
Astrocytes are the most abundant glia in the central nervous system that play a significant role in disease. Recently, it roles of synaptic plasticity in neuropathological damages have been questioned whether the structural and functional plasticity of synapses contributes to the pathogenesis of Parkinson’s disease. The regulation of synaptic plasticity by astrocytes has also been widely researched based on astrocytes regulate synaptic plasticity by releasing Adenosine triphosphate, glutamate, and D-serine. We discuss the possible role of astrocytes in the regulation of synaptic plasticity, which may provide a new direction to Parkinson’s disease treatment.
Parkinson’s disease (PD) is a complex neurodegenerative disease [1, 2, 3] with
an unknown etiology. The disease is characterized by several pathological
manifestations and symptoms that affect bodily systems [4]. Patients with the
disease have irreversible functional alterations in the nerve cells, resulting in
selective, progressive loss of dopaminergic neurons in the nigrostriatal pathway
[1, 2, 5]. The gradual dopaminergic denervation leads to dopamine (DA) deficit in
the striatum over the long disease course, causing complex functional deficits
within the basal ganglia network. The progressive degeneration retards the
voluntary movement, resulting in symptoms of rigidity, tremor, and bradykinesia
[6], which worsen as the dopaminergic denervation advances. Nonmotor symptoms,
such as psychiatric and cognitive impairment, sleep disorders, autonomic
dysfunctions, and gastrointestinal dysfunction [7], have an earlier onset, and
more are disabling than motor symptoms [8, 9, 10, 11]. Extensive research over
several years has highlighted oxidative stress [12, 13], inflammation [14, 15],
accumulation of altered proteins [16, 17],
excitotoxicity [18], endoplasmic reticulum
stress [19, 20] and mitochondrial dysfunction
[21, 22] as potential molecular mechanisms of the disease (The possible
mechanisms are shown in Fig. 1). Recently, the significant role of synaptic
plasticity in the pathological processes of PD has been identified and validated.
Neurons use most of the energy at the synapse. The onset and progression of PD
can be explained by the inability of neurons to experience synaptic plasticity
[23]. The ultrastructural features of pre-and post-synaptic neuronal elements at
the remaining corticostriatal and thalamostriate axo-spinous synapses appeared to
undergo complex remodeling, conceding with increased synaptic activity striatum
of 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP)-treated Parkinsonian
monkeys [24, 25]. In addition, glial reactions play a crucial role in PD.
Astrocytes may confer neuroprotective effects by releasing glial transmitters,
such as glial-derived neurotrophic factor (GDNF) [26], mesencephalic
astrocyte-derived neurotrophic factor (MANF) [27], and ciliary neurotrophic
factor (CNTF) [28]. A relative increase in the level of inflammatory cytokines,
senescence markers, and metalloproteinases from
astrocytes was observed in post-mortem substantia nigra specimens of five
patients with PD compared with that in the specimens of five control subjects
[29]. Astrocytes produce increased amounts of
proinflammatory cytokines in response to inflammatory stimulation by LPS,
IL-1
Transmitters released by astrocytes that regulate synaptic activity are concluded: (1) ATP [33, 34, 35, 36, 37, 38, 39, 40], (2) Purines [41, 42], (3) Glutamate [43, 44], (4) D-serine [45, 46, 47, 48, 49, 50, 51], (5) NO [52], (6) BDNF [53, 54], (7) S100b [55], (8) Thrombospondin (TSP-1) [56, 57]. In brain diseases, particularly in neurodegenerative conditions, astrocytes become highly reactive, and the process is known as astrogliosis. In this process, astrocytes undergo genetic [58] and morphological modifications [59]. Two subtypes, A1 reactive astrocytes and A2 reactive astrocytes, are formed [60, 61, 62, 63]. A1 reactive astrocytes lose their normal function and gain toxic functions. Co-culturing of human dopaminergic neurons with A1 astrocytes activates neuronal apoptosis, increasing cell death by 25% [64]. In contrast, A2 reactive astrocytes can upregulate many neurotrophic factors [56, 65, 66, 67, 68], contributing to the survival and growth of neurons or synaptic repair.
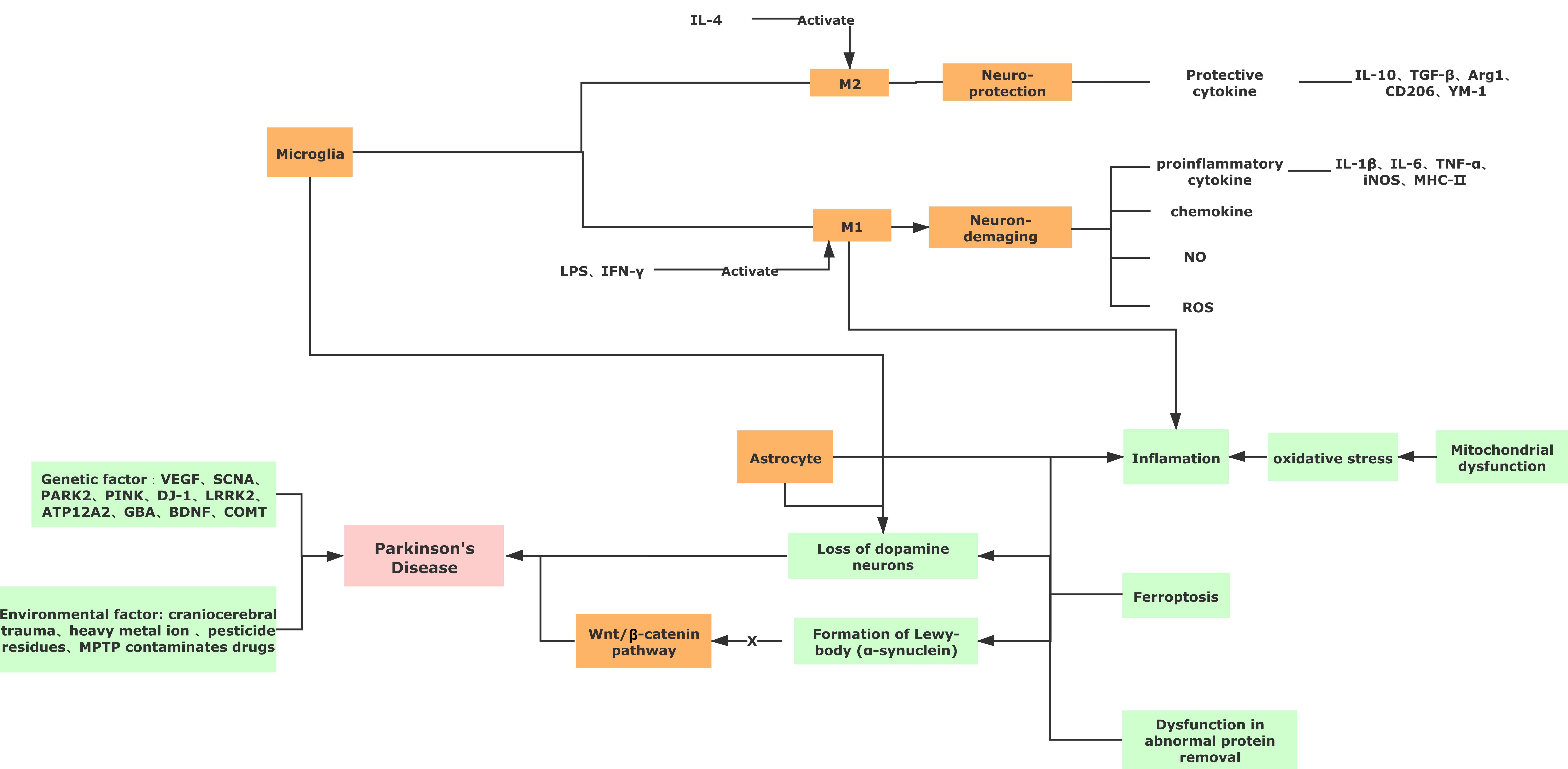 Fig. 1.
Fig. 1.Possible pathogenetic mechanisms of Parkinson’s disease have been studied.
Synaptic plasticity is the ability of synapses to change the strength of their connections. The strength of a synapse refers to the response generated in a postsynapse due to presynaptic activity [69]. Plasticity can occur in a short time and involves molecular mechanisms that influence the synaptic strength and remove, modify, or add synaptic connections [70, 71, 72, 73, 74]. Two forms of classical synaptic plasticity, namely long-term potentiation (LTP) and long-term depression (LTD) [75], depending on the synaptic activation of N-methyl-D-aspartate receptors (NMDARs), are regarded as the foundation of learning and memory [73, 76]. The highly dynamic astrocytes play various roles in the central neural system (CNS) by regulating the blood-brain barrier (BBB) [77], providing structural and metabolic support [78, 79], maintaining ionic homeostasis [80] and secreting neurotrophic factors, such as brain-derived nutritional factor (BDNF) and glial-derived neurotrophic factor (GDNF). It is estimated that multiple neuronal cell somas, 300–600 dendrites and more than 100,000 synapses can be ensheathed by just a single cortical astrocyte in the mouse brain. The number is even larger in primates and humans, reaching 2 million synapses [81, 82]. Hence, the concept of ʻtripartite synapses’ (TS) was proposed, wherein astrocytes integrate process and exchange information with pre-and post-synaptic neuronal elements [83]. In TS, this type of interaction with neurons requires physical contact between astrocytes and synaptic spines. Electron microscopic reconstructions revealed that 57% of the spines in a mature hippocampus are associated with astrocytes [84]. The size and morphology of the spine regulate the synaptic strength and determine the efficiency of synaptic transmission. For instance, large, mushroom-shaped spines contain functional synapses and are more stable, whereas thin filopodia-shaped spines are relatively unstable and nonfunctional [85]. Alterations in astrocytic processes are coordinated with the stabilization of larger spines [86]. Astrocyte processes are essential to the maturation and regulation of newly forming spines. Thus, the contact between astrocytes and spines improves both morphological maturation and the life of spines [87]. Dendritic spines are vulnerable to structural changes during aging and neurologic diseases [88, 89, 90, 91, 92, 93, 94, 95]. Compared with presynaptic terminals, astrocyte processes prefer to localize near dendritic spines [96]. Thrombospondins (TSPs) released by astrocytes are involved in the formation of excitatory synapses [56]. The reductive levels of TSP-1 can change the dendritic spine structure and decrease the number of spine and synapses [97]. Moreover, astrocytes regulate neurite formation and spine density by releasing BDNF in vitro [53].
In addition, astrocytes can secrete various factors such as glutamate, ATP and D-serine, which directly regulate the formation of synapses [79]. Hence, astrocytes are considered to have a central role in the regulation of synaptic plasticity.
Astrocytes are involved in regulating the synaptic function [98, 99]. The
passive homeostatic regulation of synaptic function by astrocytes is well
recognized. In addition, astrocytes sense synaptic activity and respond to
neurotransmitters released by these synapses and, in turn, release
gliotransmitters to regulate synaptic transmission and plasticity [100, 101]. The
release of Ca
Glutamate is converted to glutamine in astrocytes by glutamine synthetase, and glutamine is then recycled to neurons to synthesize glutamate that facilitates the regulation of synaptic transmission [115]. For instance, astrocytes release glutamate, which acts on the pre- [116, 117, 118] and post-synaptic [119, 120, 121, 122] sites to alter synaptic transmission and neuronal excitability at both excitatory [43] and inhibitory [123] synapses. Synapses are dynamic structures in terms of morphology, biochemistry, and function. Most excitatory glutamatergic synapses terminate at dendritic spines undergoing actin-driven movement [124]. An interesting feature is that glutamate can control the structural characteristics of neurons. For instance, glutamate released from the presynaptic terminals maintains the spine stability [125] and leads to small protrusions from the spine when locally applied [125].
Further, astrocytes induce the remodeling of dendritic spines by regulating the extracellular glutamate level [126]. They were revealed that the synaptic strength could be increased by stimulating astrocytes directly. In addition, direct stimulation of single astrocytes in the hypothalamus [127] and hippocampus [117] induces the sustained potentiation of synapses. This type of astrocyte stimulation in the hippocampus increases glutamate release and leads to NMDA-independent LTP production [116]. Furthermore, evidence indicates that astrocytes can modulate LTP by regulating glutamate levels at the synapse and by releasing cofactors of glutamate receptors at the neurons [128].
Moreover, astrocytes can regulate the rate of uptake and release of glutamate
[119, 128, 129, 130], and glutamate combines with the AMPA and NMDA receptors on
the post-synaptic neurons to induce LTP [130, 131]. This type of functional
expression of LTP is due to increased exocytosis of native AMPA receptors to the
membrane surface and their recruitment into excitatory synapses [132]. The
AMPA-type glutamate receptors (AMPARs) are glutamate-gated ion channels that
induce many fast excitatory synaptic transmissions in the brain [133]. Alteration
in the number, composition and biophysical properties of the AMPARs in the
post-synaptic membrane is the primary mechanism of synaptic strength regulation
[134]. During LTP induction, presynaptic input stimulation on a neuron’s
postsynapse terminates post-synaptic Ca
Adenosine triphosphate (ATP), released by astrocytes and neurons in the
pathophysiological environment and during neural activity, modulates the synaptic
strength and plasticity by activating ionic P2X and metabolic P2Y receptors. ATP
released by astrocytes regulates neuronal excitability by stimulating the purine
receptors [144]. Moreover, ATP released by astrocytes is degraded to adenosine,
which participates in the regulation of activity-dependent heterosynaptic
depression at excitatory synapses [41, 100]. Some studies have indicated that ATP
released by astrocytes leads to highly efficient glutamatergic synaptic
transmission in the paraventricular nucleus; however, it causes low synaptic
efficiency in the CA1 region of the hippocampus [144, 145]. ATP from both glia
and neurons can activate the post-synaptic P2X, wherein the synaptic currents
mediated by the NMDAR decreased significantly [146]. LTP in CA1 neurons was shown
to be decreased in P2X4
NMDA-dependent LTP and LTD have been widely studied, and the NMDA receptors are
regarded as the crucial components of LTP [135, 153]. The activation of NMDA
receptors requires glycine and D-serine in addition to glutamate [46, 154]. A
product of serine racemase, D-serine, was first identified in most astrocytes and
was considered an endogenous ligand for the glycine-binding site of the NMDA
receptor. Addition of D-amino acid oxidase to the cerebellar slices to deaminate
D-serine significantly reduces the NMDA receptor-dependent synaptic transmission
[155]. D-serine induces synaptogenesis in embryonic cortical neuron culture
induced by TGF
The effects of astrocyte-derived D-serine on synaptic plasticity and
astrocyte-derived amino acids were shown to participate in the LTP [157]. LTP
ability of the cultured neurons was recovered by adding D-serine to the
astrocyte-conditioned medium [157], and the recovery process required interaction
between the astrocytes and neurons. Yang et al. [157] demonstrated that
hippocampal neurons cultured with astrocytes could undergo LTP, whereas LTP could
not be induced in neurons cultured in astrocyte-conditioned media without
astrocyte contact. However, the induction of NMDA-dependent LTP was limited by
D-serine released by the hippocampal astrocytes [48]. Christian Henneberger
reported that the clamping of internal Ca
Moreover, blocking glial cell activation with fluoroacetate, a metabolic inhibitor, was shown to block LTP in the prefrontal cortex due to decreased extrasynaptic D-serine [158]. In contrast to the findings above, [159] reported that the astrocytes release L-serine, converted to D-serine by serine racemase in the neurons, and the latter modulates synaptic plasticity. To confirm this finding, Benneyworth et al. [160] demonstrated that LTP and NMDAR currents are significantly reduced in neuronal serine racemase conditional knockout mice but not in astrocytic serine racemase conditional knockout mice. Therefore, these findings highlight that astrocytes possibly play direct and indirect roles in the D-serine regulation of LTP.
PD is a neurodegenerative disorder characterized by the gradual loss of
dopaminergic neurons in the substantia nigra pars compacta (SNpc), which
decreases DA input to the striatum and hence causes motor dysfunctions [161]. A
decrease in LTP activation has been observed in PD models, alleviated by the DA
precursor treatment [162, 163]. Short- and long-term changes in corticostriatal
synaptic plasticity may be involved in PD [162, 164]. Some researchers have
described two classic forms of synaptic plasticity, LTP and LTD, at the
corticostriatal synapse in medium spiny neurons in vivo [165, 166]. In
the early stages of PD, extracellular DA was shown to be reduced by 40% in
PINK1-knockout heterozygous mice; although these mice did not demonstrate any
motor symptoms, LTP was alternatively impaired [167]. At the onset of motor
dysfunctions in patients with PD,
Synaptic plasticity occurs in dendritic spines under physiological conditions [171]. Thus, the alterations of spines in PD are related to synaptic plasticity damage. Significant striatal spine loss induced by nigrostriatal dopaminergic lesions has been observed in various PD animal models [25]. The neurotoxin MPTP causes the alternative loss of dopaminergic neurons in the midbrain area of both humans and animals [172, 173]. The MPTP-treated mouse is a common PD animal model [174]. Some studies have found that MPTP treatment can impair LTP [175, 176]. Toy et al. [177] suggested that MPTP injection decreases, whereas exercise increases the dendritic spine density. Motor training has alleviated PD symptoms by promoting synaptic formation and increasing dendritic arborization in the motor cortex [178, 179]. The classic synaptic plasticity forms of LTD and LTP also influence PD symptoms by mediating changes in dendritic spines. In adult mice with dopaminergic nerve degeneration in the motor cortex, LTP loss was accompanied by an increased rate of spine elimination [180].
In addition to motor dysfunctions, cognitive deficits in PD, including learning and memory impairment, seriously impair patients’ life quality and social functions, which cause a great deal of psychological and economic burden to their families. The hippocampus is crucial for spatial and episodic memory formation and novelty detection [181, 182]. Most studies on PD cognitive impairment have focused on the hippocampus. Abnormalities in the hippocampus’s structure and function were observed in sporadic patients and patients with genetic predisposition with PD, suggesting an association of the organ with memory deficits in PD [183, 184, 185, 186]. In addition, the hippocampus is associated with memory deficits [184, 185, 187, 188, 189]. Like motor regulation, cognitive regulation in the disease is based on DA, which conforms to the classical PD pathology. Reportedly, the changed habituation to a new environment in PD animals due to impaired hippocampal LTP can be reversed by the systemic L-DOPA treatment [190]. Moreover, LTP in the CA1 hippocampus is reduced in both genetic and neurotoxic PD models [190]. This alteration of CA1 LTP is accompanied by hippocampal-dependent learning deficits in both 6-OHDA-lesioned and mutant animals [191].
PD is a neurodegenerative disease associated with DA depletion and progressive loss of dopaminergic neurons. The primary treatment strategy involves external DA supplementation to alleviate the functional impairment of the basal ganglia induced by DA deficiency. However, none of the currently available PD therapies can slow down or impede the disease progression without dopaminergic neuron recovery. Therefore, the development of effective therapeutic strategies is essential to prevent disease progression. The cell-replacement therapy has been advancing considerably that uses the fetal midbrain tissue as the source of midbrain dopamine neurons (mDAs) for transplantation [192]. However, technical difficulties in obtaining sufficient graft tissues, ethical considerations, and rejection of cells limit the use of this therapy [193, 194, 195]. Alternative cell sources, such as stem cells or reprogrammed cells, have also been considered [196, 197]. The therapeutic effects of embryonic stem cells (ESCs), neural stem cells (NSCs), and bone marrow mesenchymal stem cells (BMSCs) have been widely studied [198, 199, 200].
Nevertheless, ethical consideration, differentiation, and risk of tumor limit the application of NSCs and ESCs. The complex brain environment also interferes with the survival and function of BMSC-derived transplanted neurons [201, 202, 203]. Astrocytes have been studied as potential candidates to improve the hostile brain environment for neural transplantation. Astrocyte cografting can significantly improve the survival rate of dopaminergic neurons and the behavior of PD rats [201]. Stem cell transplantation and cell reprogramming are also the proposed alternatives. In these processes, induced dopaminergic neurons, which are the phenotypically specific and functional DA neurons obtained directly from somatic cells, are used. The induced dopaminergic neurons share morphological characteristics similar to those of resident dopaminergic neurons in the brain. They produce and release DA, survive in the brain, and stimulate the target regions [204, 205, 206, 207, 208]. Astrocytes are considered potential cells for reprogramming due to their particular role in PD. In vitro studies have demonstrated that astrocytes can be converted into functional neurons [209, 210, 211, 212]. Novel strategies, such as direct reprogramming of resident astrocytes in vivo [212], by combining gene- and cell-based therapy have been developed. These strategies can be applied to convert reactive astrocytes in the injured brain and degenerative diseases into functional neurons in vivo and are being developed further [213]. In addition, compensating dysfunctional astrocytes is an attractive choice. Functional astrocytes can be obtained from embryonic glial restricted precursor cells (GRPCs) and human pluripotent stem cells [214, 215, 216]. In vivo transplantation of embryonic GRPC-derived astrocytes could release trophic factors and antioxidants in the striatum, improved behavioral deficits, and restored TH expression in PD rats [217]. Mouse astrocytes could be transformed into dopaminergic neurons in vivo, and this strategy could be used to treat animal models of PD [218].
In addition, as mentioned earlier, damage to LTP and LTD in PD is parallel to DA
depletion and the occurrence of PD dyskinesia. Indeed, damage to LTP precedes the
onset of PD symptoms. Astrocytes can affect the synaptic plasticity of neurons
and improve LTP and LTD by releasing Ca
In conclusion, further research is required to explore effective treatment strategies to delay or stop PD progression. Advances in the transplantation technology and regulation of synaptic plasticity by astrocytes can be leveraged to develop new strategies to prevent disease progression at an early stage. We believe that this approach can provide a potential research direction for future studies on PD.
AMPARs, AMPA-type glutamate receptors; ATP, Adenosine triphosphate; BBB,
blood-brain barrier; BDNF, brain-derived nutritional factor; BMSCs, bone-marrow
mesenchymal stem cells; CNS, central neural system; CNTF, ciliary neurotrophic
factor; DA, dopamine; ER, endoplasmic reticulum; GABA,
YQZ and KRL read the literature together and wrote the review. YQZ is the first author. KRL is the corresponding author.
The content of the article does not involve any experiments, and there are no ethical issues.
We thank three anonymous reviewers for their excellent criticism of the article.
Regulation of GABAA-mediated tonic inhibition on cognitive impairment in Parkinson’s disease and its mechanism 2018A0303130259.
The authors declare no conflict of interest.