
We examined synergistic effects of inhibiting reactive oxygen species generated from the mitochondria and from nicotinamide adenine dinucleotide phosphate oxidase on neurotoxicity. Primary hippocampal neurons were exposed to amyloid β, and the cells were treated with diazoxide or/and diphenyleneiodonium chloride. We found that the cell viability was decreased significantly after exposure to amyloid β for 72 h with higer reactive oxygen species and malondialdehyde levels, higher caspase-3 and cleaved caspase-3 levels and lower B-cell lymphoma 2 (Bcl-2) level. Both diazoxide and diphenyleneiodonium increased cell viability by inhibiting the increase in reactive oxygen species and caspase-3 activity as well as the decrease in Bcl-2 induced by amyloid β. The combination of diazoxide and diphenyleneiodonium exhibited better protective effects compared to a single treatment. In conclusion, the activation of a mitochondrial potassium channel in combination with the inhibitor of nicotinamide adenine dinucleotide phosphate oxidase exhibit synergistic protective effects against amyloid β neurotoxicity.
The main hallmarks of Alzheimer's disease (AD) are senile plaques (SPs) (Butterfield et al., 2013). The deposition of neurotoxic amyloid-beta (Aβ) protein can aggravate reactive oxygen species (ROS) production and exacerbate oxidative stress (Song et al., 2018; Xie et al., 2018). Notably, mitochondrial ATP sensitive potassium (MitoKATP) channel, which can be activated by diazoxide, depolarizes mitochondrial membranes, inhibits the release of ROS and maintains mitochondrial stability (Deryagin et al., 2017). Diazoxide not only resists apoptosis of PC12 cells induced by rotenone but also inhibits hydrogen peroxide-induced apoptosis of cerebellar granule neurons by stabilizing mitochondrial membrane potentials (Tai et al., 2003). We had confirmed that the activation of MitoKATP by diazoxide could inhibit oxidative stress and apoptosis of cholinergic neurons induced by Aβ1-42 (Fu et al., 2014).
It has been shown that nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) contributes to ROS production in AD (Arafdar and Pula, 2018). For example, the silencing of p47phox or blocking NADPH oxidase 2 (NOX2) inhibited superoxide production in cultured neurons and mouse hippocampus induced by N-methyl-D-aspartate (NMDA) receptor (Brennan et al., 2009).
The existence of two main sources of intracellular ROS indicates possible interaction between the mitochondria and NOX (Zinkevich et al., 2017). Nevertheless, the synergism between two sources of intracellular ROS in the development of AD remains unclear. In this study, we hypothesized that the ineffectiveness of anti-oxidative therapy for AD might, in part, result from synergistic effects of intracellular ROS. This study aimed to investigate whether activating mitoKATP channels combined with diphenylene iodonium (DPI) could exhibit synergistic protection on Aβ1-42 induced neurotoxicity.
Protocols for animals were approved by Animal Use and Care Committee (Approval No. 20170605). Hippocampal neurons were isolated and cultured from embryonic Sprague-Dawley rats following previously described protocols (Shen et al., 2006). Aβ1-42 (Sigma, St. Louis, MO, USA) was dissolved in dimethyl sulfoxide (DMSO, Sigma) at 0.5 mol/L, then diluted in phosphate-buffered saline (PBS). Neurons were treated differently as follows: 1) Control, 2) Aβ1-42 (2 μM Aβ1-42 treatment for 24 or 72 h), 3) Diazoxide (Sigma) + Aβ1-42 (500 μM diazoxide pretreatment for 1 h followed by Aβ1-42 for 24 or 72 h), 4) diphenylene iodonium (DPI) (Sigma) + Aβ1-42 (1 μM DPI pretreatment for 1 h followed by Aβ1-42 for 24 or 72h) and 5) diazoxide + DPI + Aβ1-42 (diazoxide pretreatment for 1 h, followed by 1 h of DPI pretreatment and 24 or 72 h of Aβ1-42 treatment). Each of these groups was then divided into two subgroups for evaluation at either 24 or 72 h.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) kit (Cat# ST316, Beyotime Biotech, Shanghai, P. R. China) was used to evaluate the viability of neurons. Neurons were incubated with MTT (0.5 mg/mL) at 37 °C for 2 h, and formazan was dissolved for measurement at 570 nm.
Cells were incubated with 100 μM dichloro-dihydro-fluorescein diacetate (DCFH-DA) (Beyotime, Shanghai, China) for 30 min, and then extracellular DCFH-DA was washed away. Cellular fluorescence was measured on a CytoFluor4000 reader at 488 nm (excitation) and 525 nm (emission). Cellular malondialdehyde (MDA) was measured using the MDA assay kit (Cat# S0131, Beyotime Biotech, Shanghai, People’s Republic of China) at 532 nm.
Proteins were extracted from the neurons and measured by using BCA Kit (Pierce). After sodium dodecyl sulfate-polyacrylamide gel electrophoresis, the proteins were transferred to nitrocellulose membranes. Membranes were incubated with following antibodies at 4 °C overnight: goat anti-rat Bcl-2 and goat anti-rat Bax (1 : 400; Santa Cruz Biotechnology), goat anti-rat Caspase-3 and goat anti-rat cleaved caspase-3 and (1 : 100; Cell Signaling, Danvers, MA, USA) or goat anti-rat β-actin monoclonal antibody (1 : 1,000; Cell Signaling). Membranes were washed with TBST (0.05% v/v Tween-20 in TBS) and then incubated with horseradish peroxidase-conjugated antibody (1 : 3,000) at room temperature for 1 h. The membranes were exposed to chemiluminescence kit (Millipore, Billerica, MA, USA). Optical density was calculated with Quantity One software (Bio-Rad, Shanghai, P. R. China).
Data are presented as mean±standard deviation (SD) unless indicated otherwise and analyzed with GraphPad Prism 7.0 software (GraphPad Inc., San Diego, CA, USA). The differences were analyzed by one-way analysis of variance (ANOVA) followed by Scheffe’s post hoc test. P < 0.05 indicated significant.
To examine the effects of diazoxide and DPI on the viability of primary hippocampal neurons, we performed MTT assay. We found that Aβ1-42 significantly inhibited the viability of neurons compared to control (P < 0.05 for 24 h, P < 0.001 for 72 h). Pretreatment by diazoxide or DPI significantly increased cell viability compared to Aβ1-42, while pretreatment with both diazoxide and DPI led to higher cell viability (Fig. 1).
 Figure 1.
Figure 1.Diazoxide and DPI pretreatment improved the viability of neurons exposed to Aβ1-42. Primary neurons were treated by diazoxide (DZ, 500 μM), Aβ1-42 (2 μM), or/and DPI (1 μM). Cell viability was assessed by MTT assay (n = 3). *P < 0.05; ***P < 0.001 vs. control; #P < 0.05 vs. Aβ1-42 + DZ + DPI, &&P < 0.01, &&&P < 0.001 vs. Aβ1-42 group.
Next, we assessed the effects of diazoxide and DPI on MDA and ROS levels in primary neurons exposed to Aβ1-42. Exposure to Aβ1-42 for either 24 h or 72 h significantly increased MDA and ROS levels. Pretreatment with diazoxide or DPI significantly decreased MDA and ROS levels, while pretreatment with both diazoxide and DPI inhibited Aβ1-42-induced oxidative stress to a greater extent (Fig. 2).
 Figure 2.
Figure 2.Diazoxide and DPI pretreatment reduced ROS and MDA in neurons exposed to Aβ1-42. PC12 cells were treated by diazoxide (DZ, 500 μM), Aβ1-42 (2 μM), or/and DPI (1 μM). Values are mean±SD (n=3). *P < 0.05; **P < 0.01, ***P < 0.001 vs. control; #P < 0.05 vs. Aβ1-42 + DZ+ DPI, && P < 0.01, &&&P < 0.001 vs. Aβ1-42 group.
To explore the mechanism underlying neuroprotection of diazoxide and DPI, we detected the levels of caspase-3, cleaved caspase-3, Bax, Bcl-2 in neurons exposed to Aβ1-42. Twenty-four h treatment with Aβ1-42 significantly increased the levels of caspase-3, cleaved caspase-3, and Bax, while decreased Bcl-2 level, all of which could be completely reversed by pretreatment with diazoxide or DPI or the combination of both (Fig. 3). Similarly, 72 h treatment with Aβ1-42 increased caspase-3 and cleaved caspase-3 levels while reduced Bcl-2 level and Bcl-2/Bax ratio, all of which were partially reversed with diazoxide or DPI, while pretreatment with both diazoxide and DPI significantly inhibited Aβ1-42-induced changes of apoptosis-related proteins (Fig. 4).
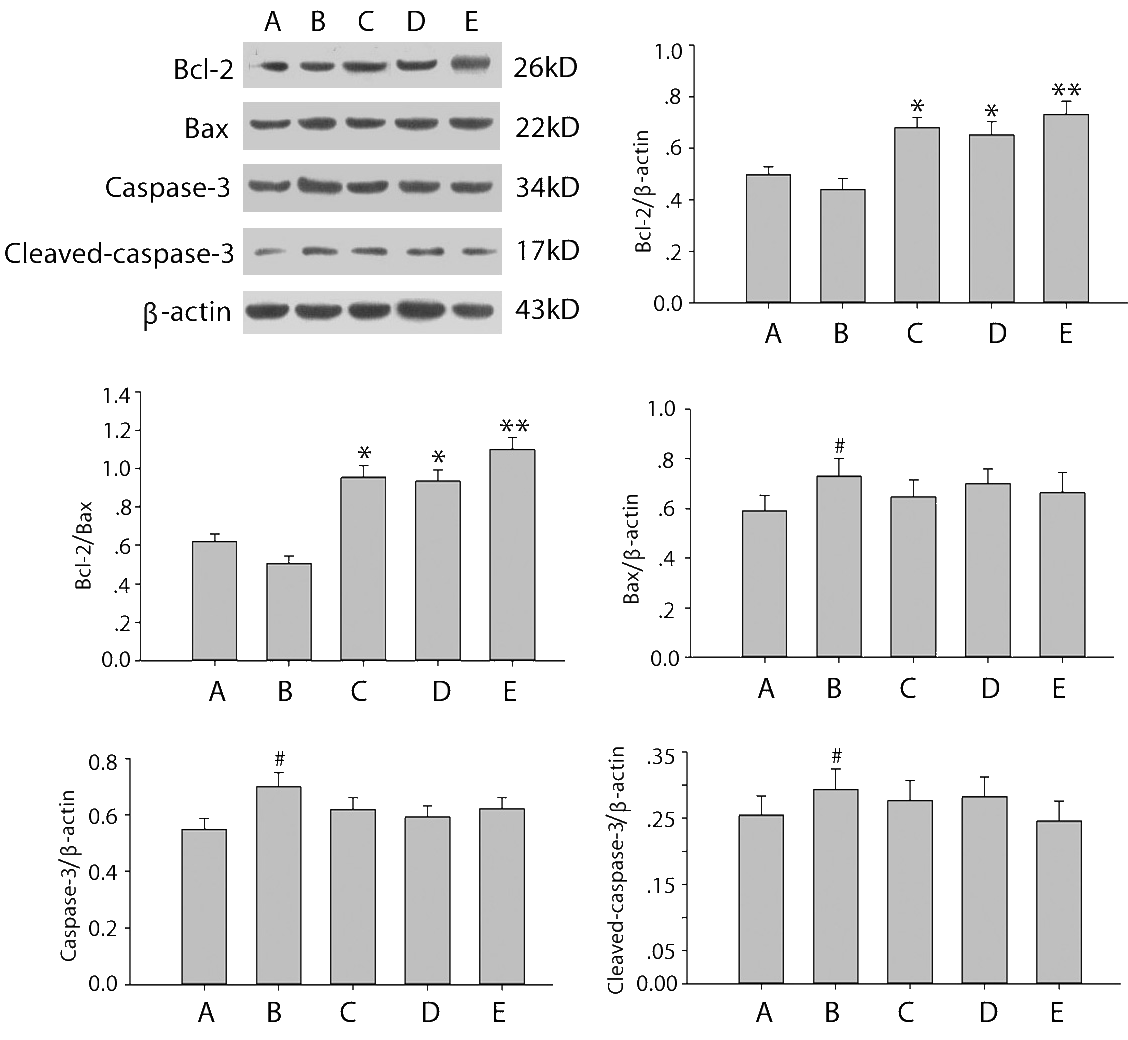 Figure 3.
Figure 3.Relative protein expression levels and Bcl-2/Bax ratios in neurons exposed to Aβ1-42 for 24 h. Protein levels were normalized to β-actin. A: Control, B: Aβ1-42, C: Aβ1-42 + diazoxide (DZ, 500 μM), D: Aβ1-42 + DPI (1 μM), E: Aβ1-42 + DZ (500 μM) + DPI (1 μM). Values are mean±SD (n=3). #P < 0.05 vs. control; *P < 0.05, **P < 0.01 vs. Aβ1-42.
 Figure 4.
Figure 4.Relative protein expression levels and Bcl-2/Bax ratios in neurons exposed to Aβ1-42 for 72 h. Protein levels were normalized to β-actin. A: Control, B: Aβ1-42, C: Aβ1-42 + diazoxide (DZ, 500 μM), D: Aβ1-42 + DPI (1 μM), E: Aβ1-42 + DZ (500 μM) + DPI (1 μM). Values are mean±SD (n=3). #P < 0.05, ##P < 0.01, ###P < 0.001 vs. control; *P < 0.05; **P < 0.01, ***P < 0.001 vs. Aβ1-42 group.
We found that Aβ1-42 significant increases ROS and MDA levels, leading to oxidative stress and apoptosis, consistent with previous studies (Butterfield et al., 2013; Swomley et al., 2014). Mitochondria play crucial role in ROS production (Villa-Hernández et al., 2018). The neuroprotective effects of activated MitoKATP mainly involve its ability to sustain mitochondrial depolarization, limit ROS overproduction, maintain ATP levels, and increase catalase and SOD2 (O-Uchi et al., 2014). Our results in this study further substantiate the neuroprotective effects of MitoKATP pre-activation. Specifically, primary neurons were resistant to Aβ1-42-induced neurotoxicity after diazoxide pretreatment, as demonstrated by decreases in intracellular ROS and MDA levels, decreases in Bax and caspase-3 levels, and increase in Bcl-2 level. Such protective effects are associated with anti-oxidative stress and anti-apoptosis. However, we found some differences in the protective effects of diazoxide and DPI at 24 h vs. 72 h. This may be due to higher Aβ1-42 neurotoxicity after cells were exposed longer at 72 h vs. 24 h. This is a limitation of this study because we did not examine more time points after Aβ1-42 exposure and diazoxide/DPI treatment in primary neurons. Also, animal studies are important to verify the protective effects of diazoxide and DPI in vivo.
Under abnormal conditions of excessive activation, large amounts of ROS can be produced, resulting in oxidative stress damage (Carvalho and Moreira, 2018; Ewald, 2018; Montes et al., 2019). It has been reported that NOX, in particular, NOX2, is implicated in AD (González-Reyes et al., 2017; Jiang et al., 2016). For example, a neocortical infusion of Aβ1-40 within wild-type mice can lead to ROS production and cerebral vascular regulation disorders, whereas the infusion of Aβ1-40 into Nox2-/- mice failed to increase ROS levels (Park et al., 2005). Moreover, when Tq2576 mice with APP Swedish mutation were hybridized with Cybb-/- mice with loss of NOX2 function, they showed significantly reduced oxidative stress within the brain (Park et al., 2008). Notably, results from our study demonstrate that NOX2 inhibitor DPI inhibited Aβ1-42 induced neurotoxicity, reduced intracellular ROS and MDA levels, and changed caspase-3, Bcl-2, Bax levels, all of which are involved in stress-induced oxidative effects and mitochondria apoptosis.
Mitochondria and NOX represent two main sources of intracellular ROS, and their interaction has been suggested (Sies, 2017). In 293T cells, serum deprivation led to an acute production of mitochondrial ROS and NOX1 activation. Mitochondrial ROS is produced within minutes of serum deprivation, and this initial release of ROS activates NOX1. The maintenance of ROS production in the later phase requires the activation of NOX1 (Lee et al., 2006). Ang II caused mitochondrial damage in endothelial cells, which was inhibited by NOX blocker apocynin (Rey et al., 2001). These data indicate a feedback loop between mitochondria and NOX. Our results show that diazoxide and DPI can individually decrease Aβ1-42-induced oxidative stress, and the combined effects of two treatments enhanced the degree of oxidative stress inhibition, indicating synergistic effects resulting from their combined application.
Based on our results, we propose that Aβ1-42 increases mitochondrial ROS levels, which activate NOX2 to increase cytoplasmic ROS production. As a result, NOX derived ROS increases mitochondrial dysfunction and mitochondrial ROS production. Intervention with either diazoxide or DPI can disrupt this vicious cycle chain to exert a certain amount of neuroprotection. However, the combined application of diazoxide and DPI may effectively block this cycle to produce significant neuroprotective effects.
This research was supported by Shandong Provincial Natural Science Foundation (ZR2016HL16).
The authors declare no conflict of interest.
QF, NG, FC, GX, and HH performed the experiments and analyzed the data, QS and GM designed the study and wrote the manuscript.