





 , Xiaoming Xin 1,5,*
, Xiaoming Xin 1,5,*1 Shanghai University of Medicine and Health Sciences, Shanghai 201318, P. R. China
2 Tai'an City Central Hospital, Taian 271000, P. R. China
3 Shanghai University of Traditional Chinese Medicine, Shanghai 201318, P. R. China
4 School of Pharmaceutical Science, Taishan Medical University, Taian 271000, P. R. China
5 Affiliated Health School of Shanghai University of Medicine and Health Sciences, Shanghai, 201318, P. R. China
#These authors contributed equally.
Abstract
We investigate whether β-carotene, a known natural antioxidant, can reduce oxidative stress induced by traumatic brain injury. In addition, we investigated the underlying mechanism of traumatic brain injury focusing on the NF-E2-related factor (Nrf2) pathway. A controlled cortical impact model was used to mimic traumatic brain injury. Using this model, we evaluated brain edema, lesion volume, neurologic deficits, reactive oxygen species, and the expression of Nrf2-related protein markers. The results of our study demonstrated that cognitive performance and neural functions were improved with β-carotene administration. In addition, β-carotene reduced brain edema and reactive oxygen species levels after traumatic brain injury. Nrf2 nuclear accumulation was increased and was accompanied by decreased Keap1 expression. The expression of quinone oxidoreductase 1, a target gene of the Nrf2 signaling pathway was increased. However, lesion volume was not significantly reduced after β-carotene treatment. Taken together, our data demonstrated that β-carotene administration was neuroprotective and alleviated oxidative stress by modulating the Nrf2/Keap1mediated antioxidant pathway in the traumatic brain injury model.
Keywords
- Traumatic brain injury
- β-carotene
- oxidative stress
- Nrf2-ARC signaling pathway
- mouse model
Traumatic brain injury (TBI) resulting from external mechanical forces are a major health concern (Fernández-Gajardo et al., 2014; Shen et al., 2016; Tsai et al., 2013). The physical impact results in primary injury, structural and functional damage. Soon after the primary insult, secondary injury manifests, which includes inflammation, oxidative stress from blood metabolites and iron toxicity. This eventually leads to neuronal degeneration and further apoptosis (McGuire et al., 2018). Despite increased focus on TBI, no effective treatment strategies are available to date (Agoston and Langford, 2018). Hence, finding new therapies that are effective in preventing secondary TBI injury needs to be developed.
A significant increase in free radical production is one of the main reasons for secondary TBI injury, which leads to neuronal tissue and vasculature damage. Studies have demonstrated that different mechanisms contribute to secondary injury, such as oxidative stress, hypoxia and edema. (Campolo et al., 2013; Kabu et al., 2015; Fehily and Fitzgerald, 2017) All these results in significant increase in free radical production resulting in secondary TBI injury. Increased levels of reactive oxygen species (ROS) have been observed in brain tissue and cerebral spinal fluid from both animal models and TBI patients (Shi et al., 2018). A significant increase in free radical production leads to functional impairment of neurons (Bolduc et al., 2018). Although the mechanism of TBI is unclear, several previous studies have demonstrated the involvement of oxidative stress and apoptosis (Cheng et al., 2016; Liu et al., 2018). Several signaling pathways are involved in regulating oxidative stress after TBI (Sun et al., 2017; Fu et al., 2018). Nrf2 signaling pathway is activated after oxidative stress to regulate downstream target genes, such as HO1 (Hall et al., 2018; Zhang and Wang, 2018). HO1, a rate-limiting enzyme for heme degradation, modulates oxidative stress and inflammation, and is cytoprotective. Hence, reducing oxidative stress is hypothesized to be beneficial for TBI patients.
Several studies have indicated that β-carotene, an antioxidant, has a protective effect on numerous diseases, such as cardiovascular disease, cancer and age-related degenerative disorders (Eggersdorfer and Wyss, 2018; Grodstein et al., 2007; Zhang et al., 2016). β-carotene functions as a free radical scavenger with chainbreaking antioxidant properties (Yusuf et al., 2012). Previous studies have demonstrated that β-carotene is converted into retinol in the body, and its antioxidant activity plays an important role in pre venting neurodegenerative disorders, including Alzheimer's disease (Bitterman et al., 2017; Obulesu et al., 2011).
It is unclear whether β-carotene can prevent oxidative stress by activating Nrf2 signaling after TBI. Hence, we investigated the neuroprotective role of β-carotene in alleviating oxidative stress.
6-8 weeks old mice (Male, C57BL/6) were housed with free access to food and water on a 12 h light/dark cycle in a pathogenfree environment. All animal experiments were approved by the ethics committee of Shanghai University. One hundred and eight mice were used in the present study. Ten mice died post-surgery, and the survival rate after surgery was 95.2%. After surgery, mice were randomly divided into the following groups: (1) Control (n = 12); (2) sham (n = 10); (3) TBI (n = 16); (4) TBI + vehicle (n = 14); (5) TBI + β-carotene (10 mg/kg) (n = 14); (6) TBI + β-carotene (20 mg/kg) (n = 14); (7) TBI + β-carotene (30 mg/kg) (n = 14); (8) TBI + β-Carotene (50 mg/kg) (n = 14). 3 h after TBI, β-carotene was administered to mice by gavage and then once daily for one week. Data analysis and experimental treatments were blinded. β-carotene (Sigma-Aldrich Co., purity > 95%) was administrated by gavage at the given dose (10 mg/kg-50 mg/kg) 3h after TBI, and then everyday afterwards.
TBI Surgery was performed with certain modifications based on previous published method by Flierl et al. (2009). Briefly, mice were placed onto a platform and anesthetized with isoflurane (4.0% for induction, 1-2.0% for maintenance, Baxter Healthcare Co. USA). The scalp was incised at the midline and craniotomy was established approximately 1 mm lateral to the midline. Then a 200 g weight was dropped onto the skull from a height of 2.5 cm. The sham-control group only received craniotomy without blunt force weight injury. During the study period, the body temperature of mice was maintained at 37 ± 0.5 ℃ using a warming blanket until mice displayed normal motor activity.
Six neurobehavioral tests which included body symmetry, gait, climbing, front limb symmetry, grip test and compulsory circling, were graded from 0 to 4 as described by previously (Bermpohl et al., 2006; Zhao et al., 2015). Neurologic deficits were assessed using a 24-point scoring system. The wire-hanging test was performed to evaluate grip strength, balance, and endurance. Briefly, mice had to suspend their bodies using their forelimbs on an iron wire (heigh: 50 cm, length: 55 cm). The hind limbs of the mice were taped together to prevent grabbing to the wire mess, and a pillow was positioned to prevent injury from falls. The duration of the suspension time were recorded.
Extravasation of Evans blue (dye) was used to evaluate BBB permeability. 2% Evans blue was intravenously injected 2 h prior to sacrifice on day7 post-treatment. Mice were then transcardially perfused with PBS followed by 4% paraformaldehyde. Each tissue sample was immediately homogenized and centrifuged after transcardially perfused with 4% paraformaldehyde. The absorbance value of the supernatant was recorded at a wave length of 620 nm (UV-1800 ENG 240 V; Shimadzu Corporation, Japan). The wet weight (ww)-dry weight (dw) method was used to evaluate the effect of β-carotene on brain edema, as described previously (Xu et al., 2013). In brief, the brains were excised and then the brain stem and cerebellum was removed. The wet weight (ww) of the remaining ipsilateral tissues were weighed and recorded. Then the dry weight (dw) of the hemisphere was weighed after drying for 24 h at 100 ℃. Water content was calculated using (ww - dw)/ww × 100%.
Malondialdehyde (MDA) and Superoxide Dismutase (SOD) levels were measured based on the manufacturer’s protocol (Nanjing Jian cheng Biochemistry Co., Nanjing, China). Briefly, tissue samples were ground and then centrifuge at 12,000 × g for 15 min. The supernatants were transferred to a new tube and then protein concentrations were determined using the bicinchoninic acid (BCA) method. Samples from the different groups were used to prepare the working solution. The blank and standards were prepared according to the manufacturer’s instructions. Absorption was measured using a microplate spectrophotometer at a wavelength of 532 nm. Enzyme activity was calculated using the following equation: MDA content (nmol/mgprot) = (ODsample - ODcontrol) / (ODstandard - ODblank) × Concentration standard sample / Concentration protein sample. For the SOD assay, the SOD detection buffer and working solution was prepared according to the kit’s manufacture’s instruction. Protein samples were added to the working solution at 37 ℃ for 40 min. Absorbance was measured at 550 nm. Enzyme activity was calculated using the following equation: SOD enzyme activity units (U/mgprot) = 2 × (ODcontrol - ODsample) / ODcontrol × reaction volume (ml) / sample volume (ml) / protein concentrition sample (mgprot/ml).
Brains were incubated in 4% paraformaldehyde overnight and then transferred to 15% and 30% sucrose in PBS for an additional 24 h. Frozen serial coronal brain sections were sliced at 30 µm using a cryostat. At least ten sections from each treatment group were used for Nissl staining. Damaged neurons were morphologically shrunken or contained vacuoles, whereas normal neurons were relatively large, had a full soma and round, large nuclei. After section-staining, images were captured at 100 × using the Olympus microscope (BX43, Japan). All images were analyzed using the Image J software. The number of damaged neurons from each section per mouse (three 100 × fields per section, three sections per mouse) were then calculated.
Immunochemistry was performed as described previously (Cheng et al., 2015). In brief, tissue antigen retrieval was performed following deparaffinization and rehydration using an alcohol gradient. Sections were then incubated overnight at 4 ℃ with the following primary antibodies; (anti-Neuronal nuclei 1 : 400; Abcam), anti-Nrf2 1: 200; Abcam), and then incubated with Alexa Fluor 488-conjugated secondary antibody (1 : 1000; Thermo Fish). TUNEL kit (Roche Diagnostics, Indianapolis, IN, USA) was used to measure cell apoptosis. The number of TUNEL-positive cells were counted and then analyzed (three 100 × fields per section) using the Image J software.
Proteins were extracted using the Protein Extraction Kit (Beyotime Biotech Inc., Nantong, China) and then separated with 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDSPAGE). The proteins were then transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were then blocked with 5% non-fat milk blocking buffer. Membranes were then incubated with the following primary antibodies; anti-Bcl-2 (1 : 1000), antiBax (1 : 1000), anti-Caspase-3 antibodies (1 : 500) (Proteintech Group, Wuhan, China) and anti-Nrf2 (1 : 800), anti-HO1 (1 : 800), anti-NQO1 (1 : 800) (Abcam, Britain).
Total RNA was extracted from the ipsilateral cortex using the RNAiso Plus kit (Takara Bio, Dalian, China). Total RNA was then immediately reverse-transcribed to cDNA using the PrimeScript RT reagent kit (Takara Bio). Gene-specific primers were designed using PubMed GenBank and synthesized by Sangon Biotech (Shanghai, China). Primer sequences are shown in the Table 1.
| Gene | Primer sequence | |
|---|---|---|
| NQO1 | F | 5′-CATTCTGAAAGGCTGGTTTGA-3’ |
| R | 5′-CTAGCTTTG ATCTGGTT-GTCAG-3’ | |
| HO1 | F | 5’-ATCGTGCTCGCATGAACACT-3’ |
| R | 5’-CCAACACTGC-ATTTACATGGC-3’ | |
| 18S | F | 5’-GGACACGGACAGGATTGACA-3’ |
| R | 5’-GACATCTAAGGGCATCACAG-3’ |
Gene expression levels were measured by qRT-PCR using the Fast7500 System (Applied Biosystems, USA). Experiments were performed in triplicate using three different tissue samples. Transcriptional levels of 18S was used for normalization.
Data analysis was performed using SPSS 18.0 statistical software (SPSS Inc., Chicago, IL, USA). Data were expressed as mean ± SEM. Behavioral tests among the treatment groups were analyzed using two-way ANOVA. For histological and immunohistochemistry data, one-way AVOVA was used to compare multiple groups. Bonferroni post hoc analysis was used for multiple comparisons. Differences between two groups were analyzed using two-tailed Student’s t-test. Values of p < 0.05 were considered statistically significant.
We administered four doses of β-carotene (10, 20, 30, 50 mg/kg) after TBI in order to determine the optimal β-carotene dose. We performed behavior test including gait, body symmetry, climbing, grip test, front limb symmetry and compulsory cir cling for the different treatment groups. Neurologic deficit scores were significantly lower in the dosed groups (20 mg/kg, 1d: 14.2 ± 0.75; 3d: 12.9 ± 0.83; 7d: 11.8 ± 1.53) (30 mg/kg,1d: 13.1 ± 1.73; 3d:11.8 ± 1.24; 7d: 10.3 ± 1.55) (50 mg/kg, 1d: 12.3 ± 1.34; 3d: 11.8 ± 1.24; 7d: 11.6 ± 1.49) using the behavior test at day1, 3 and 7 post TBI versus the non-dosed TBI group (1d: 16.7 ± 0.9; 3d: 15.7 ± 1.1; 7d: 15 ± 0.89) (p < 0.05). However, a dose of 10 mg/kg of β-carotene (10 mg/kg, 14 ± 0.89) only slightly improved neurologic deficit on day 7 compared to non-dosed TBI group (TBI, 15.2 ± 0.89) (p = 0.021). The neurologic deficit score was lower in the 50 mg/kg group (12.38 ± 0.38) compared to the 30 mg/kg group (9.43 ± 0.45) on day7 (p = 0.032). The neurologic deficit score in the TBI group (1d: 16.7 ± 0.9; 3d: 15.7 ± 1.1; 7d: 15 ± 0.89) showed no difference compared to the vehicle-treated group (1d: 16.4 ± 1.62, p = 0.63; 3d: 15.2 ± 1.4, p = 0.41; 7d: 14.6 ± 0.8, p = 0.33) (Fig. 1 A-C).
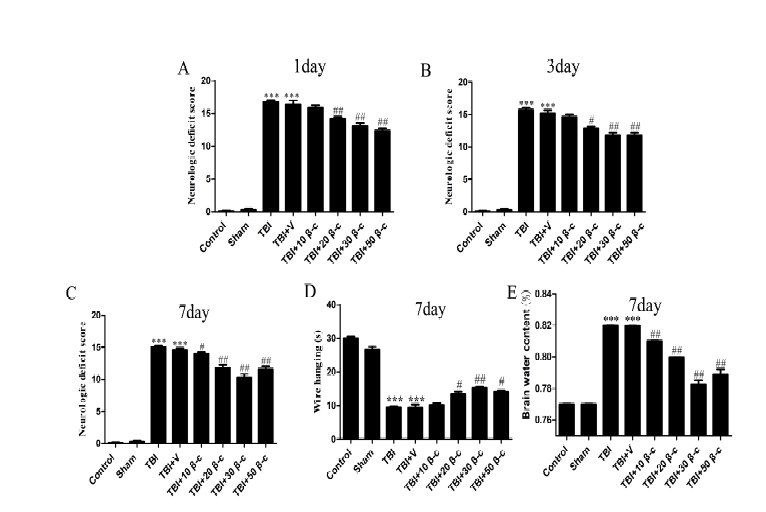 Figure 1.
Figure 1.β-carotene supplementation improves neurological function, brain edema after TBI. Mice received 10, 20, 30, and 50 mg/kg of β-carotene or vehicle (corn oil) 30 min after TBI. Neurologic deficit score were examined at 1 day, 3 days, and 7 days after TBI, but brain water content and wire hang test were measured at 7 days after TBI. (A, B) The high dose of β-carotene (20, 30, and 50mg/kg) supplementation improved neurological functions 1 day after TBI (##p < 0.001), while the neurological functions in 10 mg/kg of β-carotene group will be improved 7 days after TBI (##p < 0.01). n = 10 per group. (C) The performance of the 30 mg/kg of β-carotene was better than 50 mg/kg at 7 days. n = 10 per group. (D) β-carotene treatment increased the latency to fall in the wire-hanging test compared with that in the TBI and TBI + vehicle groups at 7 days after TBI. n = 10 per group. (E) There is the lower of brain water content in the sham or TBI + β-carotene groups than that in the TBI and TBI + vehicle groups. In addition, the dose of the 30 mg/kg of β-carotene was the best optimum concentration. n = 4 per group. Data are presented as mean ± SEM. ***p < 0.001 vs. control or sham; #p < 0.01, ##p < 0.001 vs. TBI or TBI + vehicle group.
Brain water content is an important indicator for TBI prognosis (Cheng et al., 2015). At day7 post TBI, the control (77.1 ± 0.71%) and sham group (76.9 ± 0.44%), had higher brain water content compared to the TBI group (81.9 ± 0.09%) (p = 0.00014, p = 0.00011). The brain water content for the different β-carotene doses (20 mg/kg: 79.05 ± 0.25%, p = 0.0011; 30 mg/kg: 78.6 ± 0.31%, p = 0.0013; 50 mg/kg: 78.7 ± 0.51%, p = 0.0025) was significantly lower compared to the TBI group (81.9 ± 0.09%) (Fig. 1 D). The brain water content in the 50 mg/kg β-carotene was similar to the 30 mg/kg β-carotene dosed group (p = 0.772). Based on the data above, the 30 mg/kg dose was the most efficacious, and was used in all subsequent studies.
We next measured the extravasation of Evans blue to evaluate BBB permeability. Our data indicated that the control and sham group had lower Evans blue dye extravasation in the cortex compared to the TBI group at 7 days post TBI. However, compared to the TBI group (131.49 ± 8.28) or TBI + V (125.6 ± 8.05) group, there were no differences in Evans blue dye extravasation in the 30 mg/kg β-carotene dosed group (120.3 ± 8.17) (30 mg/kg β-carotene vs. TBI, p = 0.26; 30 mg/kg β-carotene vs TBI + V, p = 0.54) (Fig. 2 A-B).
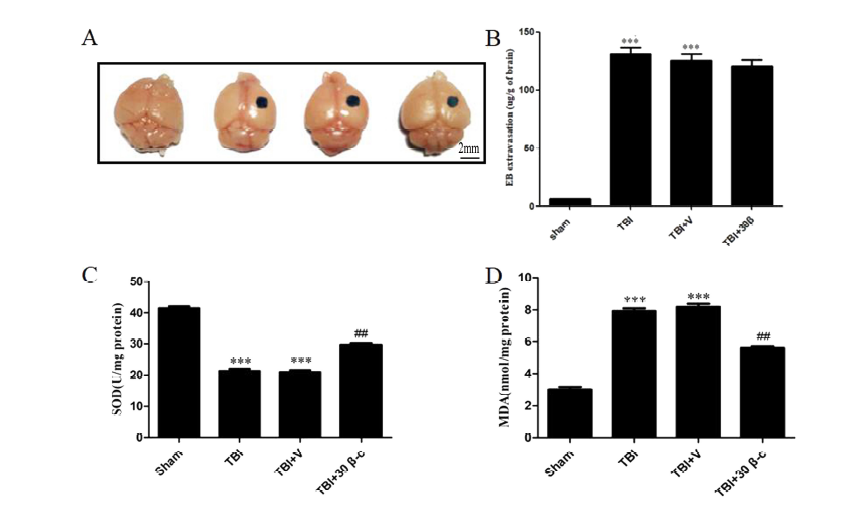 Figure 2.
Figure 2.β-carotene improves BBB permeability and against oxidative stress caused by TBI in brain tissue. (A, B) There are more Evans blue dye extravasation in the cortex 7 days after TBI compared with the sham group (***p < 0.001), but there are no significantly decreased Evans blue dye extravasation in 30 mg/kg β-carotene group compared with the TBI and TBI + V group (p > 0.05). (n = 4 per group). (C) The level of SOD in the cortex increased remarkably 7d after 30 mg/kg β-carotene treatment. (D) The content of MDA was significantly decreased by β-carotene treatment after TBI. ***p < 0.001 vs. Sham, ##p < 0.001 vs. TBI and TBI + V.
Lipid peroxidation and antioxidant enzyme activity (SOD and MDA) was then measured in brain tissues. Our data demonstrated that SOD activity in the TBI group (21.8 ± 0.38 U/mg) or TBI + vehicle (20.49 ± 0.22 U/mg) group were lower compared to the sham group (41.3 ± 0.51 U/mg) (sham vs. TBI, p = 0.0002; sham vs. TBI + V, p = 0.0002) (Fig. 2 C). In addition, the TBI (7.8 ± 0.15 nmol/mg) and TBI + vehicle (7.82 ± 0.32 nmol/mg) group had a higher MDA levels compared to the sham group (3.1 ± 0.18 nmol/mg) (sham vs. TBI, p = 0.0012; sham vs. TBI + V, p = 0.0014) (Fig.2D). Interestingly, SOD levels were higher and MDA levels lower in the TBI + 30 mg/kg β-carotene group (SOD: 29.1 ± 0.14 U/mg; MDA: 5.8 ± 0.21 nmol/mg) compared to the TBI group (sham vs. TBI, p = 0.0002; sham vs. TBI + V, p = 0.0002) (Fig. 2 C-D). Our data demonstrated that β-carotene, to a certain extent had antioxidant capacity.
We next evaluated whether β-carotene had a protective effect against TBI-induced neuronal apoptosis. Neuronal apoptosis in lesioned cortices was measured using TUNEL immunofluorescence and Nissl staining. The number of TUNEL-positive apoptotic cells were significantly higher in the TBI group (30.8 ± 2.14%) compared to the sham group (7.21 ± 0.45%) 7 days post TBI (p = 0.002). However, apoptosis was significantly reduced in the TBI + 30 mg/kg β-carotene group (15.29 ± 1.57%) compared to the TBI group (30.8 ± 2.14%, p = 0.001) (Fig. 3 B, D). In addition, western blot analyses demonstrated that the expression of Bax and caspase-3 in the TBI group was higher compared to the TBI + βcarotene group 7 days post TBI 7. Additionally, compared to the TBI group, Bcl-2 levels, an anti-apoptotic factor, was increased in the TBI + β-carotene group (p = 0.038) (Fig. 3 E-G). The above data suggests that β-carotene had a neuro protective effect in lesioned cortex.
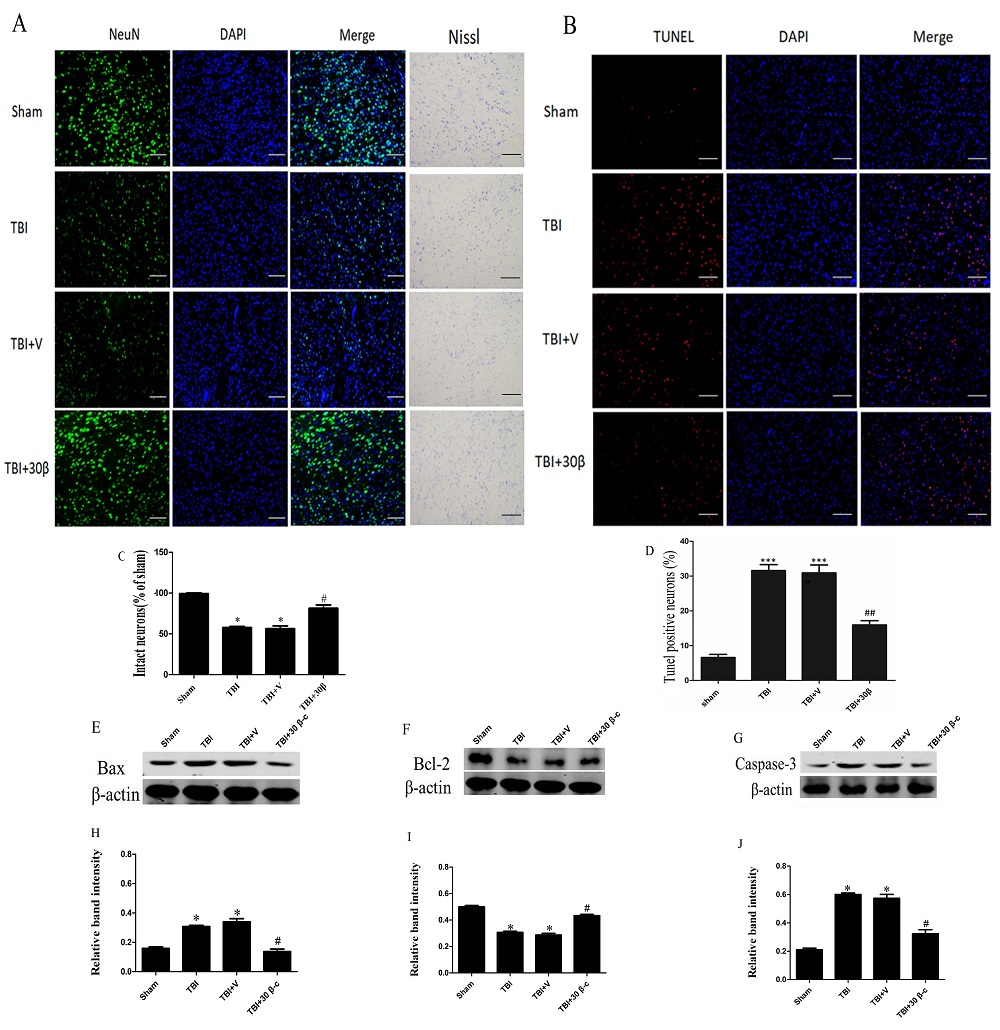 Figure 3.
Figure 3.TBI-induced neuronal apoptosis was decreased by β-carotene treatment 7 days after TBI. (A,C) Immunofluorescence staining and Nissl staining showed that the number of intact neuron in β-carotene group is more than that in TBI group,*p < 0.01 vs. Sham, #p < 0.01 vs. TBI and TBI + V. (B, D) The result from TUNEL staining showed that the number of TUNEL-positive neurons was less in the TBI + 30mg/kg β-carotene group (15.22 ± 2.13%) than that in the TBI group (32.13 ± 3.31%) or TBI + V(31.22 ± 3.08%) , ***p < 0.001 vs. Sham, ##p < 0.001 vs. TBI and TBI + V. Representative microscopy images of TUNEL-positive neurons are shown; (E-G) Representative immunoblots showing the protein levels of Bax (E) and Bcl-2 (F) and cleaved caspase-3 (G) in the sham, TBI, TBI + vehicle, and TBI + β-carotene groups at 7 days after TBI. (H-J) The relative protein expression was shown by densitometry analysis. *p < 0.001 vs. Sham, #p < 0.001 vs. TBI and TBI + V. Values are expressed as mean ± SEM (n = 3 per group). Magnification: 100×, Scale bars = 50 µm.
Nrf2 is induced after oxidative stress. Immunofluorescence staining demonstrated that β-carotene could induce Nrf2 nuclear translocation in the TBI group (Fig. 4 A). In addition, western blot assays demonstrated increased Nrf2 translocation into the nucleus and lower Nrf2 levels in the cytoplasm in the β-carotene treated group compared to the TBI + vehicle group (p = 0.012, Fig. 4 B, C). We also found that the Keap1 expression was decreased after β-carotene treatment (Fig. 4 D). These data demonstrated that Nrf2 signaling was activated by β-carotene.
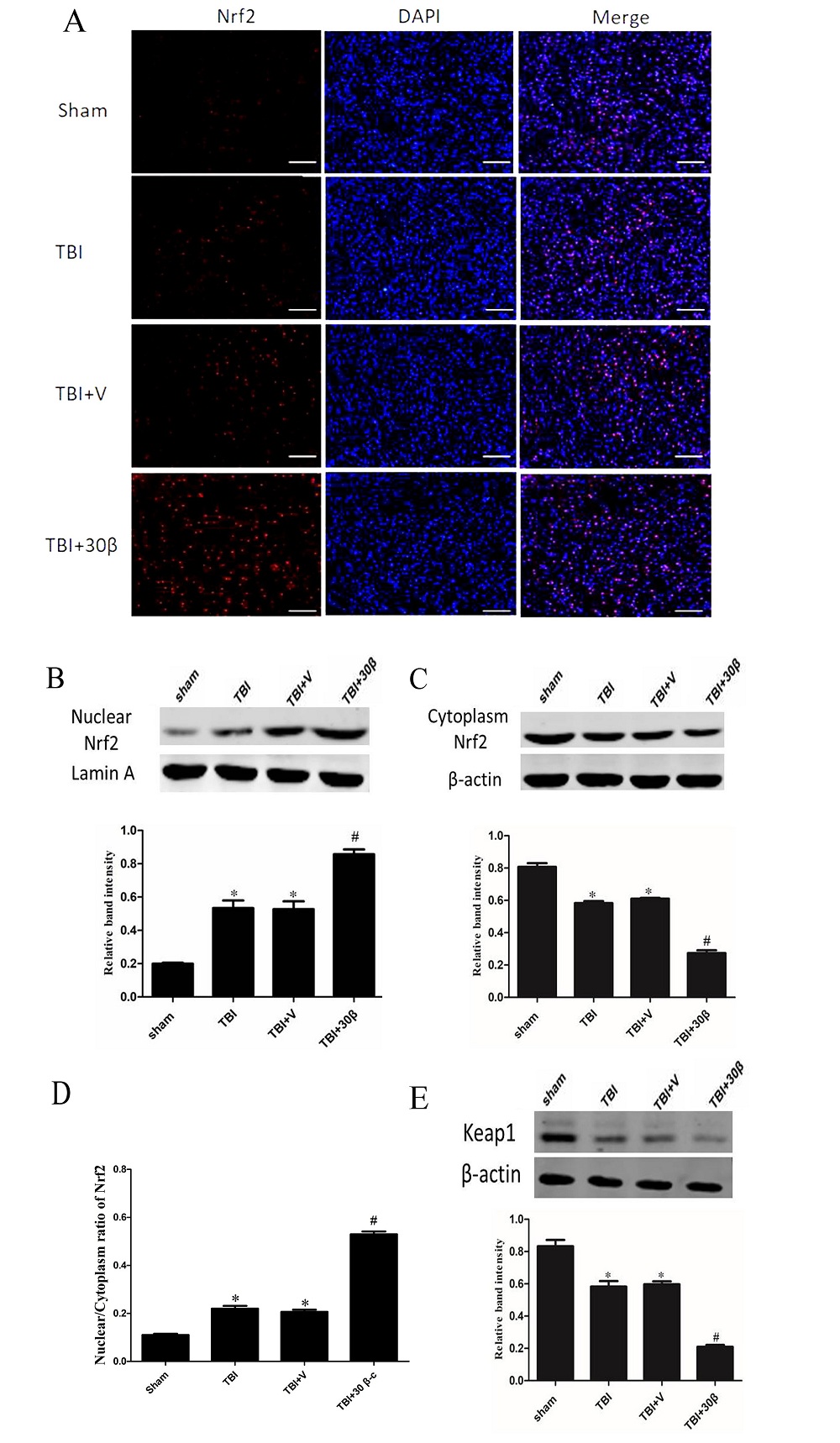 Figure 4.
Figure 4.β-carotene increased the nuclear location of Nrf2. (A) Representative immunofluorescence staining of Nrf2 after β-Carotene treatment in mice with TBI. Scale bar = 50 µm. n = 3 per group. (B, C) The expression of Nrf2 in nucleus and cytoplasm were measured respectively by Western blot 7 days after TBI in different groups. β-carotene significantly increase the nucleus/cytoplasm ratio of Nrf2. (D) The nuclear/cytoplasm ratio of Nrf2. (E) The protein level of Keap1 were measured by Western blot in different groups. Data are presented as mean ± SEM. p < 0.01 vs. sham group; #p < 0.01 vs. TBI and TBI + vehicle group. Magnification: 100 ×, Scale bars = 50 µm.
We found that β-carotene could increase Nrf2 activity and was neuroprotective after TBI. We next determined whether HO1 and NQO1 expression, i.e. the downstream targets of Nrf2, was increased after β-carotene treatment. Our data demonstrated that NQO-1 and HO-1expression were both upregulated after TBI both at the protein and mRNA levels (p = 0.029). NQO-1 and HO-1 expression levels were higher after β-carotene treatment compared to the TBI group *p = 0.025 and #p = 0.014, (Fig. 5 A-D). These results demonstrate that β-carotene could activate the Nrf2 signaling pathway, alleviate acute oxidative stress and was neuroprotective.
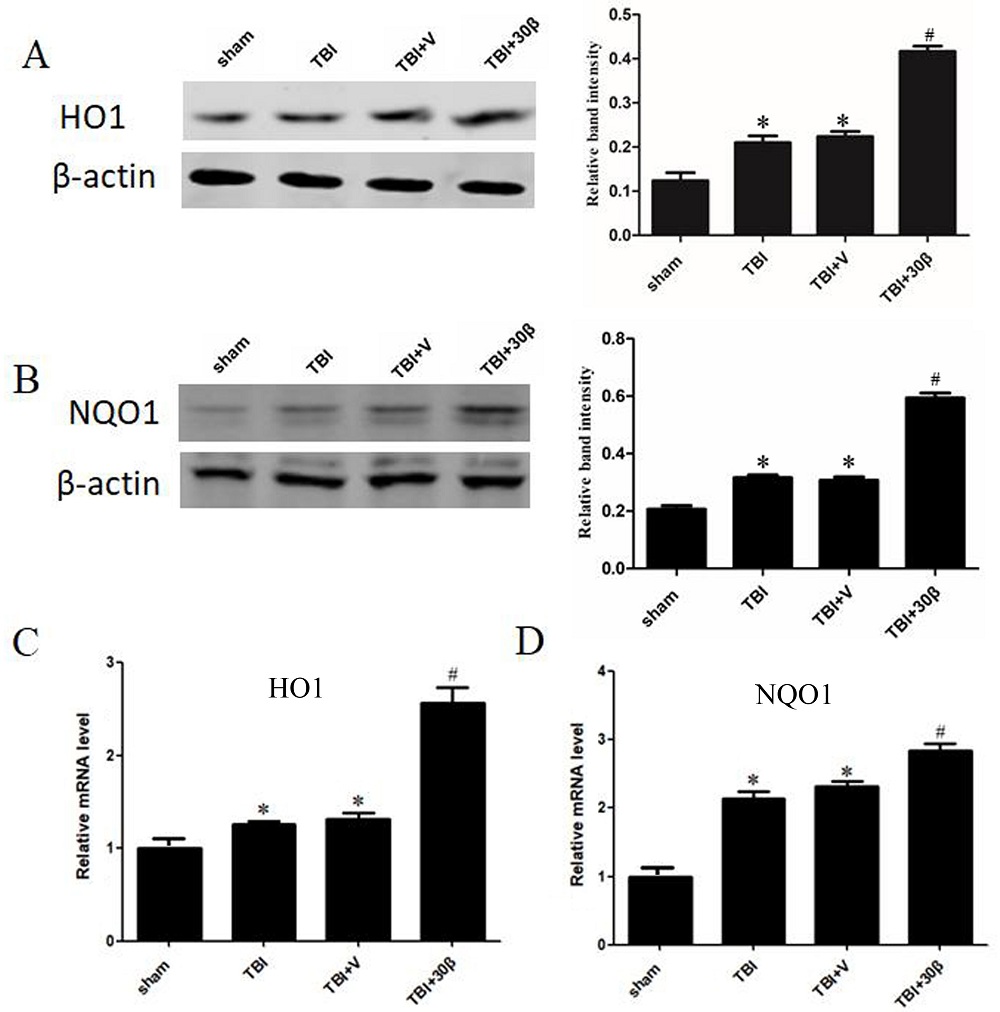 Figure 5.
Figure 5.β-carotene can promote the expression of NQO1 and HO1 after TBI in transcription and translation level. (A, B) The expression of HO-1 and NQO-1 protein was slightly upregulated after TBI; Then, β-carotene administration further to increase the expression of HO-1 and NQO-1 in brain tissue. Actin was used as a loading control. Relative band intensity means NQO-1 or HO-1/β-actin. (C, D) The mRNA level of NQO-1and HO-1 also increased after β-carotene treatment. n = 3 per group. Data are presented as mean ± SEM. #p < 0.01 vs. TBI + vehicle group; *p < 0.05 vs. sham group.
The antioxidant properties of β-carotene have been demonstrated in previous studies (Obulesu et al., 2011; Zhu et al., 2016). We demonstrated that in TBI mouse models, β-carotene could (1) to a certain degree, reduce brain injury and improve neurological function after TBI (Fig. 1); (2) significantly reduce cell death induced by TBI and increase neuronal survival in the peri-injury region (Fig. 3); (3) promote Nrf2 nuclear translocation and subsequently increase the expression of downstream target genes (including NQO1 and HO-1), which in-turn reduces keap1 expression (Fig. 4, 5); and (4) reduces oxidative brain damage, and increases SOD activity (Fig. 2).
Oxidative stress can play several physiological roles (i.e., cell signaling) (Pizzino et al., 2017). Low/moderate ROS levels are considered essential for neuronal development and function, whereas excessive levels are harmful (Li et al., 2013; Romanucci et al., 2015). The brain is more sensitive to oxidative stress compared to other tissue because of the high levels of polyunsaturated fatty acids in the brain (Sultana et al., 2013). Studies have demonstrated that secondary injury after primary injury induced by TBI is specifically associated with oxidative stress (Cornelius et al., 2013). Results from our study demonstrated that MDA levels in brain tissue was increased in the TBI group compared to the sham group. The initial injury induces neuronal apoptosis, while secondary brain injury exacerbates neuronal death by oxidative stress and is the major contributing factor (Jin et al., 2011). Our results demonstrated that 30 mg/kg β-carotene significantly reduced brain water content (Fig. 1 D) and neuronal death at 7 days post-TBI in mice (Fig. 3A, B). This dose improved neurological function (Fig. 1 A-C). Neuronal death (including apoptotic and necrotic neurons) at the site of injury and surrounding tissue occurs subsequently after TBI. TUNEL-positive cells were observed at 7 days post TBI, and were reduced in TBI mice administered 30 mg/kg β-carotene compared to mice in the TBI group (Fig. 3B, D). In addition, western blot analysis demonstrated that the expression of caspase-3 and Bax was decreased after β-carotene treatment compared to mice in the TBI group (Fig. 3E-G). Our data provides direct evidence for the protective role of β-carotene in neurons after TBI.
Numerous studies have demonstrated that the pleiotropic transcription factor Nrf2 is important for regulating antioxidant enzymes, as demonstrated in several CNS diseases (i.e., Parkinson’s disease, Alzheimer’s disease (AD), depression, and memory loss) (Pizzino et al., 2017; Ding et al., 2014; Arent et al., 2011). Nrf2 translocates to the nucleus and binds to antioxidant response elements (AREs) to reduce oxidative stress (Vriend and Reiter, 2015). Cheng et al. (2016) found that Nrf2 knockout mice had worse outcomes compared to wild-type mice, while activation of Nrf2 could up-regulate the expression of antioxidant enzymes and alleviate brain injury after TBI. Our results also showed that β-carotene promoted Nrf2 transfer from the cytoplasm to the nucleus after TBI (Fig. 4A-C). NQO1 and HO1 are downstream target genes of the Nrf2 pathway necessary to reduce oxidative stress. Correspondingly, cytoplasmic expression of HO1 and NQO1 are increased after β-carotene administration (Fig. 5A-D). Hence, as demonstrated by us and previous studies, the activation of the Nrf2-ARE pathway provides protection against brain injury.
In summary, we demonstrated that β-carotene alleviates brain injury after TBI by modulating the Nrf2/Keap1-mediated antioxidant pathway. Our study had several limitations. First, the use of Nrf2 knockout mice to investigate the mechanisms involved in β-carotene-mediated Nrf2-ARE signaling pathway was not performed. Second, we only focused on the Nrf2 signaling pathway, and did not investigate other signaling pathways that may be involved in TBI and β-carotene-mediated signaling. Third, additional in vitro and in vivo experiments are needed to confirm the direct protective effects of β-carotene supplementation on neuroprotection. In vitro 3D brain models offers great promise for understanding the nervous system (Lee et al., 2017; Sloan et al., 2018). Hence, our future plans are to use 3D brain organoid models to confirm the neuroprotective effects of β-carotene.
TBI: Traumatic brain injury; Nrf2: NF-E2-related factor; ROS: Reactive oxygen species; HO-1: Quinone oxidoreductase 1; NQO1: NAD(P)H quinone dehydrogenase 1; BBB: Bloodbrain barrier; ROS: Reactive oxygen species; MDA: Malondialdehyde; SOD: Superoxide Dismutase; TUNEL: Terminaldeoxynucleoitidyl Transferase Mediated Nick End Labeling; qPCR: Real-time quantitative polymerase chain reaction.
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed (Ethical approval number: HMMP-2017-012).
This work has been supported by grants from key special project of collaborative innovation of Shanghai University of Medicine and Health Sciences (E1-0200-18-201001 to Xiamin Hu); Seed fund of Shanghai University of Medicine and Health Sciences (E3-0200-17-201113 to Xiaoming Xin); Innovation and entrepreneurship training program for Students majoring in pharmacy of Shanghai University of Medicine and Health Sciences.
The authors declare no competing interests.