1. Introduction
Hiccup (Latin, singultus) is caused by an involuntary periodic
contraction of the diaphragm followed by the glottis’s closure. The inspired air
meeting a closed glottis causes the familiar hiccup sound. Hiccupping lasting
longer than four weeks is considered chronic. Treatment resistance (obstinate
hiccup) is defined as a lack of response to many (mostly three) successive
pharmacological treatments attempt. Generally, the longer the hiccupping
duration, the less amenable it will be to interventions [1].
Singultus is not a disease but a symptom. The most commonly encountered
hiccup is that of idiopathic origin. While many drugs have been tried off-label
in hiccup therapy, chlorpromazine is the only FDA approved drug for this purpose.
In contrast, only a few drugs (benzodiazepines, barbiturates, alcohol, and
steroids) and the phenyl-piperazine atypical antipsychotic aripiprazole are
well-established hiccup inducers [1].
Since the initial observation made by Behere [2] that aripiprazole can induce
hiccups (in his case, associated with hyponatremia), an abundance of reports on
hiccups associated with aripiprazole treatment emerged [3, 4, 5, 6, 7, 8, 9, 10, 11],
and on persistent hiccups associated with switching antipsychotic treatment to
aripiprazole [12, 13, 14, 15].
More recently, brexpiprazole was introduced into clinical practice [16]. In contrast
to the structurally closely related aripiprazole, brexpiprazole was not
associated with this particular adverse drug reaction. Having two very similar
drugs that differ in their ability to induce hiccup represents a unique
opportunity to gain insight into the receptors involved in the symptom’s
pathophysiology.
To identify the critical difference responsible for the discrepancy, we
performed a literature search (PubMed and public domain sources) and retrieved
and compared the PK/PD data and properties of the two piperazine antipsychotics
at serotoninergic, alpha-adrenergic and dopaminergic receptors.
While aware of the limitations of comparing receptor affinities/intrinsic
activity values-even more so when obtained from different sources using different
methodologies-(as they have considerable confidence intervals) and inferring
biological effects based on such data, it is currently the only practical
available option for our analysis [17, 18].
2. Methods
To identify the critical difference responsible for the discrepancy, a
literature search (PubMed and public domain sources) was performed, and
pharmacokinetics and pharmacodynamic (PK/PD) data/properties of the two
piperazine antipsychotics at serotoninergic alpha-adrenergic and
dopaminergic receptors retrieved and compared. Due to the limited number of
publications containing pharmacokinetic and or pharmacodynamic details on
aripiprazole and brexpiprazole, no filters were applied.
As a matter of terminology -as used by us-when comparing K values of the
two drugs, very (strong) high affinity implies subnanomolar K values,
high-affinity K between 1 and 10 nM, moderate K between 10 and 50 nM,
low affinity is K between 50 and 100 nM and very (weak) low affinity for
K higher than 150 nM. For K values above 90% of the therapeutic
plasma range (700 nM for aripiprazole and 300 nM for brexpiprazole), no effect
via the respective receptor is assumed. The inhibition constant (K) is
calculated based on the following: K = IC for noncompetitive
inhibition, K = IC/2 for competitive inhibition, and K values
range from IC to IC/2 for mixed inhibition, according to the
equation [19]: K = IC/(1 + ([L]/Kd) where [L] is the concentration
of ligand (nM), and Kd is the affinity constant (nM).
For comparison purposes K ratios are given as K aripiprazole/K brexpiprazole, i.e., [K(/)]; a value 1 indicates lower
affinity of aripiprazole for the respective receptor while a value 1
indicates higher affinity of aripiprazole for the respective receptor. Divergent
or significantly different effects are assumed when the ratio is either
10 or then 10. The same applies when only one of the drugs has a
K within the therapeutic range [22]. K is used to describing the binding
affinity that a molecule has for an enzyme or receptor. The half-maximal
inhibitor concentration IC is more reflective of the inhibitor’s
functional strength, but both factors in the drug’s concentration inhibit the
activity. Efficacy is the relationship between receptor occupancy and the ability
to initiate a response. As a matter of terminology -as used by us-when comparing
K (binding affinity constant) values of the two drugs, very
(strong) high affinity implies subnanomolar K values,
high-affinity K between 1 and 10 nM, moderate K between 10
and 50 nM. Low affinity is K between 50 and 100 nM and very (weak) low
affinity for K higher than 150 nM. For K values above 90%
of the therapeutic plasma range (700 nM for aripiprazole and 300 nM for
brexpiprazole), no effect via the respective receptor is assumed because such
supratherapeutic (possibly toxic) levels are not clinically targeted.
The daily dose range for aripiprazole (MW 448) is 10-30 mg with a dose-dependent
therapeutic plasma range of 100-350 (ng/mL) [223-780 nM]; for the more potent
brexiprazole (MW 434), the daily dose range is 2-4 mg with a (dose-dependent)
therapeutic plasma range of 40-140 (ng/mL) [92-323 nM] [20]. For K
values above 90% of the therapeutic plasma range (700 nM for aripiprazole and
300 nM for brexpiprazole), no effect via the respective receptor is assumed.
The brain aripiprazole concentration is approximately 0.6-0.9 of the respective
plasma concentrations [21], while brexpiprazole is 0.2-0.41 (1ABILIFY® A. Bristol-Myers Squibb Company. U.S. Food and Drug Administration website Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2005/021713s004,021436s007lbl.pdf (Accessed: 07 May 2020)). These ratios
are difficult to interpret, as they do not compare C plasma with
C brain.
The abstracts of the 60 documents (search 2 and 4) were evaluated by two of the
authors (GAP & EA) for the likelihood the paper containing relevant
pharmacokinetic and pharmacodynamics (pK/pD) data (affinity, IC,
EC, receptor occupancy). The consensus papers were mined for the targeted
data. To complement the information thus obtained, a series of databases and
other public domain publications (European Medicines Agency (EMA), 2018; Drugs
FDA; Lundbeck; American Psychiatric Association; PubChem Database) were also
consulted. Sources are cited wherever they are used.
3. Results
3.1 Serotonin (5-HT) receptors
5-HT (K 5-HT 2): Brexpiprazole,
despite an affinity one order of magnitude higher
([K(/)] 1.7/0.12 14) than
aripiprazole, displayed comparable receptor occupancy and an only slightly lower
efficacy [Emax expressed as a percentage of the effect of serotonin =
0.6 vs. 0.7] [23]. Both drugs are less efficacious than the
endogenous neurotransmitter serotonin (E 1), thus acting as
partial agonists. The two drugs have very similar effects via this
receptor, even though some argue that brexpiprazole is a full 5-HT
receptor agonist [24, 25] (Table 1).
Table 1.The affinity ratio between Aripiprazole and Brexpiprazole K() and effect at different serotonin receptors and transporter.
|
ARI Therapeutic Range 223-780 nM |
BREX Therapeutic Range 92-323 nM |
|
|
|
K ARI nM |
K BREX nM |
K() |
Effect |
| 5-HT |
1.3
[23] |
0.12
[23] |
14 |
partial agonism |
| 5-HT |
830
[27] |
32
[23] |
26 |
antagonism |
| 5-HT |
68
[27] |
Not available |
|
|
| 5-HT |
8000
[27] |
Not available |
|
|
| 5-HT |
4.7
[23] |
0.47 |
10 |
antagonism |
|
|
IC 6.5
[23] |
|
|
| 5-HT |
0.36
[58] |
1.9
[23] |
0.2 |
antagonism/inverse agonism |
| 5-HT |
15
[27] |
34
[27] |
0.4 |
antagonism |
| 5-H |
630
[27] |
Not available |
|
Possiblydivergent; only ARI |
| 5-H |
1240
[27] |
140 |
8.9 |
possibly divergent; only BREX |
| 5-H |
570
[27] |
60 [23] |
3.6 -9.5 |
weak antagonism |
|
219
[59] |
|
|
|
| 5-H |
39
[27] |
3.7
[23] |
10 |
weak partial agonism |
| SERT |
95 (IC) |
29 (IC) |
3.3 [IC ()] |
inhibition |
| : serotonin transporter |
|
|
|
| Committee for Medicinal Products for Human Use. Rxulti (Brexpiprazole) Assessment report. 2018. Available at: https://www.ema.europa.eu/en/documents/assessment-report/rxulti-epar-public-assessment-report_en.pdf. |
5-HT (K 5-HT 4): While aripiprazole
is unlikely to significantly affect this receptor (K outside of
the therapeutic range, brexpiprazole will act as an antagonist. One can speculate
that brexpiprazole as an antagonist will have some vasodilating effect (Table 1).
5-HT (K 5-HT 12): The effects of
brexpiprazole and aripiprazole at 5-HT are measured via studying
DOI-(2,5-dimethoxy-4-iodoamphetamine)-induced head twitches (mediated via
5-HT): brexpiprazole and aripiprazole inhibited DOI-induced head twitches;
maximum responses (Mean S.E.M.) for brexpiprazole and aripiprazole were;
99 0.9, and 91 3.6, respectively. Brexpiprazole, despite an
affinity about one order of magnitude higher
([K(/)] 3.4/0.47 7) than
aripiprazole, displayed comparable 2A receptor occupancy [23]. As with all
atypical antipsychotics, both drugs act at this receptor as antagonists.
The two drugs have similar effects via this receptor [26]. Thus, it is unlikely
that interaction with this receptor might explain the different ADR profile of
the two drugs (Table 1).
5-HT (K 5-HT 9): Brexpiprazole,
despite a lower 2B affinity than aripiprazole
([K(/)] 0.36/1.9 0.2)
displayed comparable IC 10-15 nM [16, 23, 27]. While
K is used to describing the binding affinity that a molecule has
for an enzyme or receptor.
The half-maximal inhibitory concentration IC is more reflective of the
inhibitor’s functional strength, but both factors in the drug’s concentration
inhibit the activity. The drugs act at this receptor as antagonists/inverse agonists and have similar effects via this
receptor. Thus, it is unlikely that interaction with this receptor might explain
the different ADR profile of the two drugs (Table 1).
5-HT (K 5-HT 5): Binding affinity of
brexpiprazole at the 2C receptor was lower than that of aripiprazole:
[K(/)] 15/34 0.4. Despite
that, the two drugs showed similarly low efficacies (E 10%)
compared with serotonin. Both drugs act as weak partial agonists, de facto
antagonists [26, 27] (Table 1).
5-HT (K 5-HT 200): K of
aripiprazole at this receptor is K 630 110 nM [27]. In
comparison the endogenous agonist 5-HT has a K = 200 nM [28]. No
data for brexpiprazole could be found, nor any indication that the drug would
interact with this receptor. Only aripiprazole likely has a week effect mediated
via this receptor (Table 1).
5-HT (K 5-HT 120): There is no
available data suggesting any effect of the two drugs via this receptor.
5-HT (K 5-HT 200): Binding affinity of
aripiprazole at this receptor is negligible with a K 1.240 280 nM, above the upper limit of the therapeutic plasma
range [27]. Brexpiprazole K 140 nM2 (2REXULTI® B. Otsuka and Lundbeck. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/205422Orig1Orig2s000PharmR.pdf (Accessed: 13 May 2020)). The only
brexpiprazole likely has an effect mediated by this receptor (Table 1).
5-HT (K 5-HT 65): K of
aripiprazole 214 (reported range from 161 17 [29] to 570
95 nM [27, 29] and 780 [30]). The affinity of the weak
antagonist brexpiprazole is similar to that of serotonin (58 nM), but the
receptor occupancy is low at 20-40%. K(/)
3.5. Both drugs act as weak antagonists (Table 1).
5-HT (K 5-HT 2-8): The affinity of the
endogenous agonist serotonin for the 5-HT receptor is (strong) high [31, 32]. Aripiprazole, a very weak partial agonist with an E 10% (of
5-HT effect), has a K 39 nM and a receptor occupancy
in the 15-30% range [27, 31, 33]. Brexpiprazole, a weak partial agonist, has a
K of 3.7 nM and a receptor occupancy in the 10-56% range3 (3Committee for Medicinal Products for Human Use. Rxulti (Brexpiprazole) Assessment report [Internet]. 2018. Available at: https://www.ema.europa.eu/en/documents/assessment-report/rxulti-epar-public-assessment-report_en.pdf) [34]. K(/) 10 (Table 1).
At the 5-HT receptor, while aripiprazole has a (weaker) lower affinity
than brexpiprazole, both drugs have similar effects (weak partial
agonist, de facto antagonist) [26] (Table 1).
SERT (K 5-HT 500) [35]: Aripiprazole has an
IC for the serotonin transporter 95 nM. Brexpiprazole is a
SERT inhibitor with an IC of 29 nM but a low ligand displacement ability
(65% at 10 M)2 (2REXULTI® B. Otsuka and Lundbeck. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/205422Orig1Orig2s000PharmR.pdf (Accessed: 13 May 2020)) (Table 1).
3.2 Alpha () adrenergic receptors
Brexpiprazole is a highly affine adrenergic receptor antagonist
[K 15 nM; K 17
nM; K 0.59 nM] comparable to mirtazapine
(K 20 nM; K 88
nM; K 18 nM) [23, 30]. Aripiprazole has a
slightly lower affinity for adrenergic receptors
(K 74 nM; K 100
nM; 37 nM) [30]. K () for the
-adrenergic receptors is 5; for
6 while for 63.
Brexpiprazole has an IC of 63 nM at receptors [23]
(Table 2).
Table 2.The affinity ratio between Aripiprazole and Brexpiprazole
K() and effect at adrenergic
receptors and transporter.
|
K ARI nM |
K BREX nM |
K() |
Effect |
| α |
74
[30] |
15
[23] |
5 |
antagonism |
| α |
100
[30] |
17
[23] |
6 |
antagonism |
| α |
37
[30] |
0.59 IC 63
[23] |
63 |
antagonism |
3.3 Dopamine (DA) receptors
D (K DA 2-8): Aripiprazole has a
K 265 nM (reported range 200-2500 nM) [19, 27, 29]; brexpiprazole has a K 1502 (2REXULTI® B. Otsuka and Lundbeck. U.S. Food and Drug Administration website. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/205422Orig1Orig2s000PharmR.pdf (Accessed: 13 May 2020).
K(/) 265/160 1.7. Both
drugs have similar agonist effects [36, 37] (Table 3).
Table 3.The affinity ratio between Aripiprazole and Brexpiprazole and
effect at different dopamine receptors and transporter.
D (K DA 2): Aripiprazole has a
K 2590 1350 nM, well above the upper limit of
the therapeutic plasma range [27]. Similar values [K
1.670 nM] have been reported by [38]. For brexpiprazole receptor, occupancy is
66% at 1 mM (Table 3).
D (K DA 12 4) [39]:
Aripiprazole, a partial agonist with an Emax 60% (of D effect), has
a K 0.32 nM. Brexpiprazole, also a partial agonist,
has a K 0.3 nM and an Emax 43%.
K(/) 1. At the D receptor,
both drugs have similar effects (partial agonist) (Table 3).
D (K DA 3 1) [39]:
Aripiprazole, a partial agonist with an Emax 30% (of DA
effect), has a K 0.8 nM. Brexpiprazole, a weak partial
agonist, has a comparable affinity (K 1.1) but a lower intrinsic activity Emax 15%.
K(/) 0.8. At the D receptor,
while both drugs have qualitatively similar effects (partial agonist),
aripiprazole has a somewhat higher intrinsic activity [26] (Table 2).
D (K DA 6 2) [39]:
Aripiprazole has a K 44 nM and an Emax of 20-30%
[40]. Brexpiprazole has a higher affinity K 6 nM.
K(/) 7. Both drugs have a
partial agonist effect at this receptor (Table 3).
DAT (K DA 29 4) [41]:
Brexpiprazole is a negligible inhibitor of the dopamine transporter with a
reported IC 950 nM, well above the upper limit of the
therapeutic plasma range; the same applies for aripiprazole (K 3220 660) [27] (Table 3).
4. Discussion
To identify differences that might explain the differential effect of the two
drugs on hiccup, the affinities of aripiprazole and brexpiprazole for the various
serotoninergic, alpha-adrenergic and dopaminergic receptors, as well as the
respective transporters, were compared. The drugs are very similar in their
pharmacodynamics at most receptors. They are generally considered partial
agonists at 5-HT and D receptors and antagonists at the 5-HT
receptors; brexpiprazole has a somewhat lower intrinsic activity at the D
receptor and acts as a more potent 5-HT agonist and as a stronger
5-HT antagonist [17]. Furthermore, both drugs appear to have additional
characteristics beyond partial agonist at D2 receptors, including biased agonism
[42, 43]. The term biased agonism (functional selectivity), initially
introduced in [44], describes the phenomenon that a ligand preferentially
activates one of several signaling pathways. In contrast, another agonist in the
same system and acting on the same receptor preferentially activates another
pathway.
Both are dopamine system stabilizers, i.e., to have either an agonist or
antagonist effect depending on the levels of exogenous dopamine (antagonist at
high dopamine concentration vs. agonist at low dopamine concentration [42, 45]).
Aripiprazole and brexpiprazole function as both a presynaptic D agonist and
postsynaptic D antagonist; furthermore, their partial agonistic activity
5-HT receptor plays a role in modulating among other dopamine releases
reviewed by [42]. Effects mediated via the major psychosis relevant receptors
(5-HT, 5-HT and D) are similar and do not provide in our view
the explanation for the appearance of a hiccup with aripiprazole treatment.
Different effects appear possible at the 5-HT receptor where aripiprazole
has a K within the therapeutic plasma concentration and the
5-HT- and 5-HT receptor where brexpiprazole has a Kwithin the therapeutic plasma concentration.
5-HT receptors are the only ionotropic serotonin receptors.
5-HT receptors are located (mainly) on sensory vagal nerve endings and play
a vital role for vagal afferent input originating from organs cranial to the
Cannon-Böhm-point (the gastrointestinal tract fewer parts of the colon and
rectum, lungs and heart). The central terminals of vagal afferents exhibit
5-HT receptors that increase glutamatergic synaptic transmission to
second-order neurons of the nucleus tractus solitarius [46]. Experimental
compounds with 5-HT blocking properties increase the heart rate by
decreasing vagal afferent input and efferent output; this is compatible with data
showing that 5-HT receptors excite vagal afferent neurons by a
glutamate-dependent mechanism [47, 48]. Blockade of these receptors by 5-HT
antagonists (setrons) is used clinically for control of emesis.
A single anecdotal mentioning of the negative impact of setrons on a patient
with chronic hiccup was published by one of the authors (GAP) [49]. Some anecdotal reports claim that
setrons cause hiccups 4,5 (4Kantrowitz M. Chemo Hiccups: Causes of and cures for chemo hiccups. 2009. Available at: http://www.kantrowitz.com/cancerpoints/hiccups.html. 5Theriot J, Wermuth HR, Ashurst JV. Antiemetic Serotonin-5-HT3 Receptor Blockers. StatPearls. Treasure Island (FL) 2020.), but overall, the evidence is relatively sparse.
The possibility of aripiprazole affecting this receptor (while the same does not
apply to brexpiprazole) is nevertheless intriguing as-if confirmed
experimentally- it might explain their differential effect on hiccup.
Mechanistically an inhibition of this receptor would be expected to lower vagal
efferent output, which is believed to favor hiccup development [50].
5-HT receptors (Gi protein-coupled) are virtually unexplored due
to a lack of selective ligands [51]. As Glennon [52] points out, “the
discovery of a therapeutically useful function for the receptors” is still
outstanding. The possibility that brexpiprazole might affect this receptor
(while the same does not apply for aripiprazole) cannot be inferred much at this
point.
5-HT receptors (Gi protein-coupled) are the traditional target
of the triptan class of drugs. Triptans act as agonists at 5-HT and
5-HT receptors at blood vessels and nerve endings in the brain and induce
vasoconstriction. The only brexpiprazole has a K within the
therapeutic range (antagonist; vasodilation). The significance or lack thereof is
difficult to interpret.
Overall, we failed to identify the one receptor that might explain the different
effect of the examined drugs on hiccup; it appears likely that the different
effect is the consequence of synergism of several smaller effects at more than
one receptor. The most consequential concerning neurotransmitter release is the
central alpha adrenergic receptor.
Brexpiprazole, similar to mirtazapine, is a highly affine
antagonist [ K 15 nM; K 17 nM; 0.59 nM] [17]. Antagonism of the
- receptors, which function mainly as inhibitory autoreceptors
and heteroreceptors, enhances transmitter release and favors neurotransmission,
notably central 5-HT receptor-mediated. Mirtazapine has been said to be a
functional “indirect agonist” of the 5-HT receptor [53].
Mirtazapine’s K for -adrenergic receptors is
20 nM, while aripiprazole’s K for
receptors [ K 74 nM; K 100 nM; 37 nM] [30].
K(/) for the -adrenergic
receptors is, therefore, 5; for 6 while for 63. While we postulated
divergent or significantly different effects when the ratio is either
10 or than 10 a K(/)
63 at indicates possibly higher efficacy for brexpiprazole.
Oosterhof et al., 2014, 2015 argue that brexpiprazole is a full
5-HTreceptor agonist, possibly due to a combined
antagonist-HT agonist effect [24, 25]. Such an effect combination would be
quite similar to that seen with the azapirone derivative tandospirone, where
tandospirone is an HT partial agonist while its primary metabolite
(1-pyrimidinyl-piperazine; 1-PP) is a centrally acting, the high-affinity
-adrenergic antagonist (K 10-40 nM)
[54, 55]. Interestingly, tandospirone was successfully used to treat hiccup [56]
(See also Fig. 1).
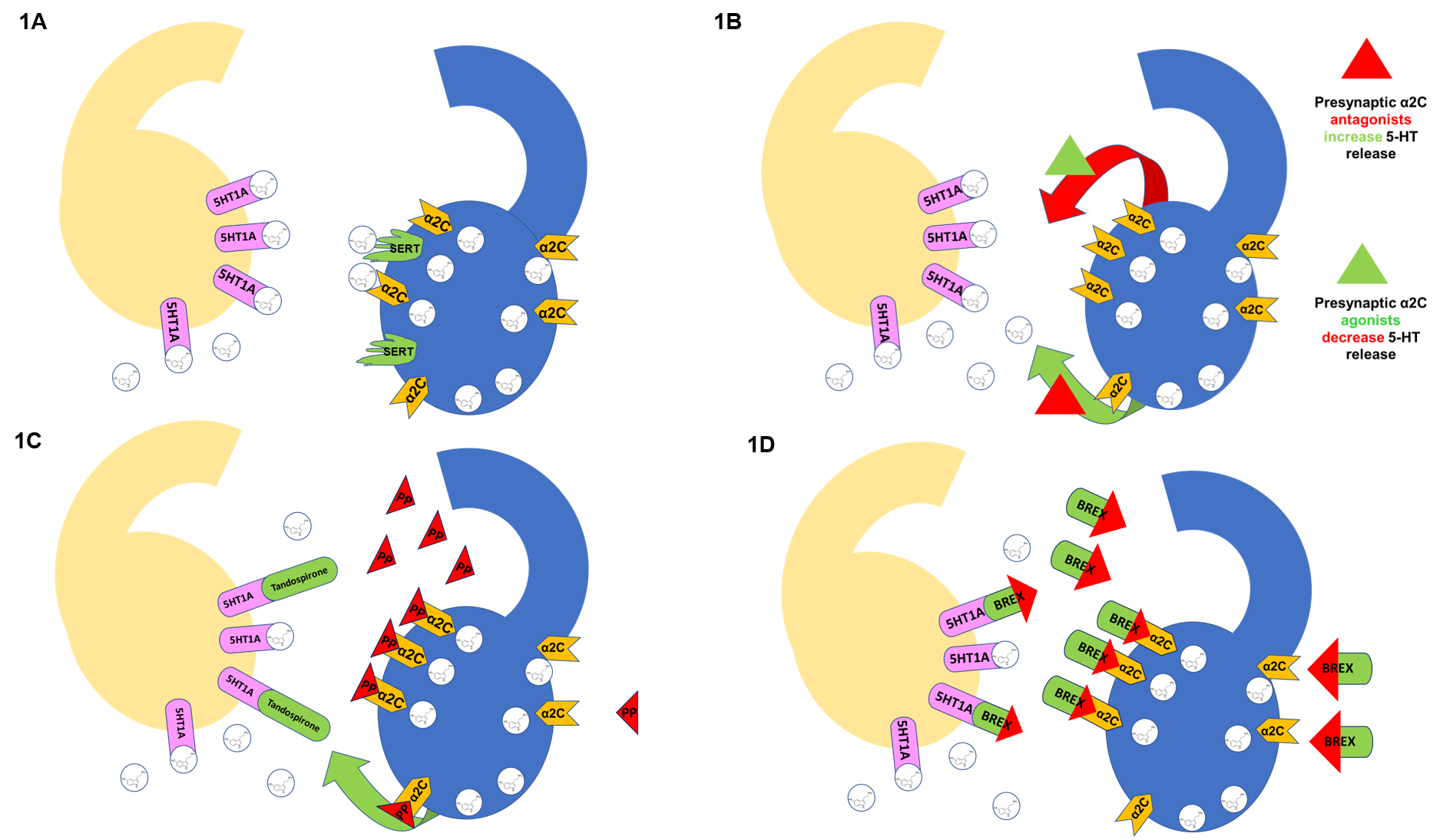 Fig. 1.
Fig. 1.
A schematic illustration showing receptor-drug interaction. 1A. Blue Neuron: adrenergic heteroreceptors control
neurotransmitter release (i.e., 5-HT). Once released, 5-HT docks at postsynaptic
receptors such as 5-HT on the yellow neuron. Activation of presynaptic
by the neurotransmitter reduces further release while SERT
removes 5-HT from the synaptic cleft.
1B. Activation of presynaptic by the neurotransmitter
or drug with agonist effect (green triangle) reduces further release (red arrow).
In contrast, the drug with antagonist effect (red triangle) increases
neurotransmitter release (green arrow).
1C. The primary metabolite of tandospirone (1-PP) is a highly affine
adrenergic antagonist, increasing serotonin release and
augmenting postsynaptic effects. Tandospirone is a 5-HT partial agonist;
the combined effect of drug and metabolite is direct and indirect agonism at
5-HT (mirtazapine-like effect).
1D. Brexpiprazole is a highly affine adrenergic
antagonist, thus increasing serotonin release and augmenting postsynaptic
effects. At the same time, brexpiprazole is also a 5-HT partial agonist;
the combined effect at the two receptors is direct and indirect agonism at
5-HT (mirtazapine-like effect).
For an overview of receptors, see the recent review from Brian
Harvey’s group [57].
Activation of 5-HT receptors enhances vagal activity; therefore,
5-HT agonists (brexpiprazole) would be unlikely to favor hiccup
development. In contrast, 5-HT partial agonists (week antagonists) such as
aripiprazole might not offer the same benefit [50].
5. Conclusions
It is unlikely that a unique receptor-drug interaction could explain the
different effects of the examined drugs on hiccup. The different effect is most
likely the consequence of several smaller effects at more than one receptor.
Brexpiprazole is a highly affine (potent) antagonist and,
therefore, also an indirect 5-HT agonist. In contrast, aripiprazole is a
partial 5-HT agonist (weak antagonist) and an HT antagonist.
Activation of 5-HT receptors enhances vagal activity while HT
blockade reduces it. Vagus nerve activation is therapeutic for hiccups. A
definitive answer continues to be elusive.
Abbreviations
5-HT, 5-hydroxytryptamine, serotonin; CNS, Central nervous system; DA, Dopamine;
DAT, Dopamine transporter; EC, concentration of a drug that gives a
half-maximal response (nM); E, concentration of a drug that gives a
maximal response (nM); IC, half maximal inhibitory concentration (nM);
K, Inhibitor constant (nM); SERT, Serotonin transporter.
Author contributions
Georg Petroianu: planning and conducting the review, literature search,
interpreting the literature, and drafting the manuscript; Eman Alefishat:
planning and conducting the review, literature search, interpreting the
literature, and drafting the manuscript; Lujain Aloum: literature search,
interpreting the literature, and drafting the manuscript; Ovidiu Baltatu:
literature search, interpreting the literature, and drafting the manuscript.
Ethics approval and consent to participate
Not applicable.
Acknowledgment
We express our sincere thanks to the reviewers for their instructive comments that improved the final version.
Funding
This research received no external funding.
Conflict of interest
The authors declare no conflict of interest.
Consent for publication
All authors have read and approved the manuscript for publication.


