
Academic Editor: Ulises Gómez-Pinedo
The innate immune system primarily gets triggered by microbe infiltration, injury, stress, aging, and brain disorders. The hyperactivation of the innate immune system and neuroinflammatory reactions contributes to chronic age-related neurodegeneration. The mechanism for activation of the immune pathway is conserved between Drosophila melanogaster (D. melanogaster) and human being. Thus, D. melanogaster can serve as a model organism to decipher the cellular and molecular mechanism between infection and neurodegenerative diseases. In D. melanogaster, prolonged protective, excessive neuroinflammatory responses in the brain lead to neurodegeneration through antimicrobial peptides mediated neurotoxicity. The prolonged inflammation in the microglial cells helps in the progression of neurodegenerative disease. Therefore, the connection between inflammatory mechanisms in the brain and neurodegeneration pathogenesis in D. melanogaster is systematically reviewed.
Neurodegeneration is a sequel of the synaptic transmission failure and death of neuronal cells in the brain. Neurodegeneration is seen among patients suffering from neurodegenerative disorders and older adults. During neurodegeneration, autoinflammation occurs, and such a condition is referred to as “inflammaging”. Inflammation protects the host from microbe infection/injury by activating microglia and astrocytes in the central nervous system (CNS) [1]. Chronic inflammation alters tissue homeostasis and may culminate in neurotoxicity [2]. An infection in the CNS can also stimulate the local immune response by elevating the cytokine level. Such infection often results in meningitis [3], encephalitis [4], Alzheimer’s disease (AD) [5] and other neurological disorders.
Drosophila melanogaster (D. melanogaster) shares striking similarities with vertebrates in the context of neural proliferation and brain circuit formation [6]. Therefore, D. melanogaster is widely used to decipher the pathways involved in microbes and parasite infection [7, 8, 9]. The molecules and signaling pathways involved in D. melanogaster and mammalian innate immune response are evolutionarily conserved [10]. Like mammalian systems, cytokine dysregulation and neurodegeneration are also observed in D. melanogaster to respond to microbes’ infection, tissue injury, and prolonged autoinflammatory response.
D. melanogaster is the widely used model organism to explore genetics, metabolism, and physiology. The decades of research in fly genetic have unveiled various metabolic and physiological pathways conserved with higher phylum, including humans. The adaptability, cost-effective rearing, short developmental cycle, well-characterized genomic organization, and easy access to manipulation have endorsed D. melanogaster as a principal model for groundbreaking discoveries. In addition, D. melanogaster has the advantage of sharing approximately 75% of the disease-causing genes and their function with humans [11].
Host-pathogen interaction suggests conserved innate immune function across species [12]. Vertebrates possess adaptive immune responses, which overshadow innate immune response and thus pose a significant drawback of using a vertebrate model for innate immune response studies. Since D. melanogaster does not have adaptive immunity, it is more likely used to decipher the mechanism of action of innate immunity [12]. Although humans are highly evolved organisms, they still share homology with D. melanogaster to produce antimicrobial peptides (AMPs), epithelial barriers, and phagocytosis, which are used as defense mechanisms against pathogens. In D. melanogaster, the epidermis, gut, and trachea serve as the first barrier to invading pathogens. Fly hemolymph act as the second line of defense by trapping the pathogens with the protein filaments of the clotting factors [13]. The other organs of D. melanogaster involved in the immune response are the fat body, different circulating hemocytes, and malpighian tubules. The organs involved in the D. melanogaster immune response are graphically represented in Fig. 1 (Ref. [14]).
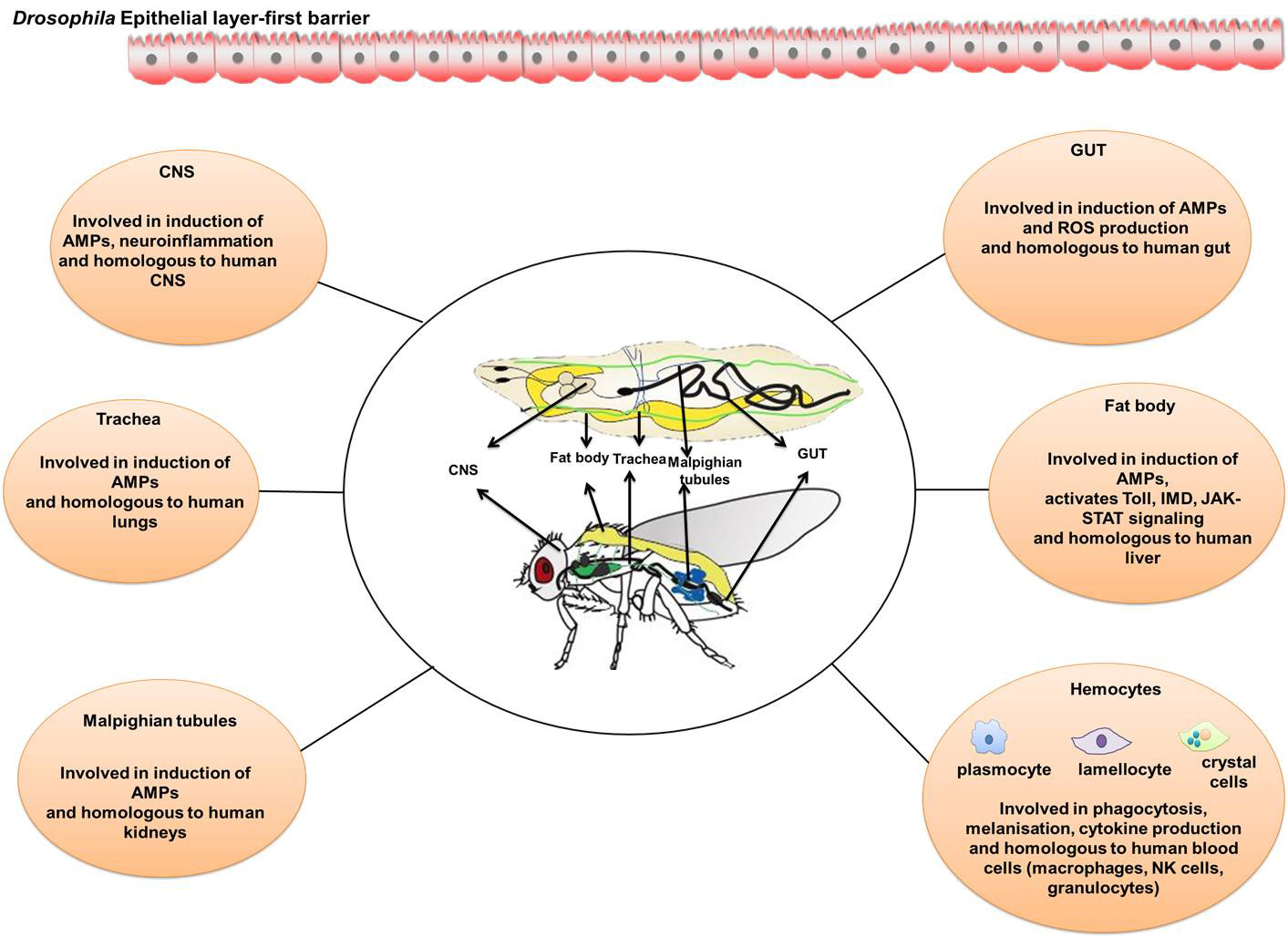 Fig. 1.
Fig. 1.Drosophila organs that are involved in the immune response. The organs of fly, which play a vital role in activating the molecules and signalling of innate immune response, have striking functional similarities with the mammals. The fly CNS, gut, trachea, fat body, malpighian tube and hemocytes mimic the human counterparts and actively participate in the triggering immune pathways, induction of AMPs and ROS, melanisation, production of cytokines and phagocytosis. The image was redrawn from Buchon et al. [14] review paper.
D. melanogaster has a multi-layered defense mechanism categorized into
(a) systemic immune response-release of NF-
In various studies, D. melanogaster is modeled to understand the immune
activation by bacteria, fungi, parasites, and viruses [8, 9, 21, 22]. D.
melanogaster Toll receptor that can evoke the immune response in fly has homolog
named Toll-like receptors in vertebrates. Also, D. melanogaster’s innate
immune system targets different classes of molecules present on the surface of
the different pathogen. For example, AMPs like drosomycin, defensin, and drosocin
respond to fungi, Gram-positive and Gram-negative bacteria, respectively [7]. The
mechanism of action of multiple signaling pathways such as JAK/STAT and AMPs
sequence is evolutionarily conserved between humans and D. melanogaster [23]. In fruit flies, stimulation of signaling cascade by pathogenic invasion
leads to activation of NF-
It has been reported that in humans, any breakdown in the innate immune system leads to several diseases, including neurological disorders [25, 26, 27]. In the mammalian system, a prolonged inflammatory response against the infection not only activates the local immune cells like microglia, and macroglia (astrocytes, pericytes and oligo-dendrocytes) [28] but also leads to infiltration of the peripheral immune cells into CNS, which results in cell/neuronal death and ultimately may result in neurodegeneration and such inflammation that occurs in response to foreign particles in the CNS is referred to as neuroinflammation.
Activated innate immune system and neuroinflammation play a vital role in the pathogenesis of neurodegeneration in mammals [29]. Besides dissimilarity between fly and mammalian brain anatomy, some crucial similarities between both structures still persists [30]. Fly CNS is formed primarily by the fusion of four ganglia namely, the sub esophageal ganglion, the protocerebrum, the deutocerebrum, and the tritocerebrum [31]. Protocerebrum, the largest ganglion covers majority of the adult fly brain and is analogous to the cerebrum of the mammals. Mushroom body, found in fly brain is associated with learning, olfactory discrimination, processing sensory inputs from olfactory and antennae lobe. This mushroom body is analogous to the mammalian hippocampus [32].
The fly glial cells are evolutionary and display some degree of morphological and functional parallelism with vertebrate microglia. Drosophila CNS does not possess oligodendrocytes but have lower proportion (10–20%) of glial cells that have functional similarity with the microglial and astroglial cells of vertebrate CNS [28, 33]. Drosophila, consist of vertebrate glial like cells and subtypes such as (i) surface, perineural, and cortex glia (Pericyte-like cells) (ii) astrocytes like glia (iii) ensheathing glia and reticular glia (microglial cells). The surface, perineural, and cortex glia form a barrier, analogous to vertebrate blood-brain-barrier (BBB), separating fly CNS from the hemolymph [34]. Fly astroglia are homologous to the mammalian astrocytes and perform variety of tasks such as metabolic, maintenance, transporting, development of dopaminergic axons, providing neurotrophic aid to fly eye and neuronal survival [35, 36, 37]. Microglia residing in mammalian CNS has a counterpart in Drosophila, namely ensheathing glia, performed a wide array of function ranging from pathogen clearance, neuronal phagocytosis, and leukocyte recruitment into the brain [38, 39]. However, other studies have defined some microglia in fly perform neurotropic and neuroprotective role similar to astrocytes and thus named as reticular glia [40, 41] (Fig. 2). Thus, both ensheathing glia and reticular glia in D. melanogaster CNS are considered as homologs on mammalian microglial cells. Microglial phenotype polarization is witnessed in the vertebrate CNS as a vital feature of innate immune system during both healthy and diseased condition. Microglia exhibits three morphological and two polarization phenotypes depending on the neurons, neighboring environment and other microglia. They were categorized as: (i) M1 microglia- round in structure and found in diseased adult CNS (ii) M2 microglia- extended process and highly ramified in morphology usually found in the healthy vertebrate CNS along neural tract and near neuropil and synapses respectively [42, 43, 44]. Drosophila also displays microglial polarization and morphologies similar to the vertebrate microglial system. Ensheathing glia are morphologically flattened cell bodies with small processes that showed striking resemblance to mammalian M1 microglia. Similarly, reticular glias are characterized by longer, ramified extensions, structurally mimicking mammalian M2 microglia in healthy CNS [38, 45].
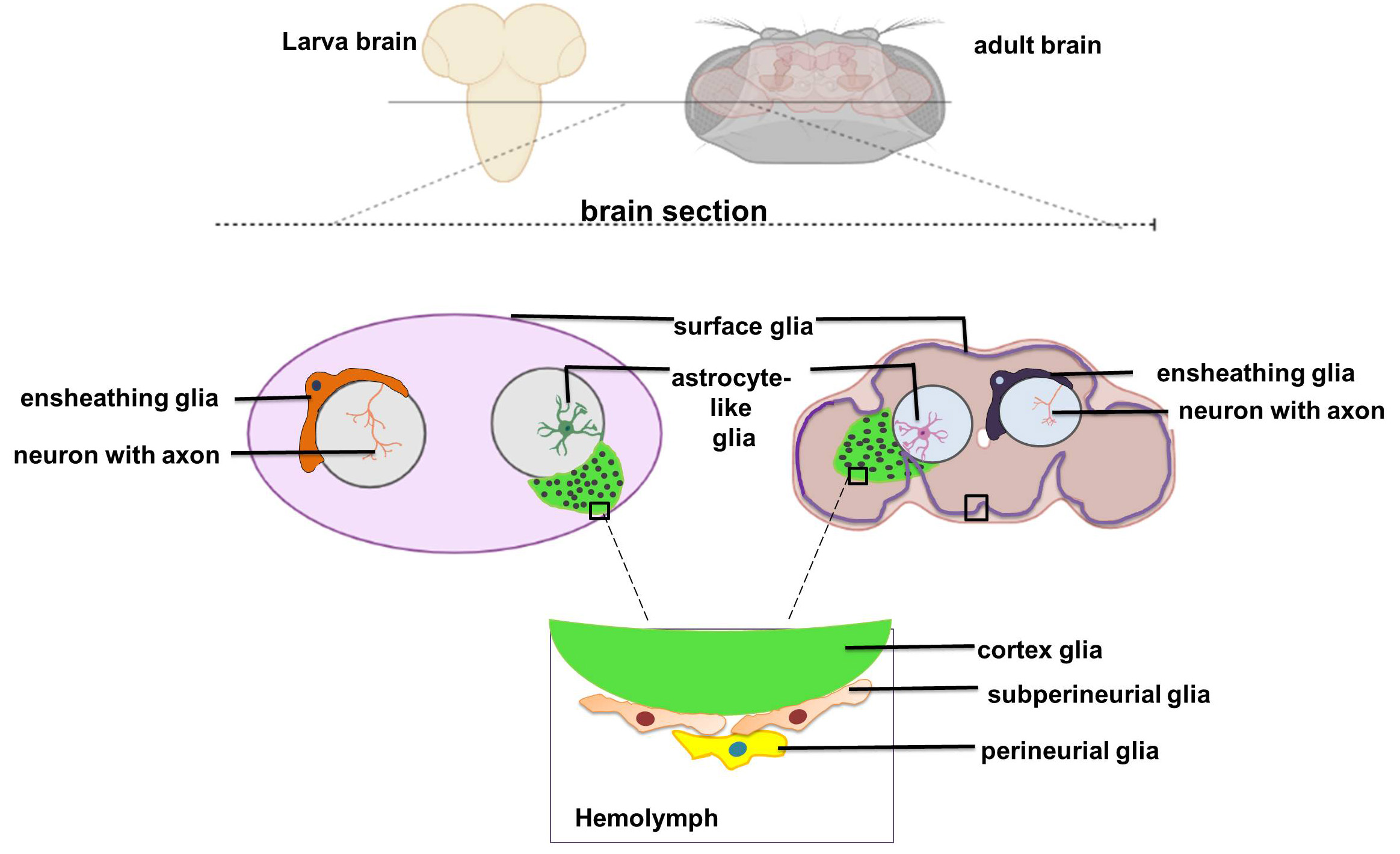 Fig. 2.
Fig. 2.Drosophila brain section showcasing the glia and subtypes. Fly CNS comprised of variety of glia. The surface glia, cortex glia, perineurial and sub-perineurial found in fly system are homologs of mammalian pericytes. Astrocyte-like cells and ensheathing glia and reticular glia display high degree conservation with mammalian astrocytes and microglia cells respectively. Drosophila glia cells display morphological and functional similarities with the mammalian system. The role of glia cells in neuroinflammation between fly and mammal is highly conserved. The image was redrawn from https://coutinhobuddlab.com/about-us/.
D. melanogaster having many striking similarities with the human immune response and glia cells, thus can be used to elucidate the host-pathogen interaction, mechanism of defense/inflammation, and also to establish a connection between the chronic infections mediated neurodegenerative disease onset/progression.
In mammals, microbial invasion is first defended by the body’s innate immune system. It serves as the first line of defense, which later activates the adaptive immunity—the second line of defense for long-term protection against pathogens [46]. Immune system activation is orchestrated by the initiation of a critical process called inflammation. Inflammation-induced by infection or injury later stimulates the production of various immune cell types and cytokines [1]. Inflammation for a short period protects the body against the infection by eradicating the pathogens, but chronic inflammatory responses have detrimental effects such as tissue/neuron cell damage. The brain was previously considered an immune-privileged organ. Still, it is now known that prolonged inflammatory response in the CNS may lead to various neurological disorders and significant mortality globally [2].
In D. melanogaster, foreign invaders such as bacteria, fungi, and
viruses can cause infection-mediated immune system activation.
Drosophila immune proteins that can recognize the bacterial components
are generally called pattern recognition receptors (PRRs) which are broadly
classified into two families: (i) the peptidoglycan recognition proteins (PGRPs)
and (ii) Gram-negative binding proteins (GNBPs) [47]. Thirteen PGRPs have been
identified in the fruit fly, out of which only three PGRPs, namely PGRP-SA,
PGRP-LC, and PGRP-LE, could recognize the invaders and increase immune
sensitivity [47]. Out of three PRRs belonging to the GNBP family in D.
melanogaster, only GNBP1 can recognize the bacterial component (LPS) and
fungal cell component (b-1, 3-glucan) when challenged by Gram-negative bacteria
and fungus [48]. The Gram-negative and Gram-positive bacterial infection
activates the Toll pathway and IMD pathway, respectively (Fig. 3). The binding of
Gram-positive bacteria or fungus initiates Toll receptor dimerization which then
recruits heterotrimeric complexes comprising Myd88 protein, Pelle, and Tube [49].
The heterotrimeric complexes mediated activation of kinases (interleukin-1
receptor (IL-1R) associated kinase (IRAK)) leads to hydrolysis of the Cactus
which in turn initiates the nuclear translocation of Dorsal and Dorsal related
immunity factor (Dif) [50]. However, both Dorsal/Dif play a crucial role in
immune response in fruit flies by activating the expression of drosomycin [50]. A Drosophila dTRAF2 (homolog of mammalian
TNF-receptor-associated factor-6 (TRAF-6) protein) interacts with Pelle to
stimulate drosomycin expression [51]. Toll is also activated in
D. melanogaster by cytokine-like protein Spaetzle (spz) [52]. IMD
signaling, provoked by Gram-negative bacteria, activates the member of
NF-
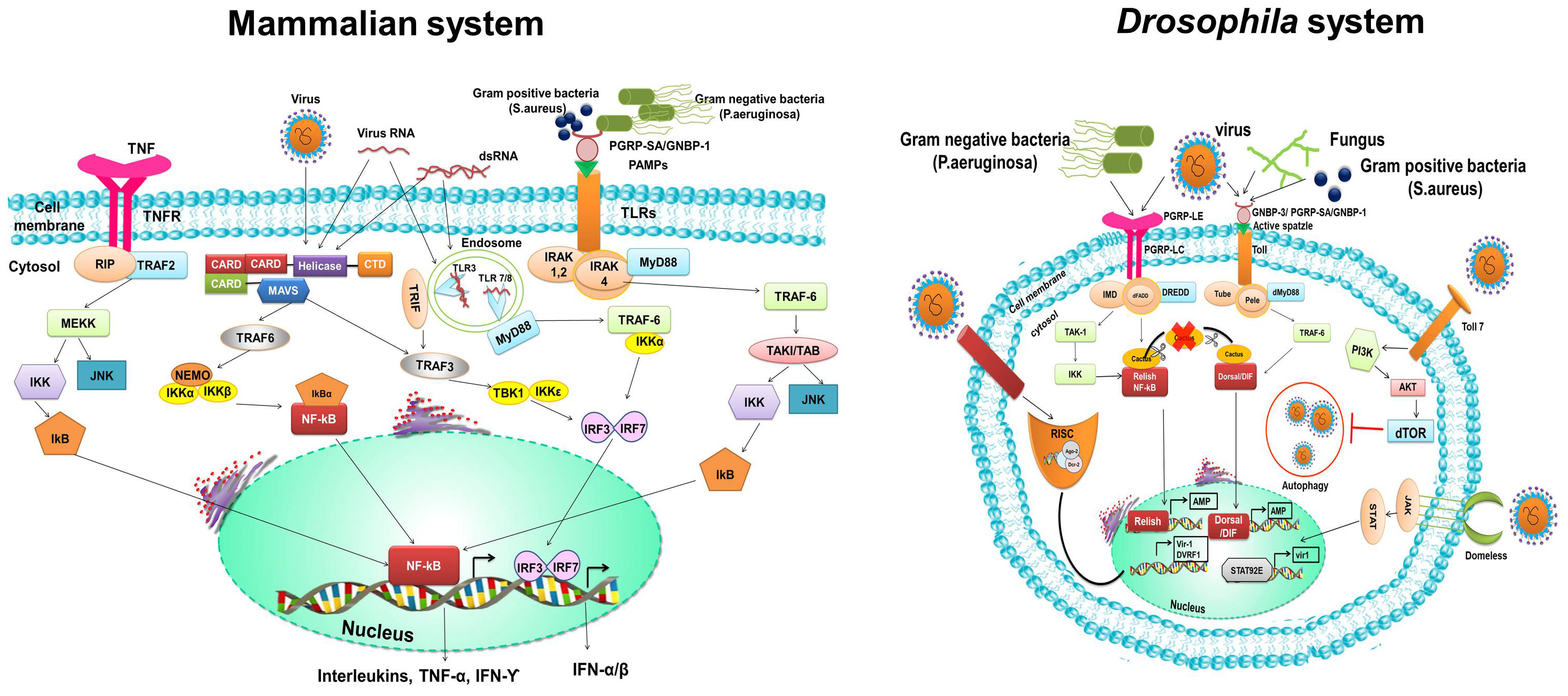 Fig. 3.
Fig. 3.Immune response upon infection. Activation of innate immune signalling such as Toll, IMD pathway and toll-7 and domeless elicited by bacterial and viral infection in Drosophila share a high degree homology with the mammalian innate immune signalling. In invertebrate, the molecules involve in innate immune signalling and mechanisms of activation of these molecules exhibit evolutionary conservation with the vertebrate except for some difference in downstream pathway of immune response. The image redrawn from Valanne et al. [55] paper.
It is well established in mammals that microbe invasion could lead to acute or prolonged inflammation in the CNS, which may later culminate in significant degeneration of specific neuronal population associated with a plethora of neurological disorders such as meningitis [3] encephalitis [56], AD [57], and PD which is presented in Table 1 (Ref. [25, 58, 59, 60, 61, 62, 63, 64, 65, 66]). Moreover, an infection caused by bacteria or the neurotrophic virus (such as the Zika virus) in the brain may lead to the onset of severe brain disorders such as schizophrenia and depression [67]. These neurotropic viruses’ replication elicits the expression of the Interferon (IFN) regulatory factors (IRFs), kappa-light-chain-enhancer nuclear factor (formed by B cell activation signaling), and the effector molecules downstream to the signaling in CNS. This immune activation causes infiltration of the microglia and astrocytes into the CNS, which recognizes the pathogen by PRRs and induces neuroinflammation [68]. If neuroinflammation continued for an extended period, it might result in neurotoxicity and neurological pathogenesis. Infection in D. melanogaster triggers a cytokine-based regulatory signal. This inflammatory response includes the production of AMPs, recruitment of hemocytes, and release of cytokines and chemokines by activated immune cells [69, 70]. These inflammatory events profoundly affect the tissue involved in the inflammatory response and neural tissues, and the animal as a whole [71]. Thus, D. melanogaster serves as an excellent model organism to delineate the role of innate immune response individually (in the absence of adaptive immune response) and inflammation in the development of neurodegenerative diseases.
| Infectious agent | Neurodegeneration | Immune response | Literature references |
| Japanese B Encephalitis | Neuronal death | Increase in pro-inflammatory mediators, iNOS, COX-2, IL-6, IL-1 |
[58] |
| Bacteroides fragilis (B. fragilis) | Sporadic Alzheimer’s disease (AD) in the brain | Generation of the inflammatory transcription factor NF- |
[25] |
| Chlamydia pneumoniae | Alzheimer’s | Enhanced cytokine levels | [59, 60] |
| Borrelia burgdorferi | Alzheimer’s, Parkinson’s disease | Higher |
[60, 61] |
| (B. burgdorferi) | Increased beta‐amyloid protein (A | ||
| Helicobacter pylori (H. pylori) | Alzheimer’s, Parkinson’s disease | Higher |
[60, 61] |
| Increased beta‐amyloid protein (A | |||
| Coronavirus | Neuro invasion, cerebral edema, neuronal degeneration, encephalitis, meningoencephalitis, acute disseminated encephalomyelitis, encephalopathy, and stroke | Excessive production of cytokines such as interleukin (IL)-1 |
[62] |
| Human herpesvirus 6 | Meningoencephalitis and leucoencephalitis, death of neurons undergoing neuronophagia | Lymphocytes and microglia activation | [63] |
| Epstein-Barr virus | Encephalopathy and acute quadriparesis with diminished reflexes, horn cell degeneration, and edema, the paralysis with diminished reflexes | EBV antibodies, IL-10 production | [64] |
| Bacterial meningitis | Neuronal loss and death, apoptosis | TLR induced microglia activation | [65] |
| Human immunodeficiency viruses (HIV) | Dementia | Activation of macrophages and migration into CNS | [66] |
The prolonged structural and functional loss of the neurons is evident when
neurodegeneration occurs in CNS. The neurodegeneration culminate in functional
and mental impairments in CNS [72]. The sources of neurodegeneration are not well
understood yet. However, one of such sources that increase the probability of
neurodegeneration is aging [73]. The neurodegeneration in the CNS could lead to
the incidence of neurodegenerative diseases such as AD, Multiple sclerosis (MS),
PD, Amyotrophic lateral sclerosis (ALS), Polyglutamine Diseases, Ataxia
Telangiectasia, Traumatic (Brain) Injury, Tauopathies, Frontotemporal Dementia,
and Progressive Supranuclear Palsy. The characteristic features of these
neurodegenerative diseases are altered and unfolded protein which leads to the
formation and aggregation of
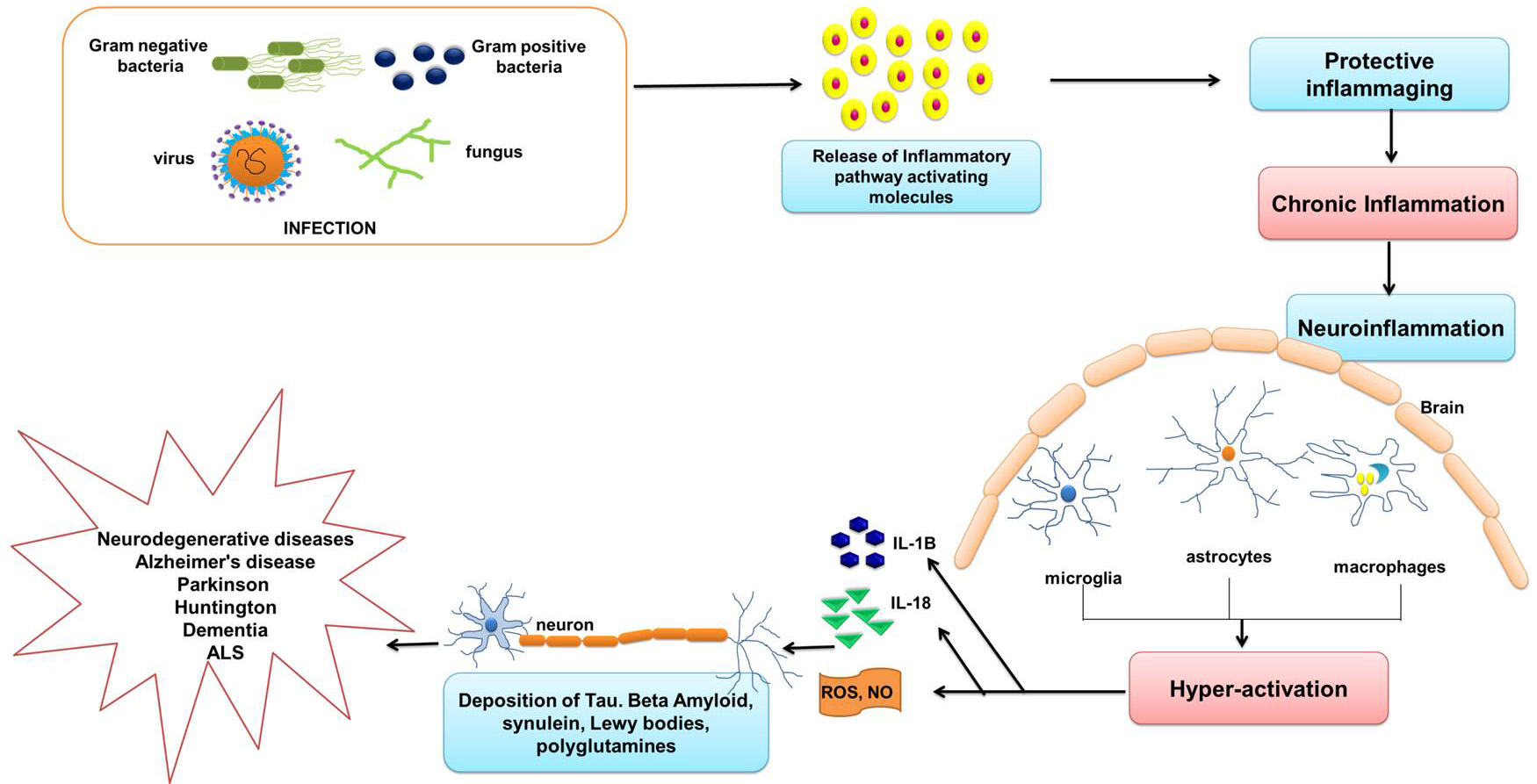 Fig. 4.
Fig. 4.Infection mediated neuroinflammation and neurodegeneration. Invasion of pathogens is a danger signal to the resting microglia. Pathogens activate the immune signalling and lead to the release of the molecules which further activates the macrophages and glia cells in the CNS. In this model, prolonged activation of the macrophages and glia cells constantly produces cytotoxic factors such as proinflammatory cytokines and ROS. These further promote damage to neurons (mainly motor neurons, hippocampal neurons, and dopaminergic) which culminate in development of neurodegenerative disorders such as AD, ALS and PD, etc. The image was redrawn from Chen et al. [75].
AD is a neuropsychiatric ailment; found most frequently in people above 65
years, has affected millions of people worldwide. The World Health Organization
(WHO) [76] has described the disease as a cognitive impairment that gradually
affects behavior, mood, memory, and learning [77, 78]. Neurofibrillary tangles
(NFTs) are derived from the paired helical filaments (PHFs), which are the
hyperphosphorylated forms of the axonal protein “tau”. Proteases cleave the
senile plaques (SPs), which are derivatives of the amyloid precursor protein
(A
In mammals, the microglia present in the brain is the key factor that links
neuroinflammation and neurodegeneration. Microglia can be activated by several
factors such as infectious agents (bacteria, viruses, fungi), advanced glycation
end products (AGE) receptors, A
The oligomer A
It has been observed that the patients with AD have an upregulated
IL-1-NF-
On the other hand, in D. melanogaster, the A
Previous reports suggest that deposits of A
PD is the second most common neurodegenerative disease after AD, accounting for
approximately 2% of the population. Patients suffering from PD have difficulty
in movement (bradykinesia), dementia such as LB dementia (DLB), multiple-system
atrophy (MSA), and neuropsychiatric dysfunction, and rest tremor, instability in
body posture, rigid movements, hallucinations, hypotension, and constipation
[96, 97, 98]. PD is a multifactorial disease caused by various factors such as the
deposition of
PD can occur from multiple damage signals such as endogenous proteins,
pathogens, toxins or toxic agents, dying neuron products, and aging. It has been
reported that the vascular channels connecting the brain to the skull employing
meninges might direct the microbes or the non-cerebral immune cells to enter into
the brain region [100] to evoke the damage signals. The inflammatory cascade
activated by damage signals causes synaptic impairment, leading to the
penetration of more inflammatory molecules to the mid-brain and triggers more
microglia production, increasing ROS and eicosanoid generation dopaminergic
neurons death that ultimately results in PD associated neurovascular dysfunctions
[101]. PD patients are found with an enhanced inflammatory response such as
activation of the peripheral lymphocytes and releasing the pro-inflammatory serum
cytokines IL-2, IL-6, IFN-
PD pathogenesis caused by infections often mediates neuroimmunomodulation.
Neuroimmunomodulation includes an increase in aggregation of substrates such as
adenosine triphosphate,
In PD-affected people, the role of lysosomal dysfunction is well studied. The
lysosomal autophagy system (LAS) and the ubiquitin-proteasome system maintain the
proper amount of intracellular
D. melanogaster also exhibits complex behaviors such as aggression,
grooming, courtship, learning, conditioning to fear, and locomotory activities
such as climbing, flying, and walking [115] which gets, impaired by PD
pathogenesis. PD mutant flies are observed with loss of DA neurons and defective
motor activity. The paraquat-induced Drosophila PD model is witnessed
with activated signaling factor of Toll, IMD and c-Jun N-terminal kinase (JNK)
[116]. Maitra et al. have also demonstrated the crucial role of Relish
to rescue mobility defects and neuronal loss in flies. Infection-mediated Relish
activation via IMD signaling leads to the induction of NF-
Mammalian mitochondria featuring multiple functions can be a central driver of diseases owing to their dysfunction caused by aging, disease (autoimmune diseases, cancer, metabolic disorders, and neurodegeneration), exposure to toxicants of the environment, and pathogenic infection. Dysfunction of mitochondria results in impaired oxidative phosphorylation (OXPHOS) and metabolism, accumulation of unfolded proteins, loss of membrane potential, and enhanced ROS generation. It regulates a wide range of cellular processes and houses the molecules involved in the antiviral and inflammasome signaling and endogenous damage-associated molecular patterns (DAMPs). These mitochondrial DAMPs engage the innate sensors/PRRs to activate pro-inflammatory and type I IFN responses [119]. Numerous studies have revealed the association of mitochondrial dysfunction with the pathogenesis of PD in humans [120]. Mammalian mitochondria also possess many sophisticated systems that participate in the proper functioning of the protein and maintain the cell’s structural integrity. These systems, comprising AAA proteases, the ubiquitin-proteasome system, mitochondrial-derived vesicles (MDVs) and mitophagy, and fission/fusion regulatory system, taken together is referred to as mitochondrial quality control (MQC) [121]. In the past few years, the role of PTEN-induced putative kinase 1 (PINK1) and PRKN in the activation of the MQC machinery in response to mitochondrial dysfunction has been extensively studied [122, 123]. Under unpleasant conditions (mitochondrial damage, mutagenic stress, and proteotoxicity), the intermembrane transport of PINK1’s N-terminus from the outer mitochondrial membrane (OMM) to the inner mitochondrial membrane (IMM) is impaired, resulting in PINK1 accumulation on the OMM. The accumulated PINK1 triggers autophosphorylation, which facilitates kinase activation and promotes binding to the Parkin and ubiquitin [124, 125]. Now activated Parkin facilitates the formation of the ubiquitin chains and attracts more Parkin to the mitochondria, thereby amplifying the damage detecting signals received by PINK1 [126]. The recruited Parkin leads to ubiquitination of many cytosolic targets such as Parkin Interacting Substrate (PARIS, ZNF746) and AIMP2, whose accumulation may cause neurotoxicity and cell death of nigral DA neurons [127, 128].
Moreover, mutations in these genes are linked to the autosomal recessive forms of PD in mammals [129, 130]. Autosomal recessive juvenile parkinsonism (ARJP) results from the dysfunctional LAS and E3 ubiquitin-ligase system produced from a mutation in the parkin gene [131]. However, the mechanism of the development of ARJP pathogenesis is still not clearly understood. Loss of function PINK1/Parkin MQC machinery may alter the correlation between CNS and peripheral immune system and evoke an adaptive immune response against mitochondrial proteins. Thus, compromised PINK1/Parkin MQC engages the peripheral immune system in an attack against CNS. Loss of Parkin impairs the generation of mitochondrial-derived vesicles (MDVs) required for bactericidal activity, resulting in the defect in clearance of infection causing chronic infection and enhanced cytokine production [132].
In D. melanogaster, PINK1/Parkin shows similarities in pathways to maintain mitochondrial fidelity with mammals but differs in its localization [133]. Greene et al. [134] studies demonstrated dysfunctional mitochondria and damaged flight muscle phenotypes in the D. melanogaster model mutant for the parkin gene. Moreover, these parkin mutant flies have also shown higher oxidative stress levels and altered levels of parkin and oxidative stress genes. Lastly, when they induced the innate immunity genes in the parkin mutant flies, the cell cycle and the endoplasmic reticulum stress regulatory pathways are altered, resulting in the inflammation-mediated ARJP pathogenesis. The PINK1/Parkin KO mutant flies are observed with a decline in male sterility and life span, impaired locomotor activity, mitochondrial dysfunction in muscle and brain, and defective DA neuron morphology [135, 136]. Loss of function of Parkin in Drosophila leads to the reduced motor activity, shrinkage of DA neurons, and decline in the level of tyrosine hydroxylase [137]. Flies with PINK1 mutation have similar phenotypic defects (impaired locomotion, defective DA neurons, and reduced life span) as that of parkin mutant flies [136]. Additionally, loss of function of PINK1 causes defective thorax phenotype in young flies (3 days old) and leads to age-dependent DA neurons deficiency in PPL1 cluster in 30 days old flies [138].
ALS is a fatal neurodegenerative disorder evident in approximately 2 people per 100,000 and usually causes the death of the patients within 3–5 years [139, 140, 141]. Men are slightly more prone to the disease than women. ALS patients exhibit weakness of limbs and are thus diagnosed with upper and lower body motor neurons defect [142]. Along with motor disorders, ALS patients are also diagnosed with dementia, sensory abnormalities, and autonomic dysfunction [143, 144, 145]. Many factors play a role in the pathogenesis of ALS, such as environmental hazards, immunological disorders, and inflammation. ALS is identified with a genetic mutation in the superoxide dismutase 1 (SOD1) [146]. The mutation in SOD1 covers only 20% of the total identified ALS cases suggesting probability of mutations in other genes. Modification of the gene encoding transactive response DNA-binding protein-43 (TDP-43), i.e., TARDBP and mutation in the gene encoding sarcoma fusion/translocation in liposarcoma, is also responsible for ALS [147].
Meissner et al. [148] reported that the endocytosed mutated SOD1, when
relocated to the cytosol, acts as a danger signal, leading to activation of the
caspase1 in the mammalian SOD1 mutant microglial cells. Then the activated
caspase-1 activates IL-1 IL-1
D. melanogaster acts as a good model to investigate the TDP-43 neurotoxicity and related disease. Zhan et al. [151] has reported the vital contribution of the leucine kinase Wallenda (Wnd) and p38 and JNK (downstream components) in the TDP-43 mediated neurotoxicity, and thus any genetic variation in the Wnd expression or its antagonist may improve the fly life-span by canceling the negative effect of TDP-43. Furthermore, overexpression p38b or loss-of-function of Basket (Bsk), a homolog of JNK, has exhibited a shorter fly life span and increased TDP-43-associated lethality [151]. In a nutshell, the cytoprotective role and cytotoxic effect of the JNK signaling and p38 signaling, respectively, have been well-studied in the D. melanogaster model. However, the conversation of the same in humans still needs to be explored.
The Polyglutamine (poly Q) diseases are of 9 types, namely; Huntington’s disease (HD), dentatorubral-pallidoluysian atrophy, spinobulbar muscular atrophy, and spinocerebellar ataxias types 1, 2, 3, 6, 7, and 17; featuring CAG-trinucleotide repeats expansion along with the open reading frame (ORF) in the corresponding genes [152]. These groups of genetic diseases are marked with the deposition of the multiple inclusion bodies comprising polyglutamine-rich proteins (insoluble) that can bring about neurodegeneration in different brain regions [153]. HD is a well-studied autosomal polyQ disease featuring CAG repeats in the Huntingtin (HTT) gene. Genetic abnormality caused due to mutated HTT subsequently causes progressive atrophy of the cortex and striatum [89].
The samples collected from plasma and affected regions of HD patient’s brains
exhibit increased TNF levels hinting at the role of inflammation (microglia cells
recruitment and proinflammatory cells activation) in producing an unpleasant
physiological state in the brain and disease progression [154]. Elevated levels
of IL-1 IL-1
To elucidate onset/progression of polyQ diseases mainly HD and spinocerebellar ataxia type 3 (SCA3), D. melanogaster is recently used as a model organism. Transgenic flies’ mutant for transgenes encoding for ATXN3 and HTT are generated to investigate cellular and molecular mechanisms of the disease [152]. Jackson et al. have reported adult fly retinal degeneration in the SCA3 and HTT mutant flies [152, 156, 157]. Thus, D. melanogaster retina can be used as a model to decipher the link between the polyQ mediated neurodegeneration in the retina and pathogenesis SCA3 and HD mutants [152]. Evolutionary conserved innate immune (Toll and IMD) pathways in D. melanogaster play a pathological role in developing polyQ-mediated neurodegeneration. These immune signalling in D. melanogaster are involved in the inhibition of the Yorkie (Yki) a transcriptional coactivator of the Hippo pathway, by accumulated polyQ, leading to enhanced AMPs expression and the onset of neurodegeneration. Altogether, this validates an interrelation between immune pathway and neurodegeneration. Dubey and Tapadia have reported that Yki in humans can negatively regulate the innate immune pathways and reduce the polyQ neurotoxicity either by overexpressing Yki or by triggering cyclin E/bantam mediated cell proliferation in the affected cells [158].
Shieh and colleagues characterized 160 genes responsible for differential expression signatures, including genes associated with innate immune responses in the fly model having CAG repeat-associated neurodegeneration. The authors have also explored a correlation between inflammation and polyQ mediated neurodegeneration as they observed overexpression of Hsp70 and AMPs, especially metchnikowin in CAG repeat fly model [159]. Involvement of Hsp70 is also found in the human polyQ and other human neurodegenerative disease suppression [160]. These works suggest that the mechanism of inflammation-mediated neuro-pathogenesis is highly conserved between flies and humans. Shieh et al. [159] also identified genes, namely; DpId, Orb2, and Tpr2 in flies which can modify the Ataxin-3 polyQ protein toxicity and CAG-repeat RNA-based pathogenicity. Altogether, these reports suggest that the genetic modifiers identified in flies can be targeted in the mammalian model to establish a relationship between RNA/protein toxicity mediated polyQ pathogenicity [157].
Mutation in the gene Ataxia telangiectasia mutated (ATM) (that encodes for a protein kinase responsible for maintaining genomic integrity) results in a recessive autosomal neurodegenerative disease called Ataxia telangiectasia (A_T), observed with clinical features such as cerebellar ataxia, immunodeficiency, occulocutaneous telangiectasia, and sensitivity towards radiation [161]. An impaired ATM gene function produces chromosomal instability, leading to dysfunctional immune response and thus activates systemic inflammatory signaling that participates in the onset of neurodegeneration, speeding up the aging process, tampering the cardiovascular system, and developing autoimmune disease similar to the pathological features of A_T [162].
McGrath-Morrow et al. [163] had identified more than 300 genes expressed in the A_T patients and showed the association of some of the identified genes with the immune/inflammatory pathway when their peripheral blood mononuclear cells were compared with the healthy control (without A_T disease). The authors have also found an increase in the level of IL-8 in serum, uncontrolled/prolonged inflammatory response, and free activation of the innate immune system, indicating the role of inflammation in the A_T pathogenesis; which is more likely to develop in malignancy/death in 4–6 years: yet to be discovered.
Petersen et al. [164] have used flies, with mutated ATM genes, as a
model to decipher the mechanism of inflammation-mediated A_T-related
neurodegeneration in the brain. Subsequently, the authors have modified the
amino-acid in the C-terminal region of the D. melanogaster ATM gene to
inhibit the protein kinase activity, and this impaired ATM in the glial cells
contributes significantly to sustained immunological responses, which in turn
impairs glial mobility or cause glial/neuronal cell death [165]. The authors have
also reported that the regulatory molecules (NF-
Traumatic brain injury (TBI) is a consequence of the primary or secondary injuries in the head due to external mechanical forces, which subsequently trigger functional defects in the individual’s behavior, cognition, and physical responses. The severity of the secondary injuries depends on how the host cellular and molecular function responds to the external mechanical stress on the brain primarily [166]. TBI is categorized into subcategories [167], such as (i) based on skull and dura condition; (ii) closed head injuries (no damage observed in dura and skull); (iii) penetrating injuries (damage observed in both dura and skull); (iv) based on the clinical characteristics (v) length and state of consciousness; (vi) incidence of amnesia and (vii) neurological disorders.
Csuka et al. [168] had reported that dysregulation of the innate immune responses via cytokines can stimulate secondary injuries in humans, indicating the role of inflammation in the pathogenesis of TBI. On the contrary, some studies on TBI patients have also reported the beneficial role of cytokines to rescue the neural system [169]. TBI patients are observed with an elevated level of TNF in the CSF which indirectly affects the patient negatively. Thus, targeting TNF serves as a potential therapeutic for TBI treatment [170].
Recently, as D. melanogaster is modeled in various studies related to inflammation and neurodegeneration, Katzenberger et al. [166] developed ‘high-impact trauma’. This adjustable device primarily imposes closed-head TBI conditions in flies. These close head-TBI fly models are found with elevated expression of genes such as ‘metchnikowin’ and ‘spz’ of Toll and IMD pathways, respectively. Consequently, hyper-activated immune pathway has also been reported in the TBI fly model, inducing damage in the neural system similar to that of aged flies undergoing neurodegeneration [171].
Unrestrained AMPs expression leads to neurodegeneration mediated vacuolar lesion
formation in the neuropil (area of nervous system comprising dendrites,
unmyelinated axons, and glial cells) of the human brain [115]. The vacuolar
lesions analogous to the brain are identified in the nervous system (neuropil
area) of the flies used as TBI model. These lesions in the TBI
Drosophila model vary from as small as 1.0
Neuroinflammation is one of the major aspects of the chronic innate immune
response in the CNS. Infiltration of foreign invaders or neuronal injury provokes
the activation of pro-inflammatory molecules secreted from the host immune system
and triggers the accumulation of microglial cells, causing a deregulated brain
tissue homeostasis, which exaggerates into neurotoxicity or neurodegeneration.
The glial cell-derived prolonged expression of proinflammatory cytokines, or AMPs
(in D. melanogaster) in the CNS cause elevated deposition of the endogenous
non-infectious ligands like tau,
CNS, central nervous system; AMPs, antimicrobial peptides; ROS, reactive oxygen
species; IRFs, Interferon (IFN) regulatory factors; PGRPs, peptidoglycan
recognition proteins; GNBPs, Gram-negative binding proteins; IL-1R, interleukin-1
receptor; DIF, Dorsal-related immunity factor; TRAF-6,
TNF-receptor-associated factor-6; spz, Spaetzle; dFADD, Fas-associated Death
Domain; DREDD, death-related ced-3/NEDD2-like protein; dTAK1, Drosophila
transforming growth factor activated kinase 1; NO, nitric oxide; GNBP-3,
Gram-negative binding protein-3; GNBP-1, Gram-negative binding protein-1; RNAi,
RNA interference; DptB, Diptericin B; AD, Alzheimer’s disease; MS, multiple
sclerosis; PD, Parkinson’s disease; ALS, amyotrophic lateral sclerosis; WHO,
World Health Organization; NFTs, Neurofibrillary tangles; PHFs, paired helical
filaments; SPs, senile plaques; A
NN did the literature review and wrote the paper. MM did drafting and critical revision of the article.
Not applicable.
NN is thankful to DST/INSPIRE Fellowship/2016/IF160257 for financial support.
This study was supported by grants from the Inspire Fellowship (DST/INSPIRE Fellowship/2016/IF160257).
The authors declare no conflict of interest.