











 , Miha Kocar 2, Andreja Eberlinc 2, Barbara Haber 1, Tomaz Velnar 1
, Miha Kocar 2, Andreja Eberlinc 2, Barbara Haber 1, Tomaz Velnar 11 Paediatric Neurosurgery Unit, Clinical Department of Neurosurgery, University Medical Centre Ljubljana, 1000 Ljubljana, Slovenia
2 Clinical Department of Maxillofacial and Oral Surgery, University Medical Centre Ljubljana, 1000 Ljubljana, Slovenia
Academic Editors: Francesco Nicita and Rafael Franco
Abstract
Background: Craniosynostosis is a rare congenital disease of the skull. They arise when one or more cranial sutures ossify prematurely. This causes an obstruction to normal brain growth and leads to specific deformations of the skull, which may result in intracranial hypertension and cognitive delay. Materials and methods: We have retrospectively analysed all children treated at the Unit of paediatric neurosurgery of the University Medical Centre Ljubljana between June 2015 and September 2020. The following items have been recorded: affected suture, underlying syndromic condition, hydrocephalus, Chiari malformation, raised intracranial pressure, age at surgery, surgical technique, need for multiple operations and surgical complications. Results: During the study period, 71 children have been treated for craniosynostosis. The median postoperative follow-up was 31 months. There were: 54.9% sagittal, 25.3% metopic, 14.0% unicoronal, 1.4% bicoronal and 1.4% unilateral lambdoid craniosynostosis. Multiple sutures were affected in 2.8% cases. 7.0% of the cases were syndromic. Overall, 74 surgical procedures have been performed: frontoorbital advancement represented 40.5% of them; biparietal remodelling 32.4%: total cranial vault remodelling 22.9%; posterior distraction 2.7%; posterior expansion 1.3%. Median age at surgery was 12.8 months. Conclusions: The treatment of craniosynostosis is surgical and requires a multidisciplinary approach, with expertise in plastic and reconstructive surgery, maxillofacial surgery and neurosurgery. The aim of surgical treatment is to release the constrictive and deformative effect that the synostosis has on skull growth. This requires a remodelling of the neurocranium and, if necessary, of the viscerocranium. Beyond aesthetic purposes, the primary aim of surgical treatment is to permit a normal development of the brain.
Keywords
- Craniosynostosis
- Intracranial hypertension
- Apneas
- Hydrocephalus
- Chiari malformation
Craniosynostosis is a rare congenital disease which occurs when one or more cranial sutures ossify prematurely [1, 2]. As a result, the child’s skull is unable to grow normally and the rapidly growing brain expands in the directions of lower resistance. The resulting head deformities are characteristic for each involved suture. Scaphocephaly (Fig. 1) is caused by a craniosynostosis of the sagittal suture and is characterised by an elongated head shape [2, 3]. Trigonocephaly (Fig. 2) is caused by a premature ossification of the metopic suture and is characterised by a triangular forehead shape [4]. Anterior plagiocephaly (Fig. 3) is caused by unilateral coronal craniosynostosis and its features are a marked asymmetry of the skull, with an upright ipsilateral forehead, protrusion of the contralateral forehead and deviation of the nasal root [5]. Brachycephaly (Fig. 4) results from the synostosis of bilateral coronal sutures and its features are a reduced antero-posterior head circumference, with flattening of the occiput and forehead [6, 7]. Last, posterior plagiocephaly (Fig. 5) is the rarest craniosynostosis and it involves one single lambdoid suture, causing an ipsilateral flattening of the occiput, with protrusion of the ipsilateral mastoid and of the contralateral occiput [2, 6, 7].
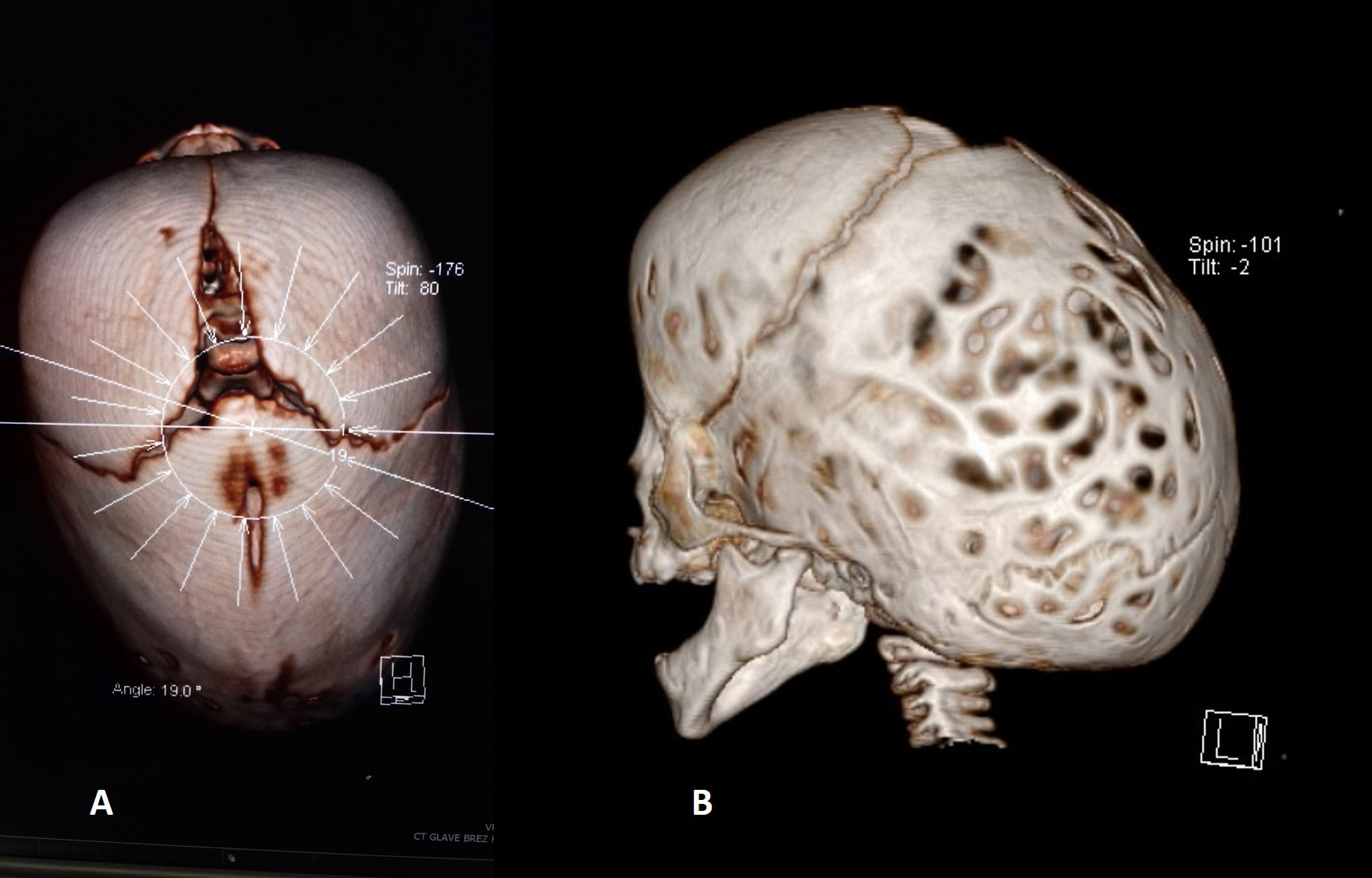 Fig. 1.
Fig. 1.CT appearance of two cases of sagittal suture synostosis. In these cases, the head is characterised by a prominent frontal bulge (A) and an elongated and narrow (B) shape.
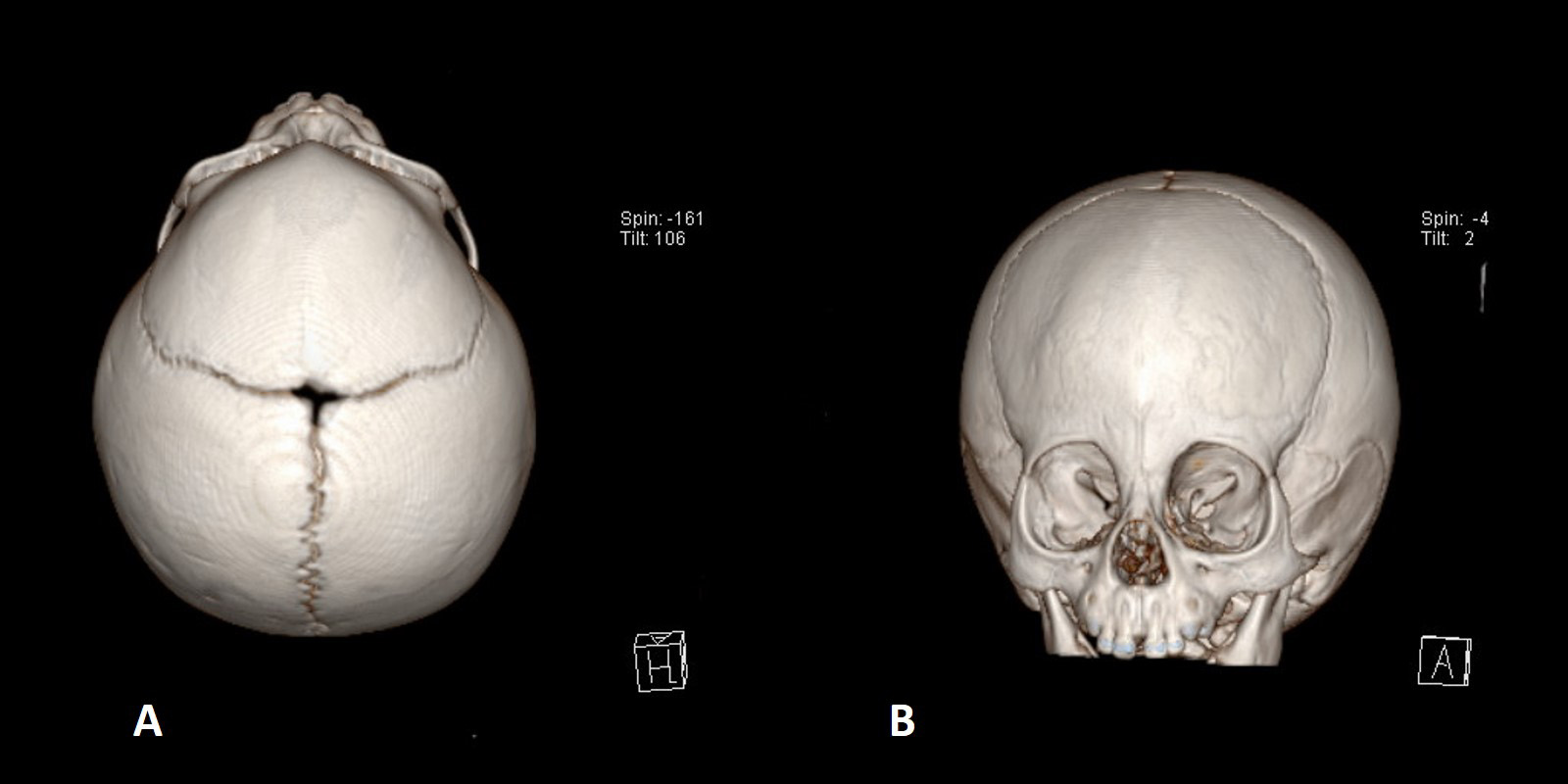 Fig. 2.
Fig. 2.CT appearance of craniosynostosis of the metopic suture: the characteristics are a triangular shape of the forehead (A) and hypotelorism (B) (reduced distance between the inner orbital angles).
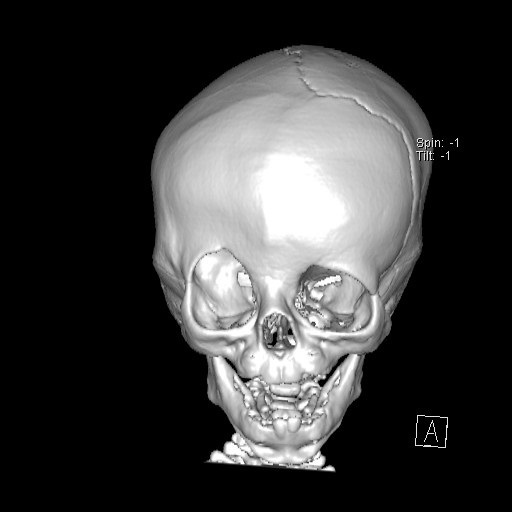 Fig. 3.
Fig. 3.CT appearance of unilateral craniosynostosis of the coronal suture: the head becomes markedly asymmetrical in these cases. The orbits, nasal root and facial axis are also distorted.
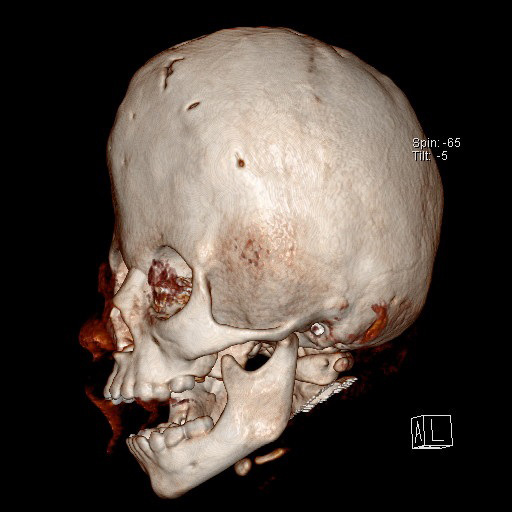 Fig. 4.
Fig. 4.Brachycephaly is caused by bicoronal craniosynostosis. Characteristic features are a short anteroposterior axis of the head, retraction of the supraorbital arch and frontal bulging.
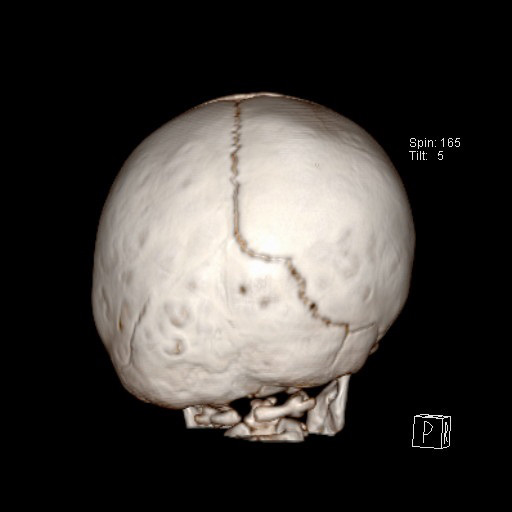 Fig. 5.
Fig. 5.Unilateral lambdoid craniosynostosis is the rarest and results in asymmetry of the entire cranial vault and retraction of the ipsilateral ear.
Syndromic craniosynostosis are more complex conditions where multiple sutures are usually involved. Apert, Pfeiffer and Crouzon syndromes are encountered most frequently. These syndromes are often associated with raised intracranial pressure (ICP), hydrocephalus and Chiari malformation [8, 9, 10, 11]. Obstructive (associated with maxillary hypoplasia and upper airway stenosis) or central (associated with Chiari malformation) apneas, exophthalmos, syndactyly and other limb malformations may also be present [8, 9, 10].
The aim of the surgical treatment is to release the neurocranium and to allow a normal brain growth. In the vast majority of non-syndromic craniosynostoses, a single skull remodelling procedure is sufficient. There are several surgical techniques, which must be correctly applied to different clinical conditions. The frontoorbital advancement (FOA) is used for metopic and coronal craniosynostosis [12, 13, 14] and the posterior vault expansions for lambdoid craniosynostosis [6, 7]. Biparietal or total cranial vault remodelling techniques are used in scaphocephaly [3, 15, 16]. In syndromic cases, Chiari malformation, intracranial hypertension, hydrocephalus and central or peripheral apneas are often present and must be considered during the planning of the correct treatment [8, 9, 10]. These complex clinical conditions need to be resolved with a number of interventions performed in a correct time sequence. Obstacles to cranial growth vary according to the syndrome and the treatment must be adapted to these differences.
The aim of this article is to review our experience with the treatment of syndromic and non-syndromic craniosynostosis between the years 2015 and 2020. The series reflects the everyday activity of a middle volume centre, which serves as the referential craniofacial centre for all children in Slovenia affected by craniosynostosis. Treatment strategies and results are shown and can serve as a basis of comparison for surgeons dealing with these cranial malformations worldwide.
All children included in the study had the diagnosis of craniosynostosis established in the period from June 2015 to September 2020 and have been surgically treated at the Unit of paediatric neurosurgery of our Institution. The following parameters were included: affected suture, underlying syndromic condition, hydrocephalus, Chiari malformation, elevated ICP, age at surgery, surgical technique, need for multiple operations, and complications of surgical treatment. All children included in the study underwent a clinical and ophthalmological examination before surgery. The diagnosis has been confirmed in all cases by imaging (ultrasound and CT).
The surgical techniques varied according to the affected cranial suture. For scaphocephaly treatment, we used a biparietal remodelling according to Renier’s technique [3]. In this technique, the parietal bones are extensively remodelled, allowing the brain to grow freely in the lateral directions and to reshape the skull. To achieve correction of the frontal bossing, we introduced the total cranial vault remodelling technique in 2018 (Fig. 6), in which not only the parietal, but also the frontal region is remodelled.
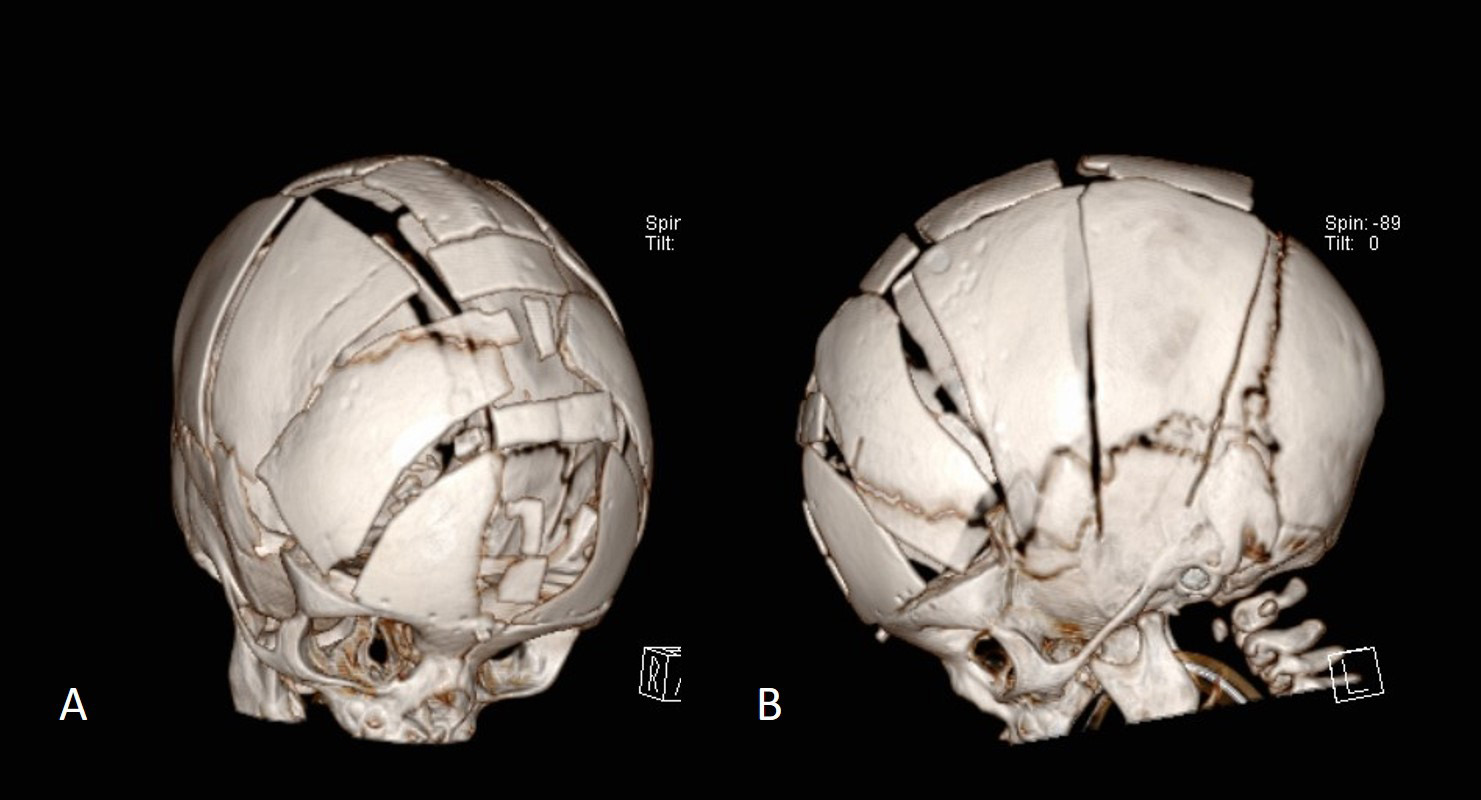 Fig. 6.
Fig. 6.Frontal (A) and lateral (B) view of a CT after total cranial vault remodelling for scaphocephaly. We do not use osteosynthetic material in this procedure because we want the growing brain itself to give shape to the released skull.
For the correction of trigonocephaly and anterior plagiocephaly we used the FOA technique [12, 13, 14]. In this procedure, a wide bifrontal flap and supraorbital bone arch are both elevated, remodelled and implanted with resorbable plates (Fig. 7). In trigonocephaly the aim is to create an upright, straight forehead and correct the hypotelorism. In anterior plagiocephaly the aim is to restore the symmetry between the left and right halves of the skull, which is usually difficult to achieve (Fig. 8).
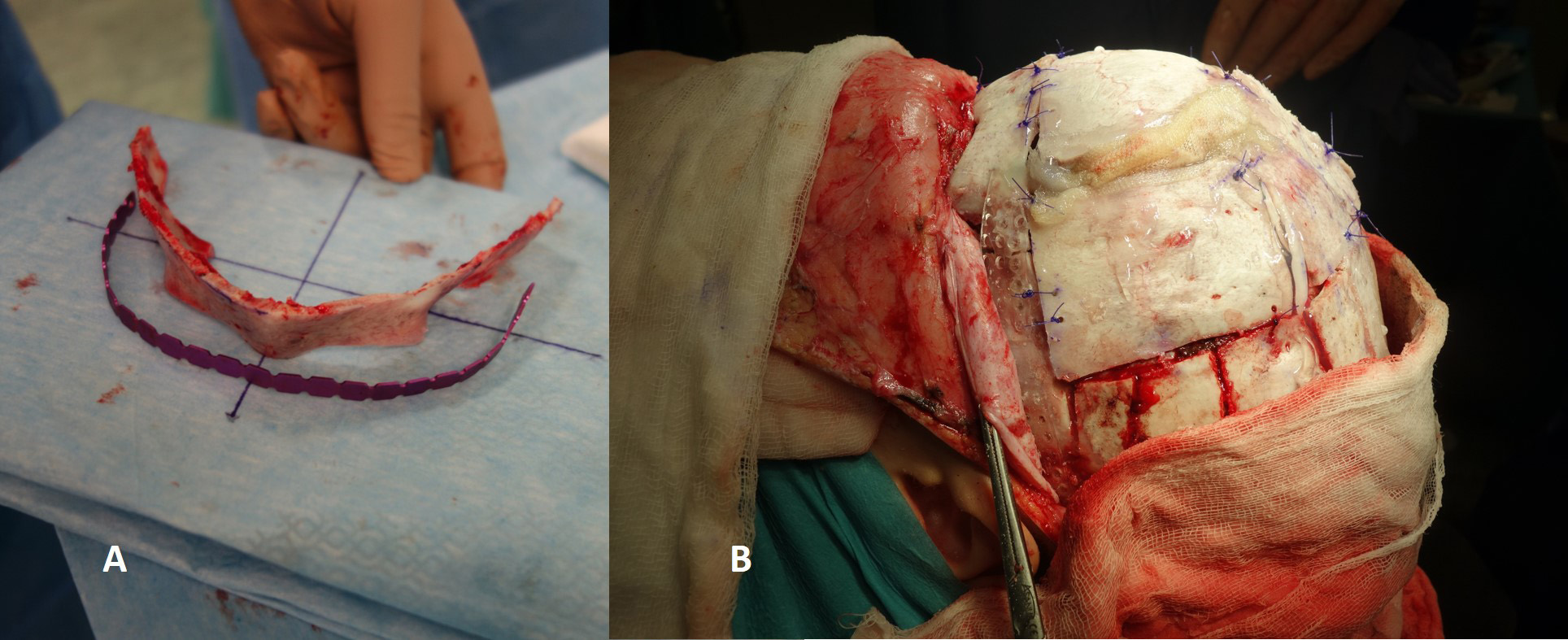 Fig. 7.
Fig. 7.In frontoorbital lengthening, the supraorbital arch (A) and the forehead must be reshaped and then fixed in the new, appropriate position with osteosynthetic material (B).
 Fig. 8.
Fig. 8.To correct trigonocephaly and anterior plagiocephaly, the entire anterior part of the cranial vault must be remodelled. This procedure is called frontoorbital remodelling.
For correction of brachycephaly we used a surgical expansion of the anterior cranial vault. In non-syndromic cases and in the absence of signs of raised ICP, only a FOA has been performed.
In the case of posterior plagiocephaly, an expansion of the temporo-parieto-occipital part of the skull on the affected side has been performed. The expansion was performed by bone remodelling and fixation.
In syndromic cases, the expansion was done in two stages, first with a posterior expansion and then, one year later, with a FOA. The posterior expansion was performed by means of posterior distraction [6, 8, 9]. In this technique, external distractors were placed on the edges of the parieto-occipital craniotomy and the distraction preceded by 1 mm per day until the final goal of lengthening the posterior part of the skull by 25–30 mm (Fig. 9) [6]. 3–6 months after completion of distraction, when ossification of the dura between the craniotomy edges has occurred, the distractors were removed. Beyond this, in syndromic cases the multiple associated pathological conditions (Chiari malformation, elevated ICP), hydrocephalus, apneas, exophthalmos) needed to be followed and eventually surgically treated.
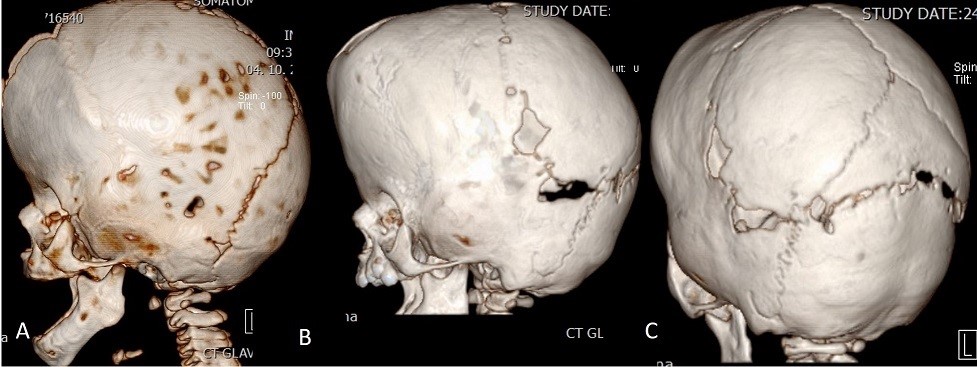 Fig. 9.
Fig. 9.The figure shows the situation before (A) and after (B, C) posterior osteogenic distraction. In (B) and (C), the distractors have already been removed and the posterior osteotomy line, which has been partially ossified, is visible and has been widened by approximately 20 mm with the help of the distractors.
The parents have given their written informed consent to be included in the study. After multidisciplinary discussion and expert recommendation, it was advised that this retrospective archive study does not need ethics approval.
From June 2015 to September 2020, 71 children with craniosynostosis have been surgically treated. Considering that the number of births is around 20000 per year in Slovenia and that all patients have been treated in our Institution, the incidence of craniosynostosis in Slovenia was 1:1500 births.
The median follow-up was of 2 years and 7 months. The patients were male in 56 cases (78.9%) and female in 15 cases (21.1%). The general demographic data of the children are summarised in Table 1. The affected sutures were: sagittal in 39 cases (54.9%), metopic in 18 cases (25.3%), coronal unilateral in 10 cases (14.0%), coronal bilateral in one case (1.4%), lambdoid unilateral in one case (1.4%). Multiple sutures were affected in two cases (2.8%), both cases were syndromic. There were 5 syndromic cases (7.0%): two Apert syndromes, one Muenke syndrome, one fronto-naso-orbital syndrome and one PTEN Hamartoma syndrome.
| Number | % | ||
| Affected suture | |||
| Sagittal | 39 | 54.9 | |
| Metopic | 18 | 25.3 | |
| Coronary unilateral | 10 | 14 | |
| Coronary bilateral | 1 | 1.4 | |
| Lambdoid | 1 | 1.4 | |
| Multiple | 2 | 2.8 | |
| Total | 71 | 100 | |
| Syndromic disease | 5 | 7 | |
| Elevated intracranial pressure | 7 | 9.8 | |
| Hydrocephalus | 1 | 1.4 | |
| Chiari malformation | 2 | 2.8 | |
In all cases, treatment was surgical. The details of the treatment modalities are shown in Table 2. A total of 74 procedures have been performed. The procedure were: FOA in 30 cases (40.5%), biparietal remodelling in 24 cases (32.4%), total cranial vault remodelling in 17 cases (22.9%), posterior distraction in two cases (2.7%) and posterior expansion in one case (1.3%).
| Num. | % | Mean age | ||
| Surgical technique | ||||
| FOA | 30 | 40.5 | 16.4 m | |
| Biparietal remodelling | 24 | 32.4 | 5.1 m | |
| Total vault remodelling | 17 | 22.9 | ||
| Posterior distraction | 2 | 2.7 | 11.3 m | |
| Posterior expansion | 1 | 1.3 | ||
| Total | 74 | 100 | ||
| 2nd surgery needed | 3 | 4 | 23.6 m | |
| Mean age in all surgeries | 12.8 m | |||
The median age at the first procedure was 12.8 months. Children with scaphocephaly were operated at a median age of 5.1 months, but in two cases, the procedure was performed in older children (10 and 8 years). In children with synostosis of the metopic or coronal sutures, the FOA was performed at a median age of 16.4 months. The three posterior expansions have been performed at a median age of 11.3 months.
A second operation was performed in three cases during the study period. One was a FOA after the posterior distraction in a child with Apert syndrome. The second was a total cranial vault remodelling performed due to persistent scaphocephalic head shape after a primary biparietal remodelling. In the third case, a biparietal expansion was performed with the aim of enlarging the intracranial volume in a child with progressive papilledema after the correction of trigonocephaly.
In 7 cases, signs of increased intracranial pressure have been recognized. Some patients had more than one of them. These were: papilledema in 2 cases, “copper beaten” appearance of the skull on X-ray (Fig. 10) in three cases and intraoperative finding of a gyral impressions on the inner table of the skull in 5 cases. Breathing problems did not occur in any case and no evidence of central apneas was present. Exophthalmos was present in a mild form in two cases of Apert syndrome, but ophthalmological problems did not occur. Hydrocephalus was present in one case, but not in association with craniosynostosis per se, but in the form of posthaemorrhagic hydrocephalus in a premature infant with an isolated sagittal craniosynostosis. Chiari malformation was present asymptomatically in two cases and craniocervical decompression was not indicated. Surgical complications occurred in two cases. In one there was a wound healing problem and the dehiscence needed to be covered with a rotational skin flap. In the other case the complication was represented by a postoperative Gram-negative sepsis. This states the overall frequency of complications in our series at 2.8%.
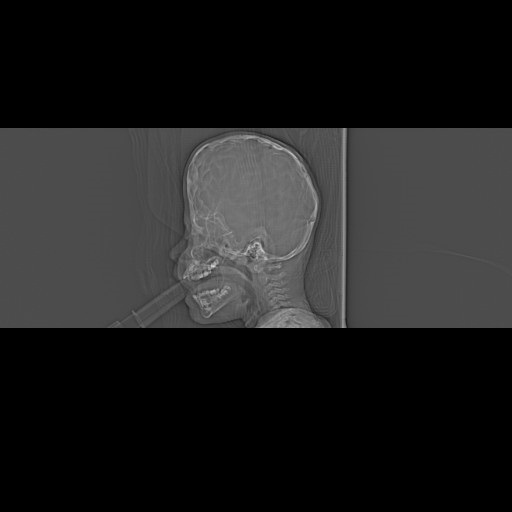 Fig. 10.
Fig. 10.The copper beaten appearance of the skull on X-Ray images is a sign of elevated ICP and can be associated with craniosynostosis. It is caused by the pressure of the cortical gyri on the cranial lamina interna.
The vertebrate skull consists of the neurocranium, which surrounds and protects the brain, and the viscerocranium, which forms the face [1, 2, 8, 9]. In children up to the age of three years, the brain can grow while remaining adequately protected mechanically. This is possible because the sutures at the junctions of the cranial bones permit a gradual deposition of bone tissue, acting as ossification centres of the skull and allowing a smooth brain development.
Craniosynostosis are defined as prematurely ossified cranial sutures that no longer function as ossification centres. At that point the growth of the skull stops and brain growth progresses in a parallel direction to the affected suture, no longer in a perpendicular direction, deforming the skull. The clinical picture varies according to the ossified suture [2].
Craniosynostosis may be isolated or associated with syndromic conditions. The incidence is estimated at 1/2100 births [1, 2]. Non-syndromic craniosynostosis account for 80–95% of all cases and syndromic craniosynostosis for 5–20% [1, 2, 9, 10]. The results of our study are consistent with this data, with an annual incidence of 1:1500 births and 7% of syndromic cases.
In terms of morphological phenotypes, sagittal synostosis is the most common (40–55%), followed by metopic synostosis (15–30%), unilateral and bilateral coronal synostosis (15–20%) [1, 2]. Lambdoid synostosis is the rarest (0–5%). Craniosynostosis of two or more sutures is rare and mostly occurs in syndromic cases [9, 10]. In our series, the distribution was similar (Table 1). Also, the predominance of male patients (78.9%) was consistent with the literature [1, 2].
In the initial phase the diagnosis of craniosynostosis is clinical and is based on the recognition of the specific deformities of the head. Confirmation of the diagnosis is ultimately radiological, by means of ultrasound, which can confirm suture ossification and head CT with 3-D reconstruction, which clearly shows the ossified suture. Ocular fundus examination is indicated to rule out the possibility of elevated ICP. MRI is rarely indicated, because many cerebral anomalies are already identifiable by head ultrasound and CT. Genetic investigations are always necessary when an underlying syndromic condition is suspected.
The treatment of craniosynostosis is exclusively surgical. The constrictive and deformative potential of the synostosis on the growth of the head, must be relieved through a surgical intervention (Table 3). This requires remodelling of the bones of the neurocranium and, if necessary, of the viscerocranium. The procedure must be carried out not only for aesthetic purposes, but above all, to allow a normal brain growth. The age at which surgical release of craniosynostosis is performed depends on the affected suture, on the eventual underlying syndrome and on the possible associated pathological findings (elevated ICP, hydrocephalus, Chiari malformation, apneas). In the vast majority of non-syndromic craniosynostosis, a single skull remodelling procedure is sufficient, but it has to be performed in ideal intervals of time. For scaphocephaly the ideal age for surgery in our experience was between three and six months of age [3, 15, 16]. By performing the procedure so early, the brain has enough growth potential to shape the skull on its own. For this reason, no osteosynthetic material is used in these operations [3]. On the contrary, FOA is ideally performed after 12 months of age, when brain growth is mostly complete [12, 13, 14]. In our experience, an intervention before nine months of age was not indicated because the growing brain would encounter a rigid, immobile barrier represented by the osteosynthetic material.
| Diagnoses | 1st operation | 2nd operation | Associated intracranial pathologies |
| Scaphocephaly | Biparietal or total cranial vault remodelling | / | / |
| Trigonocephaly | FOA | / | / |
| Anterior plagiocephaly | FOA | / | / |
| Brachycephaly | Posterior expansion | FOA | / |
| or distraction | |||
| Posterior plagiocephaly | Posterior expansion | FOA | / |
| or distraction | |||
| Syndromic craniosynostosis | Posterior expansion | FOA | CHIARI |
| or distraction | HYDROCEPHALUS | ||
| / | ELEVATED ICP |
The posterior cranial vault expansion (or distraction) should be performed early, between four and eight months of age, in both non-syndromic and syndromic cases [6, 7], to limit the increase of ICP, the development of turricephaly and the tonsillar herniation.
We used open surgical approaches in all non-syndromic cases of our cohort, despite the interest for endoscopic procedures is growing worldwide and the results are described as similar to open approaches [17]. Indeed, the endoscopy permits lesser blood loss and shorter surgical times, but the remodelling is not as wide as in open surgery and the postoperative remodelling is achieved by means of the corrective helmet [17, 18].
In syndromic craniosynostosis, the multiple associated pathological conditions (Chiari malformation, intracranial hypertension, hydrocephalus, apneas) [10] need to be resolved with a number of interventions performed in the correct time sequence [9]. The basic principles of treatment are to perform initially a posterior cranial expansion or distraction, followed by a FOA, performed one year later [9]. The third step is represented by a midface advancement through Lefort 3 osteotomies [9], which is performed later during childhood, between five and ten years of age. In the presence of severe neonatal peripheral apneas and significant exophthalmos, it is advisable to perform an early single procedure, the monobloc advancement, in which the forehead and midface are advanced or eventually gradually distracted [19, 20]. When indicated, this procedure is ideally done between 12 and 24 months of age. In our series no child needed a monobloc advancement.
Children with craniosynostosis, in particular those with an underlying syndrome, must be regularly followed throughout their childhood, to rule out the occurrence of intracranial hypertension and hydrocephalus. The follow-up requires regular controls of the ocular fundus and regular CT and MR examinations. If hydrocephalus occurs, a treatment without the insertion of a ventriculo-peritoneal shunt should be attempted [8, 9]. In this sense, cranial expansion techniques and eventually a craniocervical decompression can lower the venous hypertension, bringing to improvement of CSF resorption. If these attempts fail, endoscopic ventriculostomy should be performed, leaving ventriculo-peritoneal shunt as a last option, because any CSF drainage brings to intracranial hypotension, which interferes with favourable postoperative skull remodelling. The presence of apneas must be ruled out with regular polysomnographies. In case of early and severe peripheral obstructive apneas, a tracheotomy should be performed in the first weeks or months of life. If nocturnal polysomnography demonstrates the presence of central apnoea and if MRI confirms the presence of Chiari malformation, it is recommended to perform the decompression of the craniocervical junction.
Surgery for craniosynostosis is complex. According to the literature, complications occur in 2–8% of cases [21]. Mortality is recorded below 1% in all contemporary series [21]. Possible complications are: wound dehiscence, infection, subcutaneous haematoma, dura injury and CSF fistula. In our series, only one case of wound dehiscence (1.4%) and one case of systemic infection (1.4%) were reported. The mortality rate was 0%.
The evaluation of the success of craniosynostosis surgery is based on the aesthetic and on the functional outcome. Aesthetic outcome is difficult to assess objectively and one of the most useful methods for this purpose is the cranial index (skull width/skull length), which ranges between 76% and 78% in healthy children [22]. The closer the cranial index approaches these values after the procedure, the more favourable the aesthetic outcome. In our series the cranial measurements have not been routinely applied to preoperative and postoperative conditions and therefore these results are inconclusive, we have not presented them in this article.
Early aesthetical results are often confusing and do not reflect the definitive outcome. In anterior plagiocephaly, for example, the aim is to restore the symmetry between left and right halves of the skull, which is usually difficult to achieve (Fig. 9). Even if the early aesthetic result appears sometimes unsatisfying, the release of the craniosynostosis creates the conditions for a further gradual correction of the asymmetry in the years following the procedure [13, 14].
The cognitive outcome is assessed on the basis of neuropsychological testing performed throughout the child’s development. Despite correct surgical treatment and in the absence of raised ICP and hydrocephalus, up to 30–50% of children can present some forms of neurocognitive delay [23, 24, 25, 26]. These are particularly common in the development of speech, writing and reading, although the intelligence quotient is mostly within normal limits. Our relatively short follow-up did not allow us to set any conclusions regarding the neurocognitive conditions of the children included in the series.
In the period from June 2015 to September 2020, 71 children with craniosynostosis have been treated at our Institution. The incidence of craniosynostosis in Slovenia was 1:1500 births and was consistent with worldwide studies. Sagittal suture craniosynostosis was the most common, followed by metopic suture craniosynostosis and unilateral coronal synostosis. Coronal and lambdoid craniosynostosis were less common. Syndromic craniosynostosis accounted for 7%. Overall, 74 surgical procedures have been performed: frontoorbital advancement represented 40.5% of them; biparietal remodelling 32.4%: total cranial vault remodelling 22.9%; posterior distraction 2.7%; posterior expansion 1.3%. The surgical treatment of craniosynostosis is complex, but with careful preoperative planning and with the correct surgical techniques the complication rate is very low and the aesthetic results satisfying.
CSF, cerebrospinal fluid; FOA, frontootbital assessment.
PS analyzed the data. PS and TV wrote the paper. MK, AE and BH contributed by writing some parts of the paper.
The consent to participate in the study was obtained with the informed consent of all participants. The institutional review board of the University Medical Centre Ljubljana stated that this retrospective archive study does not need ethics approval.
We thank the Department of Pediatric Surgery for keeping the data about craniosynostosis patients and contributing to their well-being.
This research received no external funding.
The authors declare no conflict of interest. TV is serving as one of the Guest Editors of this journal. We declare that TV had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to FN and RF.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.