
Ischemic brain injury is a prevalent disease with high disability and mortality, but no efficient therapeutics for the disease are currently available mainly due to the narrow therapeutic window. The treatment of cerebrovascular disease by using herbal medicine has been applied for a long time, from which large amounts of medical experience and knowledge have been accumulated. Numerous natural bioactive compounds extracted from Chinese medicines exhibit neuroprotective activities, especially protecting the brain from ischemic injury. This review summarizes the mechanisms underlying cerebral ischemic pathophysiology, including excitotoxicity, generation of free radical, inflammation, astrocytic influence, apoptosis, blood-brain barrier dysfunction, and discusses neuroprotective activities of the representative natural bioactive compounds extracted from traditional medicinal herbs, with targeting one or more signal molecules. Confirmation of potential neuroprotective activities of bioactive compounds derived from Chinese medicine in ischemic stroke treatment is discussed.
Ischemic brain injury, a prevalent disease with high disability and mortality, is resulted from cerebral blood flow reduction caused by thromboembolic or thrombotic arterial occlusions, which are the leading cause of stroke incidences (Roger et al., 2011). The neurological deficits induced by stroke include numbness, hemiplegia, balance problems, loss of sensory and vibratory sensation, decreased reflexes, visual field defects, ptosis (of the eyelid), apraxia, and aphasia (Tsuchiya et al., 1992). Insufficient blood supply to the brain initiates a series of pathophysiological events, including inflammation, excitotoxicity, apoptosis, and oxidative and nitrative stress (Li et al., 2018). A large number of efforts have been made to design and develop neuroprotective agents to delay these processes (Green, 2008; Ly et al., 2006). However, no effective therapy is available due to the narrow therapeutic window (Christophe et al., 2017). An exception is the tissue-type plasminogen activator (tPA), which is the only Food and Drug Administration (FDA)-approved thrombolytic treatment and antithrombotic drug treatment.
The treatment of cerebrovascular disease by natural medicine has a long history with rich theoretical knowledge and treatment experience, and many natural bioactive compounds extracted from natural medicines exhibit neuroprotection against ischemic brain injury (Gu et al., 2014; Wu et al., 2010) which have been widely applied in treatment of cerebral ischemic stroke worldwide.
Herein, we outline the pathophysiology of cerebral ischemia which is shown in Fig. 1 and summarize the research progress in the development of bioactive compounds extracted from natural medicinal herbs that possess neuroprotective effects on ischemic brain injury. The effects of the individual bioactive compounds treating for cerebral ischemic injury have obtained specific achievements in recent years. However, there still exists the following shortcomings: (i) After the cerebral ischemic injury, neuronal degeneration and restore involved in numerous signaling pathways in various stages. But the molecular mechanism researches on individual compounds treating for ischemic stroke were studied targeting one single signaling pathway in most cases, which cause the synergistic biological effects of a pharmacological target in complex signaling networks challenging, yet to be fully explored. (ii) The therapeutic effects of natural compounds have been investigated in the aspects of morphology, pathomorphology, behavior, and biochemical indicator observation in most cases. However, the related research reports are still lacking in the field of molecular biology, which is relatively laggard as compared with chemical synthesis drug studies. (iii) Young animals have been commonly used in MCAO modal preparation for experimental study, ignoring the crucial impact of the aging factor on disease progression in human cerebrovascular disease, which leads to differences between the experimental results and clinical practice. (iv) The bioactive ingredients in traditional Chinese medicine are complex. Experimental research may investigate the biological effect of individual compounds acting on the specific pathological process only, but not reflect the characteristics of traditional Chinese medicine via multi-level, multi-pathway, multi-link, multi-target. Thus, typical agents targeting individual factors have limited effects on stroke treatment.
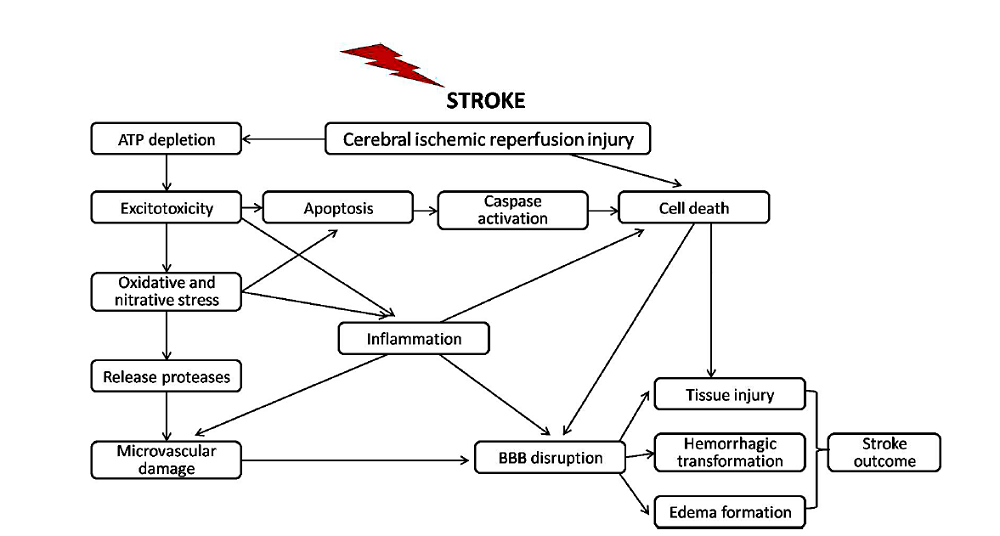 Figure 1.
Figure 1.Schematic review of the main pathophysiological mechanisms leading to cerebral ischemic reperfusion injury in acute ischemic stroke.
Excitotoxicity is a primary mechanism of neuronal injury following cerebral ischemia. It is a cell death form induced by neuronal stimulation with glutamate concentration, a consequence of the depletion of local energy supply (Wang et al., 2018). Due to deprivation of glucose and oxygen, cerebral ischemia triggers bioenergetic failure and reduction of adenosine triphosphate (ATP) generation in mitochondria (Hertz, 2008; Hertz et al., 2007; Yu and Gao, 2017). Reduced ATP production leading to plasma membrane depolarization causes an excessive influx of Ca2+ and Na+, and efflux of K+ (Khanna et al., 2014; Song and Yu, 2014) and glutamate reuptake abolishment in neurons and glial cells, both of which bring about glutamate accumulation at synapses. Glutamate accumulation further results in overstimulation of kainite, amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), and N-methyl-d-aspartic acid (NMDA)-type glutamate receptors, and the consequent influx of Na+, Ca2+, and Cl- ions through these ligand-gated channels (Chen et al., 2008; Dirnagl et al., 1999). The increased Ca2+ influx triggers a series of downstream signaling events, leading to metabolic derangements and resulting in local amplification of ischemic brain injury, referred to as excitotoxicity (Bano et al., 2005; Jeffs et al., 2007; Li et al., 2007). As described above, excitotoxicity causes ion gradients dysfunction (ionic imbalance) in ischemic injury, which leads to loss of K+ in exchange for Na+, Ca2+ and Cl- ions, accompanied with a water inflow, resulting in swelling of neurons and glial cells (Deb et al., 2010).
Another primary mechanism that contributes to tissue injury and infarct development is free radical damage (Lorenzano et al., 2018). Several mechanisms, such as NMDAR-mediated excitotoxicity, excessive Ca2+ influx, mitochondrial dysfunction, would lead to the free radical formation (Cherubini et al., 2005). Under ischemic conditions, accumulation of cytosolic and mitochondrial Ca2+ ions through the mitochondrial calcium uniporter into the matrix leads to the formation of mitochondrial permeability transition pores with the occurrence of electron leakage resulting in the generation of a substantial amount of oxygen radicals and release of pro-apoptotic molecules (Durukan and Tatlisumak, 2007; Triantafilou et al., 2013). Free radicals include two main types: reactive nitrogen species (RNS) and reactive oxygen species (ROS), both of which play critical roles in numerous pathological processes during ischemia-reperfusion (Sun et al., 2018).
When the free radicals production overpowers the endogenous radical scavenging, nitrative and oxidative stress occur (Beckman and Ames, 1998; Marrocco et al., 2017). ROS include hydrogen peroxide (H2O2), superoxide anion (O2-) and hydroxyl radicals (OH-) (Sun et al., 2018), all of which induce tissue damage through oxidative reactions with endogenous proteins, nucleic acids, lipids and carbohydrates (Adibhatla and Hatcher, 2010; Cabiscol et al., 2000; Lenaz, 2012; Shiva et al., 2004). Under normal physiological conditions, glutathione peroxidase (GPX), catalase, and superoxide dismutase (SOD) can protect the brain against ROS cytotoxicity by catalysis, and maintain a neutral balance (Robinson and Winge, 2010; Yoshioka et al., 2002). Overall, moderate oxidative stress in the ischemic penumbra can induce signaling cascades-mediated apoptosis, while severe oxidative stress in the ischemic core region can cause cell necrosis via severe cellular damages (Chen et al., 2011; Ridder and Schwaninger, 2009).
RNS include two species, NO and ONOO-, both of which can cause nitrosative stress and induce ischemia-reperfusion injury. There are three isoforms of nitric oxide synthase (NOS): neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS). Among them, nNOS and eNOS are calcium-dependent, and iNOS is calcium-independent. Generally, eNOS generated low level NO is physiological, while excess NO generated from activated iNOS and nNOS can cause further damage to the ischemic brain (Samdani et al., 1997). Overactive eNOS may be harmful upon ischemia-reperfusion, while partial eNOS inhibition may provide optimum prevention for the brain through peroxynitrite formation, which may positively affect the outcome by decreasing platelet and white blood cell clogging during reperfusion (Gursoy-Ozdemir et al., 2000).
Brain ischemia also triggers an inflammatory response followed by the generation of ROS (Kawabori and Yenari, 2015). Inflammation is crucial in the pathophysiology of ischemic stroke (Chamorro and Hallenbeck, 2006). The inflammatory responses include activation of microglia, astrocytes and endothelial cells, and infiltration of leukocytes, including monocytes and neutrophils (Huang et al., 2006; Zaleska et al., 2009). Microglia and astrocytes can be activated by ROS. Astrocytes could secret inflammation factors including iNOS, chemokines, and cytokines (Che et al., 2001; Dong and Benveniste, 2001; Yamagami et al., 1999), and they also express important costimulatory molecules and histocompatibility complex, contributing to anti-inflammatory responses (Stoll et al., 1998). Microglia play critical roles as resident phagocytic and immunocompetent cells in the brain (Morioka et al., 1993). Some studies showed that the activated microglia during ischemia by ROS is the main contributor of penumbra region secondary cell death (Danton and Dietrich, 2003; Lai and Todd, 2006); while others indicated that microglia are activated immediately following ischemic stroke stimulated by oxygen deficiency before neuronal death occurrence (Nakajima and Kohsaka, 2004). Three groups of cell adhesion molecules, including integrins, immunoglobulin superfamily, and selectins play critical roles during leukocyte infiltration into the brain parenchyma (Brouns and De Deyn, 2009; DeGraba, 1998). Activation of brain microvascular endothelial cells led by cerebral ischemia causes contacting of leukocyte with endothelium mediated by cell surface adhesion molecules via a tethering process (Kim et al., 2016). Macrophages and monocytes transmigrate from blood and enter into brain parenchyma following adhesion molecule expression at the vascular endothelium. Both microvascular obstructions induced by neutrophils and the generation of toxic mediators triggered by activated inflammatory cells can enhance the ischemia degree (Durukan and Tatlisumak, 2007).
Astrocytes are the principal glial cells in the brain, which play critical roles in neurotrophin, neural support, and neural regulation. They can coordinate changes of vascular tone induced by neuronal activity, remove excess glutamate from the synaptic cleft, prevent spillover of the transmitter, modulate excitotoxicity, help blood-brain barrier (BBB) formation and integrity maintenance, release pro- and anti-inflammatory molecules, and promote synaptogenesis (Sofroniew and Vinters, 2010). When an ischemic stroke happens, astrocytes, act as the double-edged sword, exert both beneficial and detrimental functions during an acute phase. On the one hand, stroke-induced astrocytic inflammation can make the ischemic lesion more serious. On the other hand, astrocytes can also exert neuroprotection via anti-excitotoxicity and releasing neurotrophins, thereby limiting lesion extension (Liu and Chopp, 2016). In short, astrocytes act as a balance. In response to neuronal damage, astrocytes reduce excitotoxicity and inflammation, but once the inflammation crosses a certain threshold, it can override anti-inflammatory functions (Cekanaviciute and Buckwalter, 2016). Astrocytes become reactive 2~7 days after stroke (Bao et al., 2012), and form a glial scar around the ischemic lesions in the brain as a physical and functional wall (Bidmon et al., 1998; Silver and Miller, 2004).
It is known to all that gap junctions provide ultrastructural cytoplasmic continuities to mediate intercellular communication, and these ultrastructural cytoplasmic continuities are critical for the formation of functional syncytium of astrocytes (Bennett et al., 2003). However, the role of astrocyte gap junctions in stroke remains controversial because both detrimental and beneficial substances can pass through them and exert opposite effects on stroke (Nakase and Naus, 2004). During the acute phase of ischemia, although the amounts of gap junction connections are reduced, the astrocytic gap junctional channels remain open (Cotrina et al., 1998; Martinez and Saez, 2000), allowing diffusion of pro-apoptotic factors from dying cells, leading to a secondary propagation of brain injury to the neighboring cells (Lin et al., 1998) and size expanding of ischemic lesions. In contrast, a neuroprotection function of gap junctions has been proved via stabilizing cellular calcium homeostasis and dissipating oxidative stress and thereby decreasing neuronal vulnerability to oxidative injury (Blanc et al., 1998).
Ischemia injury produces two types of cell death, including necrosis and apoptosis. Necrosis is caused by severe and irreversible deprivation of ATP, oxygen, and glucose within the core ischemic region. While a milder injury within the penumbra, which is reversible change leads to apoptosis (Datta et al., 2020). Apoptosis is triggered by either extrinsic or intrinsic stimuli (Du et al., 1996; Zaleska et al., 2009). The intrinsic stimuli for apoptosis are via a series of mitochondrial signaling pathways, and extrinsic stimuli are via several death receptors on the cell surface, including Fas (CD95/APO1), TRAIL (TNF related apoptosis-inducing ligand) receptors and TNF-α (Adams, 2003; Kroemer et al., 2007). Apoptosis acts as a balance to mitosis and can function to regulate tissue size and tumor growth related to pathologic processes (Shang et al., 2013). The process of apoptosis could involve in reperfusion by consuming cellular energy, and it is regarded as an energy-dependent process. Apoptosis results from activation of a series of detrimental biochemical events including metabolic derangements caused by ionic imbalance, leading to increased excitotoxicity, malfunction of mitochondria and endoplasmic reticulum, oxidative stress elevation leading to DNA damage, variation of proapoptotic gene expression, and activation of endonucleases and effector cysteine proteases (caspases) leading to the final genome degradation (Schaller and Graf, 2004). Apoptosis can be triggered by the generation of reactive oxygen species (Cai and Jones, 1998), and downstream signaling and enzymatic mechanisms are all regulated by oxidant activity (Dong et al., 2000). Caspases can be activated after cleavage, and activated caspases are protein-cleaving enzymes, which can modify homeostasis and repair proteins (Brouns and De Deyn, 2009). Caspases, especially caspases 1 and 3, play critical roles in ischemia-mediated apoptosis (Leist et al., 1997; Namura et al., 1998). Caspase-dependent apoptosis is initiated by the release of cytochrome C from mitochondria through activation of the apoptosome complex (Green and Reed, 1998). Caspase-independent apoptosis can delay neuronal death in ischemic stroke via the apoptosis-inducing factor in mitochondria (Cho and Toledo-Pereyra, 2008).
The blood-brain barrier (BBB) is a series of specialized structures located in the cerebrovasculature that control the molecule passage between blood and brain (Haley and Lawrence, 2017). The anatomical substrate of BBB is cerebral endothelium, and integrated BBB can protect the neuronal microenvironment. Besides the physical barrier, there is also a metabolic barrier located in the cerebral endothelium, contributing to drug and nutrient metabolism. Under the condition of cerebral ischemia, BBB integrity is damaged, leading to the entry of a substantial amount of molecules into the brain parenchyma, which further worsens brain damage (Haley and Lawrence, 2017). Although reperfusion is necessary to support the recovery of damaged neuronal tissues, it can also cause further cerebrovascular damage (Khatri et al., 2012). In a transient middle cerebral artery occlusion model, a biphasic BBB permeability increase during reperfusion corresponds to increased vasogenic edema formation, which may cause intracranial hypertension and further secondary damage (Sandoval and Witt, 2008). Early reperfusion may attenuate BBB alterations temporarily, but thrombolytic therapy or delayed reperfusion may exacerbate endothelial injury (Bang et al., 2007; Hjort et al., 2008; Kidwell et al., 2008). Also, uncontrolled entry of blood components into brain parenchyma led by BBB disruption causes red cell extravasation, resulting in a hemorrhagic transformation in the infarcted area (Brouns and De Deyn, 2009). The leaky BBB allows inflammatory cell transmigration, thereby enhancing the post-ischemic inflammatory response (del Zoppo and Hallenbeck, 2000).
Based on the pathophysiological mechanisms and potential therapeutic targets of cerebral ischemic, we next review the progress in the research of natural bioactive compounds for the treatment of ischemic stroke in the past 10 years. Many efficacious components isolated from traditional herbs have been demonstrated to exert a neural protective effect against cerebral ischemic insult in experimental research. We selected 16 suitable compounds targeting one or more targets to discuss in the following section and summarized their pharmacological mechanisms in Table 1 and Table 2.
| Compound | Origin |
Pinyin |
|---|---|---|
| Baicalin | Scutellaria baicalensis Georgi | Huangqin |
| Curculigoside A | Curculigo orchioides | Xiaomao |
| EGb761 | Ginkgo biloba | Yinxingye |
| Gastrodin | Gastrodia elata | Tianma |
| Glycyrrhizin | Glycyrrhiza glabra | Gancao |
| Gallic acid | Natural plants, tea, and red wines | / |
| Hydroxysafflor yellow A | Carthamus tinctorius L | Honghua |
| Honokiol | Magnolia officinalis | Houpu |
| Leonurine | Herba Leonuri | Yimucao |
| Paeonol | Moutan Cortex | Danpi |
| Puerarin | Puerariae radix | Gegen |
| Paeoniflorin | Paeonia lactiflora pall | Shaoyao |
| Shikonin | Lithospermum erythrorhizon | Zicao |
| Salidroside | Rhodiola rosea | Hongjingtian |
| Scutellarin | Erigeron breviscapus | Dengzhanxixin |
| Tanshinone IIA | Radix Salviae Miltiorrhiza Bge | Danshen |
| Compound | Model | Type | Time point | Path | Dosage | Targets | Major findings | Refs. |
|---|---|---|---|---|---|---|---|---|
| Baicalin | MCAO | in vivo | 2 h and 12 h after the onset of ischemia | I.P. | 100 mg/kg | MMP-9↓, occludin↓ | Neuronal damage↓, brain edema↓, BBB permeability↓ | (Tu et al., 2011a) |
| Curculigoside A | MCAO, OGD | in vivo and in vitro | 30 min after reperfusion; after OGD | I.V.; / | 5, 10, 20, 40, 80 mg/kg; 1, 3, 9, 27, 81 μM | HMGB1↓, NF-κB↓, IκB-α↓ |
Neurological deficit↓, infarct volume↓, brain edema↓, BBB permeability↓ | (Jiang et al., 2011) |
| EGb761 | MCAO | in vivo | Pretreatment and posttreament | P.O. | 100 mg/kg | HO-1↑ | Infarct volume↓, neurological deficit↓ | (Saleem et al., 2008) |
| Gastrodin | Hypoxia, NMDA treatment | in vitro | Pretreatment | / | 100, 200 mg/ml | Glutamate↓, NMDA↓ | Neurotoxicity↓ | (Xu et al., 2007) |
| Glycyrrhizin | MCAO | in vivo | 30 min after ischemia | I.V. | 2, 4, 10 mg/kg | Cytochrome C↓, caspase-3↓ | Infarct volume↓, brain edema↓, apoptosis↓ | (Gong et al., 2014) |
| MCAO | in vivo | 1, 24, 48 hours after MCAO | I.P. | 10 mg/mouse | HMGB1↓, TLR4↓, IL-17A↓ | Infarct volume↓, neuronal apoptosis↓ | (Zhang et al., 2014) | |
| Gallic acid | MCAO, Na2S2O4-inducing hypoxia/reoxygenation | in vivo and in vitro | 20 min before ischemia onset; pretreatment | I.V.; / | 0.1, 1, 10 μM ; 25 and 50 mg/kg | Cytochrome C↓, ROS↓, ATP↑ | Infarct volume↓, MPTP viability↓ | (Sun et al., 2014) |
| Hydroxysafflor yellow A | OGD | in vitro | After OGD | / | 20, 40, 80, 160, 320, 640 and 1280 μM | 1L-1β↓, TNF-α↓, iNOS↓, COX-2↓, MCP-1↓, MAPK/p38↓, ERK↓, JNK↓ | Cell viability↑, inflammation↓ | (Li et al., 2013) |
| LPS stimulation | in vitro | Cotreatment, pretreatment | / | 12-800 μM | TLR4↓, MyD88↓, NF-κB↓, JNK↓, ERK1/2↓, p38MAPK↓, BDNF↑ | Inflammation↓ | (Lv et al., 2016) | |
| Honokiol | MCAO, LPS stimulation | in vivo and in vitro | Posttreatment;cotreatment | I.P.; / | 0.1-10 μM, 0.01-10 μM | NO↓, p65 subunit of NF-κB↓, TNF-α↓, RANTES↓ | Exudation of cerebral capillary↓, brain water content↓ | (Zhang et al., 2013) |
| Leonurine | MCAO | in vivo | Posttreatment | P.O. | 15, 30, 60 mg/kg | SOD↑, GPx↑, MDA↓, ATP↓, ROS↓ | Infarct volume↓, neurological deficit↓, mitochondrial respiratory↑ | (Loh et al., 2010) |
| Paeonol | OGD | in vitro | Co-treatment with OGD | / | 0.2, 1, 5 μmol/l | NMDAR↓, Ca2+↓ | Neurotoxicity↓ | (Wu et al., 2008) |
| Puerarin | MCAO, OGD | in vivo and in vitro | 1 hour after MCAO; 12 h after OGD-R | I.G.; / | 2.62, 7.86, 23.59 mg/kg; 0.08, 0.4, 2 μM | Bcl-2↑, Bax↓, BDNF↓, Akt↑, ERK↑ | Infarct volume↓, neurological deficit↓, apoptosis of astrocyte↓ | (Wang et al., 2014b) |
| Paeoniflorin | MCAO | in vivo | 14 days after MCAO, twice per day | I.P. | 5 mg/kg | GFAP↓ | Infarct volume↓, neurological deficit↓, astrocytic activation↓ | (Guo et al., 2012) |
| Shikonin | MCAO | in vivo | Pretreatment and posttreatment | I.G. | 10 and 25 mg/kg | MMP-9↓, claudin-5↓, p38MAPK↓, TLR4↓, NF-κB↓ | Neurological deficit↓, infarct volume↓, brain edema↓, BBB permeability↓ | (Wang et al., 2014a) |
| Salidroside | MCAO | in vivo | 1 h after reperfusion of MCAO | I.P. | 50 mg/kg | CD11b↓, IL-6↓, IL-1β↓, TNF-α↓, CD14↓, CD44↓, iNOs↓, Akt↑, HIF1α↑, HIF2α↑, HIF3α↑, EPO↑ | Neurological deficit↓, inflammation↓ | (Wei et al., 2017) |
| Scutellarin | MCAO, LPS -activated | in vivo and in vitro | 2 h before and 12, 24, 36, 48, 60 h after ischemia; pretreatment | I.P.; / | 100 mg/kg; 10 ml basic medium containing 0.54 mM of scutellarin | GFAP↑, Notch-1↑, NICD↑, HES↑, nestin↑ | Reaction of astrocytes↑ | (Fang et al., 2015) |
| Tanshinone IIA | MCAO | in vivo | 10 min after MCAO | I.P. | 25 mg/kg, 40 mg/kg | Bcl-2↑, caspase-3↓ | Infarct volume↓, brain edema↓, neurological deficit↓, apoptosis↓ | (Chen et al., 2012) |
| MCAO, OGD | in vivo and in vitro | 7 days before MCAO; pre-treatment | I.P.; / | 5 mg/kg; |
Bax↓, Bcl-2↑ | Infarct volume↓, brain edema↓, neurological deficit↓, apoptosis↓ | (Zhou et al., 2017) |
I.G., intragastrical; I.P., intraperitoneally; I.V., intravenous; LPS, lipopolysaccharide; MCAO, middle cerebral artery occlusion; OGD, oxygen and glucosedeprivation; P.O., per oral
Gastrodin was extracted from the rhizome of a Chinese herbal medicine Gastrodia elata (Orchidaceae) and has been used in the treatment of epilepsy, stroke, dementia, and dizziness. Gastrodin was investigated in the treatment of hypoxia-induced neurotoxicity in rat cortical neurons by Xu et al. (2007). Results showed that gastrodin effectively promoted neuron survival, and suppressed hypoxia-stimulated enhancement of extracellular glutamate level. Furthermore, gastrodin reduced neurotoxicity induced by NMDA or glutamate in cortical neurons, and also suppressed extracellular glutamate level stimulated by NMDA. These results demonstrated the neuroprotective effect of gastrodin against hypoxia with a possible underlying mechanism via decreasing the extracellular glutamate level (Xu et al., 2007).
Paeonol, a phenolic bioactive component of Chinese traditional medicine Moutan Cortex, possesses neuroprotection against brain ischemia injury by increasing antioxidase activity, reducing oxygen-free radicals-induced damage, and inhibiting cell apoptosis in vivo. Studies by Wu et al. (2008) showed that paeonol treatment significantly enhanced cell survival, suppressed the binding force of NMDA receptors, and inhibiting the overload of intracellular Ca2+, which elucidated that paeonol exerts neuroprotective effect by restraining calcium overload and decreasing excitotoxicity (Wu et al., 2008).
EGb761 is a well-known extract from Ginkgo biloba and has antioxidant and neuroprotective activities against various neurological and cardiovascular diseases, including Alzheimer's disease, ischemia, and depression (Ahlemeyer and Krieglstein, 2003; Chandrasekaran et al., 2003). Saleem et al. (2008) used the MCAO model to evaluate both the preventive and therapeutic effects of EGb761 on stroke. Results showed that EGb761 could significantly improve the outcome in MCAO mice in terms of neurobehavioral function and infarct volume, increase de novo HO-1 in neurons, and provide neuroprotection against H2O2-induced oxidative stress injury and glutamate-induced excitotoxicity (Saleem et al., 2008).
Gallic acid (GA) is an important polyphenolic compound widely existing in tea, red wines, and natural plants (Shahrzad et al., 2001). GA was found to be able to alleviate memory deficits caused by streptozotocin by suppressing oxidative stress and activating the corresponding enzyme-dependent signaling pathways (Kade and Rocha, 2013; Mansouri et al., 2013). Sun et al. (2014) explored the effects and related mechanisms of GA both in vitro and in vivo. The study demonstrated that GA attenuated hypoxia/reoxygenation insult and protected neurons against cerebral ischemic injury via modulating mitochondria dysfunction, reducing oxidative stress and suppressing neural cell apoptosis (Sun et al., 2014).
Leonurine is an alkaloid present in Herba Leonuri, and it was reported to have uterotonic action and antiplatelet aggregation activities (Kuang et al., 1988). Loh et al. (2010) pretreated MCAO rats with leonurine orally for 7 days to verify its therapeutic potential. It is revealed that leonurine protects the cells from neuronal injury through increasing activities of antioxidant enzymes SOD and GPS and decreasing levels from the LPT marker MDA. Also, leonurine inhibited ROS production and ATP biosynthesis in terms of mitochondrial modulation. Animal studies also demonstrated that mitochondrial dysfunction was rescued by leonurine treatment (Loh et al., 2010).
Hydroxysafflor yellow A (HSYA), a major active component of the safflower yellow pigments, has been applied in the treatment of cardiovascular and cerebrovascular diseases. Research performed by Li et al. (2013) demonstrated that HSYA could improve the viability of BV2 after oxygen-glucose deprivation (OGD), decrease protein levels of COX-2 and iNOS, and mRNA levels of iNOS, MCP-1, COX-2, TNF-α and 1L-1β in BV2 microglia after OGD, and reduce p65 nuclear translocation and p38 phosphorylation in BV2 microglia after enhanced by OGD. These results suggested that HSYA could inhibit OGD-induced inflammatory responses in BV2 microglia by suppressing p38 phosphorylation and NF-κB signaling (Li et al., 2013). Lv et al. (2016) showed that HSYA could inhibit TLR4 expression and the downstream effectors NF-κB, MyD88, and MAPK-regulated proteins p38MAPK, ERK1/2, and JNK, as well as LPS-induced release of inflammatory cytokines. However, HSYA could up-regulate BDNF expression. These results revealed that HSYA could exert neurotrophic and anti-inflammatory roles by inhibiting TLR4-mediated signaling (Lv et al., 2016).
Honokiol, extracted from Magnolia officinalis has a series of bioactivities, including anti-apoptosis, anti-oxidation, and cerebral infarction reducing activities. It has been reported that honokiol inhibits inflammation in lymphocytes, macrophages, and neutrophils (Kim and Cho, 2008). Zhang et al. (2013) investigated the effects of honokiol in cerebral ischemia-reperfusion mice. They found that honokiol effectively suppressed the levels of NF-κB p65 subunit, TNF-α, nitric oxide (NO), and RANTES/CCL5. Therefore, honokiol might exert anti-inflammatory functions via inhibiting activation of NF-κB and the production of inflammatory factors (Zhang et al., 2013).
Salidroside is a bioactive component of Rhodiola rosea (Lai et al., 2015; Shi et al., 2012). Wei et al. (2017) explored the underlying mechanisms for the neuroprotective and anti-inflammatory activities of salidroside, and the results showed that salidroside induced production of HIFα subunits and EPO, mediating its anti-inflammatory effect (Wei et al., 2017).
Paeoniflorin (PF) is the principal bioactive component isolated from the root of Paeonia lactiflora pall. Studies showed that PF administration attenuated pathological damages, behavioral changes as well as cognitive impairments induced by ischemia (Chen et al., 2006; Liu et al., 2005, 2006). Guo et al. (2012) demonstrated that intraperitoneal administration of PF twice a day for 14 days displayed inhibition of astrocytic activation and proliferation against ischemic stroke via reversing the increased expression of astrocyte marker protein GFAP (Guo et al., 2012).
Scutellarin is a major active component of Erigeron breviscapus, exhibiting anti-inflammation, anti-apoptotic and antioxidant activities in an animal ischemic stroke model. Studies showed that scutellarin significantly reduced pro-inflammatory mediators both in activated microglia under MCAO-induced cerebral ischemia and in BV-2 microglia in vitro (Yuan et al., 2015, 2014). Fang et al. (2015) investigated the neuroprotective effect of scutellarin against focal cerebral ischemia in vivo and lipopolysaccharide (LPS)-activated BV-2 microglia in vitro. They treated MCAO rats with scutellarin before and after ischemic stroke, found that scutellarin can regulate the expression of nestin, GFAP, Notch-1, and proinflammatory mediators in reactive astrocytes. In vitro experiment showed that scutellarin exerted an indirect effect on astrocytes through the intermediary activated microglia. These results suggested that scutellarin exerted a neuroprotective effect by facilitating the communication between reactive astrocytes and activated microglia in cerebral ischemia (Fang et al., 2015).
Tanshinone IIA (TSA), a lipid-soluble bioactive constituent of Chinese herb Danshen (Radix Salviae Miltiorrhiza Bge), was used for the treatment of cerebrovascular and cardiovascular diseases in the clinic. Studies performed by Chen et al. (2012) showed that treatment with TSA after middle cerebral artery occlusion (MCAO) dramatically decreased brain water content and infarct volume, and attenuated neurological deficits. It also remarkably suppressed the level of cleaved caspase-3 and enhanced the protein level of B-cell lymphoma 2 (Bcl-2) in the ischemic cortex. Also, TUNEL-positive cells were found in the penumbra of the treated group. These results indicated that TSA has neuroprotective effects against focal cerebral ischemic/reperfusion injuries via suppressing apoptosis (Chen et al., 2012).
Zhou et al. (2017) studied the neuroprotective effects and action mechanisms of TSA in cerebral infarction both in vitro and in vivo, and they found a 7-day intraperitoneal administration of TSA before MCAO effectively decreased cerebral edema, cerebral infarct volume, and neurological deficits score. Cellular experiments showed that TSA significantly enhanced cell viability and suppressed cell apoptosis in neurons, and further studies showed that TSA dramatically upregulated the expression of Bcl-2 and downregulated the Bax. These results indicated that TSA has a preventive effect on cerebral infarction through inhibition of neuronal cell apoptosis.
Glycyrrhizin (GL) is an active constituent of the root of Chinese herb Glycyrrhiza glabra containing a glycyrrhizic acid. GL has a series of pharmacological activities, including anti-tumor, anti-viral, anti-inflammatory, and hepatoprotective activities. Gong et al. (2014) explored the potential neuroprotective effect of GL against ischemia/reperfusion injury and the related mechanisms. They found pre-treatment with GL remarkably decreased infarct volume and attenuated neurological deficits by inhibiting cytochrome C release and caspase-3 activity and alleviating apoptotic injury resulted from cerebral ischemia/reperfusion (Gong et al., 2014). A similar neuroprotective effect was observed by Zhang et al. (2014) with GL administration. Furthermore, they found administration of GL significantly inhibited IL-17A secretion induced by HMGB1, suggesting HMGB1 inhibition and the subsequent inhibition of IL-17A contribute to the neuroprotective effect of GL (Zhang et al., 2014).
Puerarin is a primary bioactive constituent of puerariae radix and exhibits an effect in the treatment of various diseases, including cardiovascular disease. The anti-hypertensive activity of puerarin was first demonstrated in cats in the 1980s (Lu et al., 1987). Wang et al. (2014b) explored the neuroprotective functions of puerarin, both in vivo and in vitro. They found significant astrocyte apoptosis during cerebral ischemia-reperfusion, and puerarin post-treatment could effectively suppress astrocyte apoptosis by regulating apoptosis-related signaling pathways, including PI3K/AKT, MAPK/ERK and BDNF (Wang et al., 2014b).
Baicalin is a flavonoid compound extracted from Scutellaria baicalensis Georgi with neuroprotective activity through anti-apoptotic and anti-inflammatory properties (Tu et al., 2009) via suppressing toll-like receptor 2/4 (TLR2/4)-mediated signaling (Tu et al., 2011b), and inhibiting the expression of nucleotide-binding oligomerization domain protein 2 (NOD2) and TNF-α (Tu et al., 2011b). Studies performed by Tu et al. (2011a) showed that baicalin could effectively attenuate neuronal damage and brain edema induced in a focal cerebral ischemia model. Moreover, baicalin could ameliorate BBB permeability changes induced by ischemic injury by inhibiting MMP-9 expression and occludin degradation (Tu et al., 2011a).
Shikonin, extracted from Lithospermum erythrorhizon, possesses bioactivities, including anticancer anti-inflammatory and antithrombotic activities (Assimopoulou et al., 2004; Chan, 2001). Wang et al. (2014a) investigated the potential neuroprotective effect of shikonin on stroke, and found that shikonin attenuated edema, reduced infarct volume, alleviated neurological deficit and brain leakage, decreased pro-inflammatory mediators in the ischemic cortex, enhanced claudin-5 expression, and suppressed MMP-9 over-expression in the ischemic brain. These findings revealed shikonin exerted neuroprotective activities against cerebral ischemic injury by suppressing inflammation and ameliorating BBB permeability (Wang et al., 2014a).
Curculigoside A is a bioactive compound extracted from Curculigo orchioides. Jiang et al. (2011) treated MCAO rats with curculigoside A. They found that curculigoside A inhibited phosphorylation of NF-κB and IκB-α induced by TNF-α, as well as HMGB1 expression in SH-SY5Y cells, and inhibited HMGB1 expression and NF-κB activation, decreased cerebral extravasation and attenuated histopathological damage in rats with ischemia and reperfusion. These data suggested curculigoside A played neuroprotective roles by suppressing HMGB1 and NK-κB signaling pathway and attenuating BBB breakdown (Jiang et al., 2011).
Ischemic stroke occurs when blockage of the brain artery causes a reduction of regional cerebral blood flow, which leads to selective nerve damage and triggers a cascade of pathophysiological events, including excitotoxicity, free radicals generation, inflammation, astrocytic influence, apoptosis, and BBB dysfunction. In ischemic stroke treatment, recovery of blood supply of brain tissue in the ischemic region and salvage of brain cells in the penumbra area of the ischemic brain is especially crucial for neuronal survival. Chinese medicine exhibits a particular advantage in neuroprotective effects against ischemic stroke for its availability, lower price, and less toxic effects compared with the synthetic drug (Chaturvedi et al., 1999).
Due to the complexity of Chinese herbal medicine ingredient, the possible mechanism underlying their neuroprotection is limited, which prompts the focus of scientific research to shift from traditional Chinese herbal medicine to natural bioactive compounds extracted from Chinese herbal. Although the complex pathogenesis of ischemic stroke and the multifactorial effects of natural bioactive compounds extracted from herbal medicine may bring a considerable challenge for exploring the synergistic effects of multiple mechanisms, natural compounds are the proper sources for developing novel therapeutic strategies on ischemic brain injury rescue. With the increasing application of metabolomics, proteomics, DNA microarray, LC-MS, etc. in experimental studies, comprehensive matrix analytic technologies offer new opportunities to explore neuroprotection activities of natural compounds against cerebral ischemic injury. Last but not least, although herbal medicine has a golden future for stroke treatment, a large amount of effort should be invested to transform the success of animal research to human use.
In this review, sixteen representative bioactive compounds derived from natural medicines have potential neuroprotective activities against cerebral ischemic injury by the different pathophysiologic mechanisms. They are the primary ingredients that exert neuroprotection in treatment for cerebrovascular diseases. The results reflect tremendous progress that is promising to protect cerebral ischemia by the compounds extracted from traditional Chinese medicines. Given the limitation of narrow therapeutic time window with tPA treatment, which is the standard therapeutics in acute ischemic stroke, such efforts may provide a direction for the further exploitation of traditional Chinese medicine in cerebral ischemia treatment as an adjunct therapy.
This work was supported by grants from National Natural Science Foundation of China (81503370), Shanghai Natural Science Foundation (19ZR1447900), Youth Talent Promotion Project of China Association of Chinese Medicine (CACM-2019-QNRC2-C10),Science and Technology Innovation Project of Putuo District Health System (ptkwws201908) and Shanghai Putuo District Health System Clinical Special Disease Construction Project of 2019: Stroke (2019tszb02).
The authors confirm that this article content has no conflict of interest.