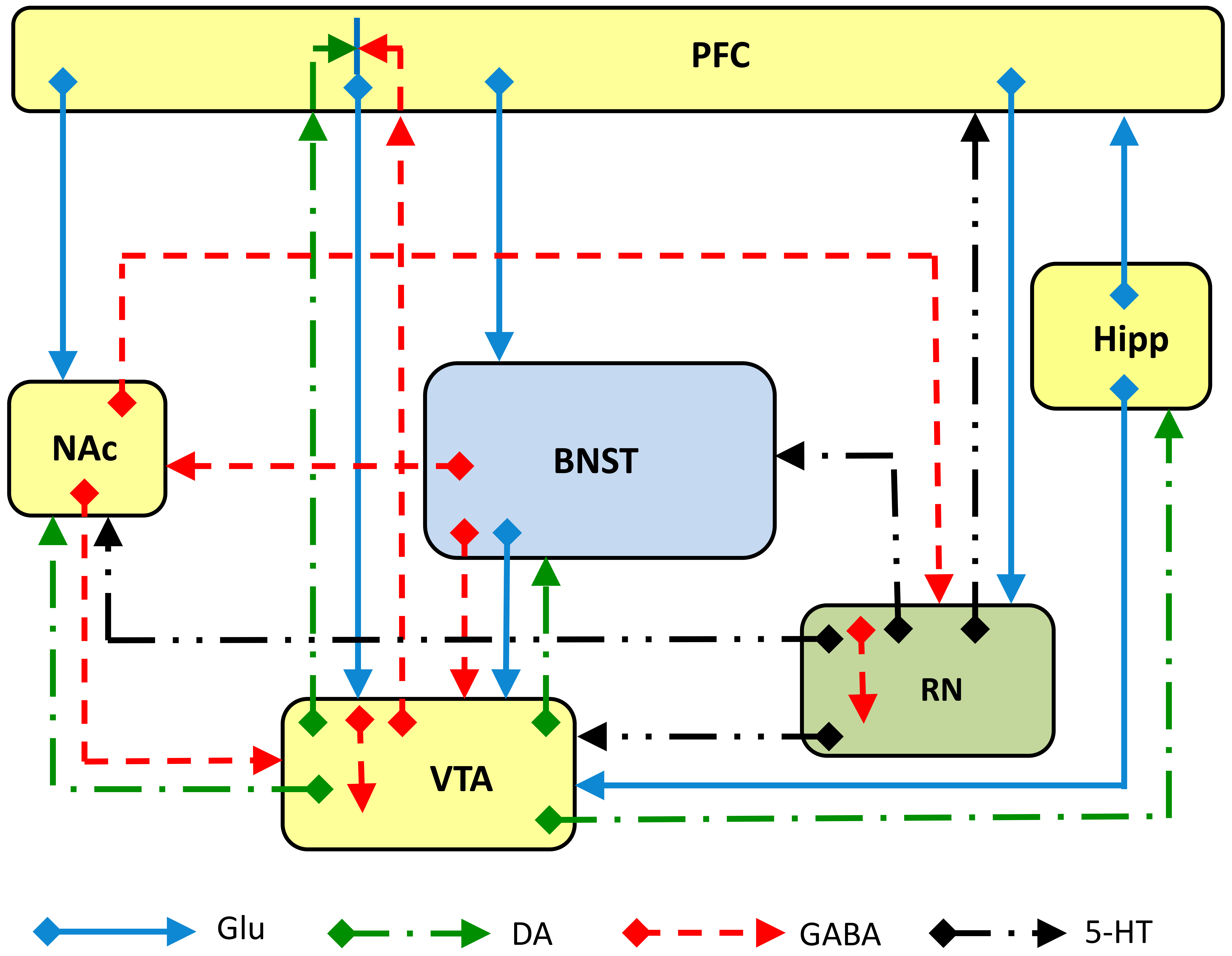 Fig. 8.
Fig. 8.Schematic representation of glutamate (Glu), GABA, dopamine (DA) and serotonin (5-HT) inputs and outputs among the bed nucleus of stria terminalis (BNST) and the brain areas involved in the “primary circuit of depression”. Some inputs and outputs of dorsal and median raphe with these brain areas are represented schematically. Other abbreviations: PFC, prefrontal cortex; Hipp, hippocampus; NAc, nucleus accumbens; VTA, ventral tegmental area; RN, raphe nuclei.
In addition, the BNST receives a dense dopaminergic innervation [186] that clearly involves the circuits that mediate reward appraisal [187, 188] and in stress-induced relapses of drug-seeking [189], thereby suggesting that drug addiction shares common neurocircuitry with depression. Part of this neurocircuitry is the BNST output to the VTA; in fact, it has been shown that GABAergic efferent from the BNST forms symmetrical synapses on VTA neurons, which are mostly GABAergic neurons [190]. The activation of this output, mediated by glutamate innervation in the BNST is likely to disinhibit VTA dopaminergic neurons; in addition, dopamine and norepinephrine projections on the BNST innervate CRF neurons that in turn innervate glutamatergic afferents on BNST projecting neurons to VTA neurons [190, 191]. Although it was previously reported that the electrical stimulation of BNST can produce both excitatory and inhibitory responses in VTA neurons in vivo [192], it was recently reported that inhibitory synaptic transmissions to BNST neurons projecting to VTA are enhanced in rats exposed to chronic stress [193].
Significantly, the 16 unique and complex sub-regions of the BNST [194, 195], and the multiple inputs from many limbic regions, mostly glutamatergic (e.g., basomedial and central amygdala, hippocampus, mPFC) but also GABAergic (from central and medial amygdaloyd nucleus [181]) and its multiple outputs (e.g., VTA, lateral hypothalamus, central nucleus of the amygdala, periacqueductal grey, parabrachial nucleus) [196, 197], does not allow us to predict whether or not ketamine, through its direct interaction with the glutamatergic transmission in the BNST, provides a substantial contribution to the manifestation of its rapid and sustained antidepressant effects. On the other hand, from the direct interactions of BNST with brain areas involved in anhedonia or in depression symptoms in general, it might be presumed that the BNST plays a central role in depression aetiology, as well as in the mechanism of action of antidepressants. This hypothesis is supported by the observation (albeit in a pilot study only) that patients affected by TRD could benefit from deep brain stimulation of the BNST [198]. Unlike other brain areas involved in depression, there have been very few investigations concerning the assessment of the BDNF expression in the BNST. In particular, what has been observed is that early-life stress alters affective behaviours in adult mice through the activation of CRF-BDNF signalling in the oval bed nucleus, which indicates enhanced neuronal plasticity [199].
We have previously observed that StAD [177] and ketamine [24] shared the ability to increase dopamine and noradrenaline extracellular concentration in the BNST; interestingly, the SSRIs citalopram or fluoxetine and the selective blockers of norepinephrine reuptake such as desipramine or reboxetine and ketamine (i.e., 10–40 mg/kg i.p.) similarly increased the output of norepinephrine and dopamine. These data have enabled us to hypothesize that a converging pathway of activation of catecholamine transmission in the BNST could well play a determining role in the effect of antidepressants, although it is hard to predict whether this effect is due to a local action or to the involvement of a complex and integrated circuit of depression.
This review has discussed the role of several brain areas, namely the mPFC, hippocampus, NAc, VTA, amygdala, LHb and the BNST, in the behavioural expression of AnMD or in the occurrence of depression in humans and in the antidepressant effects of ketamine. In particular, the evaluation of the effect of ketamine in reversing the changes in BDNF expression and release, induced by chronic stress, gives further credence to the role of BDNF in depression and in ketamine’s antidepressant effects [33, 49, 200]. A considerable amount of data has been accumulated in investigating either monoamine transmission’s role in depression [201], together with biochemical markers, transmitters and receptors in specific brain areas of stressed rodents used as AnMD [202, 203]; the limited contribution of brain images of depressed patients [204, 205], or post-mortem brain analysis of brains of suicidal subjects [83, 84, 85, 86, 206], has not really contributed to further our understanding of the aetiology of depression [207]. However, it has been frequently reported that the alteration of one specific parameter in one specific brain area is sufficient to trigger depression-linked behavioural changes [69, 76, 94, 100, 142]. Are the various existing rodent models of depression too limited to represent such a complex disorder? Or is depression simply a pathological alteration that cannot be triggered by an alteration of one single brain area or one single biochemical parameter?
Approval for the clinical use of esketamine, and the broad experimental data on ketamine’s effects and mechanism of action in AnMD has made it viable to identify glutamatergic transmission as being a key element in depression; it has also given research a further step forward in understanding the aetiology of depression. Notably, the specific change of BDNF levels observed in different brain areas of AnMD [63, 64] suggests that the response of the brain to stress challenges is a loss of connectivity, synaptic density and strength in specific brain areas such as the hippocampus and the mPFC, while in others such as the amygdale, the NAc and possibly the BNST, the increase in BDNF levels would suggest the activation and strengthening of unhealthy circuitry.
Several reports suggest that alterations and their reversal in just one of these areas could be sufficient to trigger and reverse the onset of depression. However, if we consider the strict interconnections between and among these areas and the role of glutamate in these connections, it can be assumed that a continuous flow of communication among these brain areas is necessary and sufficient for the functioning of a healthy brain. The manifestation of depression symptoms, on the other hand, can be viewed as the result of a failure of brain circuits to handle new environmental challenges. We can thus identify the mPFC, the hippocampus and the NAc/VTA as being essential components of a brain circuitry that plays a pivotal role in the expression of depression and in turn in the therapeutic effects of antidepressants; we might call it the “primary circuit of depression”. Also noteworthy is the fact that ketamine rapidly increases the BDNF and VEGF in the mPFC as well as in the hippocampus; in the latter area ketamine also increases neurogenesis [57]; Indeed, hippocampal neurogenesis is also increased by StAD through adrenergic or serotoninergic pathways, specifically involving the cAMP-PKA-CREB-BDNF pathway [208], thus providing further proof of the pivotal role of the hippocampus in depression, even though the precise role of hippocampal neurogenesis in healthy humans has not yet been comprehensively clarified [81, 209]. We have previously described the critical importance of changes that occur in other brain areas involved in depression, such as the amygdala, the LHb and the BNST (including its close connection with the paraventricular nucleus of the hypothalamus); therefore, we believe that their connections with the stations of the primary circuit of depression are of fundamental importance in mediating the flow of inputs generated by stressful environmental changes. We also believe that a healthy circuit of depression can handle these challenges whereas an inadequate resilience of this circuit can trigger the appearance of depression symptoms. Unfortunately, the limited data on the effect of ketamine on BDNF expression and release in the amygdala, the LHb and in the BNST has not enabled us to properly evaluate their role in depression and in the therapeutic action of ketamine.
To sum up, we believe that further investigations on the potential role of ketamine in reversing the molecular and circuit alteration induced by chronic stress or other biochemical challenges in specific brain area of AnMD will provide further insights required to explain the workings of brain circuits involved in the behavioural expression of AnMD. Additionally, the clinical data that will be gained thanks to the FDA approval of esketamine to be used in the treatment of depression, will soon make it possible to ascertain whether or not ketamine can indeed be a cure for depression, at least in a significant number of patients. At the same time, approval will make it easier to characterize the potential risk of its abuse.
AMPA,
EC conceptualized, wrote and revised the full manuscript. AC contributed substantially to the revision of the full manuscript.
Not applicable.
The authors acknowledge the invaluable artwork of Archt. Elisabetta Carboni, for the preparation of the figures.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.