
 , Natalia V. Naryzhnaya 1, Alexandr V. Mukhomedzyanov 1, Alla A. Boshchenko 1, Ivan A. Derkachev 1, Boris K. Kurbatov 1, Andrey V. Krylatov 1, Aleksandra E. Gombozhapova 1, Stanislav V. Dil 1, Julia O. Samoylova 1, Feng Fu 2, Jian-Ming Pei 2, Galina Z. Sufianova 3, Emiliano R. Diez 4
, Natalia V. Naryzhnaya 1, Alexandr V. Mukhomedzyanov 1, Alla A. Boshchenko 1, Ivan A. Derkachev 1, Boris K. Kurbatov 1, Andrey V. Krylatov 1, Aleksandra E. Gombozhapova 1, Stanislav V. Dil 1, Julia O. Samoylova 1, Feng Fu 2, Jian-Ming Pei 2, Galina Z. Sufianova 3, Emiliano R. Diez 41 Department of Emergency Cardiology and Laboratory of Experimental Cardiology, Cardiology Research Institute, branch of the Federal State Budgetary Scientific Institution “Tomsk National Research Medical Center of the Russian Academy of Sciences”, 634012 Tomsk, Russia
2 Department of Physiology and Pathophysiology, National Key Discipline of Cell Biology, School of Basic Medicine, Fourth Military Medical University, 710032 Xi'an, Shaanxi, China
3 Department of Pharmacology, Tyumen State Medical University, 625023 Tyumen, Russia
4 Instituto de Fisiología, FCM–UNCuyo IMBECU - CONICET-UNCuyo, 5500 Mendoza, Argentina
Abstract
Microvascular obstruction (MVO) of coronary arteries promotes an increase in
mortality and major adverse cardiac events in patients with acute myocardial
infarction (AMI) and percutaneous coronary intervention (PCI). Intramyocardial
hemorrhage (IMH) is observed in 41–50% of patients with ST-segment elevation
myocardial infarction and PCI. The occurrence of IMH is accompanied by
inflammation. There is evidence that microthrombi are not involved in the
development of MVO. The appearance of MVO is associated with infarct size, the
duration of ischemia of the heart, and myocardial edema. However, there is no
conclusive evidence that myocardial edema plays an important role in the
development of MVO. There is evidence that platelets, inflammation, Ca
Keywords
- heart
- ischemia
- reperfusion
- microvascular obstruction
- no-reflow
The term “no-reflow” was first proposed by Majno et al. (1967) [1]. These investigators found that after ischemia of the rabbit brain lasting 15 min, complete restoration of brain blood flow does not occur. A few years later, Kloner et al. [2] could demonstrate that injury of cardiac microvascular vessels is involved in the pathogenesis of no-reflow of the canine heart. In 1985, the no-reflow phenomenon was found in patients with ST-segment elevation myocardial infarction (STEMI) [3]. Investigators reported that thrombolysis could not completely restore coronary blood flow (CBF) in these patients. The duration of chest pain was less than 3 h before admission [3]. Currently, researchers often use the term “microvascular obstruction” (MVO) or the term “the slow flow phenomenon” because complete no-reflow (thrombolysis in myocardial infarction (TIMI) = 0) in the infarct-related coronary artery was detected angiographically in only 5% of patients with acute myocardial infarction (AMI) and percutaneous coronary intervention (PCI), in other patients the incomplete restoration of CBF was observed [4], where TIMI is Thrombolysis In Myocardial Infarction. It should be noted that some investigators suggested that no-reflow could be distinguished as TIMI = 0–1 or TIMI = 0–2 [5, 6, 7, 8, 9]. In this case, the terms “no-reflow” or “MVO” are used as synonyms. There is a correlation between TIMI evaluated angiographically and the MVO size measured by magnetic resonance imaging (MRI) [10, 11]. In a study of patients with AMI and PCI it was found that the duration of cardiac ischemia and infarct size are major determinant of severe MVO [12, 13, 14, 15]. MVO was found in 59% of patients with STEMI + PCI and a 3-h duration of ischemia [13]. If the duration of ischemia was 4–6 h, MVO was found in 72% of patients with STEMI + PCI [13]. The MVO area was measured by MRI [12, 14]. Infarct size in patients with STEMI and MVO was 2-fold larger than in patients without MVO [14]. MVO peaked at 07:00 a.m. [14]. Consequently, infarct size and the duration of ischemia are predictors of the development of MVO. It was reported that the MVO area is 1.9–5.4% of left ventricular (LV) mass in patients with STEMI and PCI according to MRI [11, 16, 17, 18, 19] or 22% of the infarcted myocardium [20]. According to Zia et al. [21] the MVO area is 3.1% of the myocardium by MRI. Infarct size was 13–32% of LV mass in patients with STEMI and MVO after PCI by MRI [11, 20, 22]. The intra-myocardial hemorrhage (IMH) area was 3.8% of LV mass in patients with STEMI and MVO after PCI [11]. Currently, the assessment of MVO often uses both angiography and MRI (Table 1, Ref. [5, 7, 9, 10, 11, 12, 13, 14, 16, 17, 18, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69, 70]). However, in recent years, investigators increasingly favor MRI as a more accurate method of assessing MVO. Angiographic parameters are more variable (Table 2, Ref. [5, 7, 9, 10, 11, 18, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 34, 35, 36, 38, 39, 43, 44, 71]). Sardu et al. [72] found that prediabetes promotes the disorder of acetylcholine-induced coronary vasodilation and major adverse cardiac events in patients with non-obstructive coronary stenosis. Treatment with metformin, an AMP-activated kinase activator, partially restored acetylcholine-induced coronary vasodilation and reduced the incidence of major adverse cardiac events [72]. These data indirectly demonstrated that diabetes can promote the development of MVO and metformin partially reversed this negative effect of diabetes,
Thus, the MVO area is correlated with infarct size and depends on the duration of ischemia.
| The incidence of MVO and IMH | Reference |
| The incidence of MVO in patients with STEMI and thrombolysis according to angiographic data is 12%. In the case of AMI + thrombolysis, MVO was found in 19% of patients | [43, 44] |
| The incidence of MVO in patients with STEMI and thrombolysis according to MRI is 25% | [45] |
| The incidence of MVO in patients with STEMI and PCI according to angiographic data is 12–74%. In the case of AMI + PCI, MVO was found in 2–70% of patients | [5, 7, 9, 10, 11, 14, 18, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42] |
| The incidence of MVO in patients with STEMI and PCI according to echocardiographic findings is 50% | [69] |
| The incidence of MVO in patients with STEMI and PCI according to MRI data is 37–76%. In the case of AMI + PCI, MVO was found in 25% of patients | [12, 13, 16, 17, 21, 22, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62, 63, 64, 65, 66, 67, 68, 70] |
| The incidence of a combination of MVO + IMH in patients with STEMI and PCI according to MRI findings is 36%–51% | [13, 56, 66] |
| IMH without MVO was observed in 15% of patients with STEMI and PCI according to MRI | [66] |
Note. AMI, acute myocardial infarction included STEMI and Non-STEMI; IMH, intra-myocardial hemorrhage; MRI, magnetic resonance imaging; MVO, microvascular obstruction; PCI, percutaneous coronary intervention; STEMI, ST-segment elevation myocardial infarction. MVO data after thrombolysis or PCI was included.
Microvascular obstruction was detected by MRI in 25% of patients with STEMI [45]. Ndrepepa et al. [5] reported that MVO was angiographically observed in 29% of patients with STEMI and PCI. According to Mayr et al. [65], MVO was found in 56% of patients with STEMI and PCI by MRI. MVO was diagnosed by echocardiography in 50% of patients with STEMI and PCI [69]. It was reported that no-reflow (TIMI = 0–2) was documented in 25% of patients with STEMI and PCI [7]. Microvascular obstruction was found in 25% of patients by angiography (TIMI = 0 or 2) with STEMI and PCI [9]. According to our data, the incidence of MVO is 37% in patients with STEMI and PCI by MRI data [66].
Thus, the incidence of MVO is observed in 25%–56% of patients with STEMI and PCI.
According to Abbo et al. [43], the incidence of no-reflow was 66 of 566 (11.6%) patients with AMI and thrombolytic therapy or PCI. They reported that patients with AMI and no-reflow experienced a 10-fold higher incidence of in-hospital death compared to AMI without no-reflow [43]. Cardiovascular events 6 months after AMI in patients with MVO are observed more often than in patients without MVO [45]. The in-hospital mortality rate was 14% in patients with AMI and MVO and only 3% in patients with AMI and without MVO [73]. The no-reflow phenomenon was accompanied by increased mortality for 3 years after AMI [44]. The no-reflow phenomenon was detected angiographically [44]. Patients with STEMI and no-reflow had an increased incidence of in-hospital mortality than patients without no-reflow (TIMI = 0–1) [9]. The no-reflow phenomenon was evaluated angiographically [9]. The mortality rate in patients with STEMI and MVO was greater compared to patients without MVO [14]. Adverse cardiovascular events in patients with AMI and MVO for 2 years after AMI developed more often than in patients with AMI without MVO [45]. MVO is an independent predictor of adverse LV remodeling in patients with STEMI [74]. MVO was evaluated angiographically [74]. In patients with STEMI with PCI, no-reflow (TIMI = 0–1) is a strong independent predictor of the mortality rate for 5 years after AMI [5]. No-reflow was detected angiographically [5]. Microvascular obstruction was usually accompanied by increased myocardial infarct size, a decreased LV ejection fraction, and a high mortality rate for 5 years after AMI [67]. The MVO area was measured by MRI [67]. Microvascular obstruction was associated with adverse cardiac remodeling for 8 months after AMI [68]. The MVO area were measured by MRI [68]. Major adverse cardiac events (MACE) for 6 months after STEMI was documented more often in patients with AMI and MVO [75]. No-reflow was detected angiographically [4]. MVO is a predictor of MACE in patients with STEMI and PCI [22, 59, 60, 61, 64, 76].
In summary, MVO is a common manifestation of AMI, especially in patients with STEMI. Microvascular obstruction is accompanied by a high mortality rate and is associated with MACE.
The highly effective therapy and prevention of MVO are impossible without knowledge of the pathogenesis of this pathology.
In a study performed in 2012, MVO was assessed by myocardial blush grade (MBG)
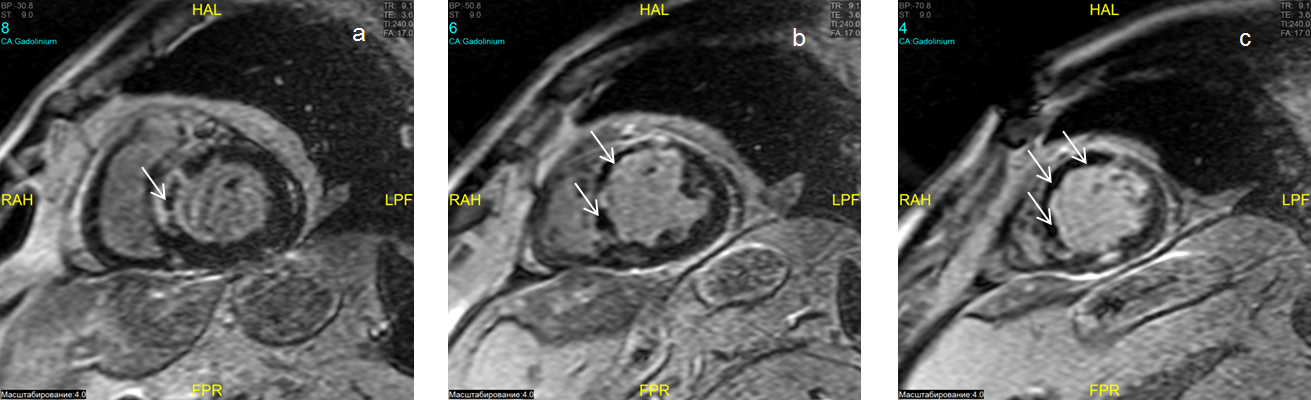
in patients with STEMI and PCI [77]. No-reflow was detected angiographically (Fig. 1). Blood samples were drawn from coronary arteries and the aorta for the
detection of microparticles. It was shown that the microparticles’ level in the
coronary artery is accompanied by MVO. It was concluded that-microparticles could be involved in the development of MVO [77]. This evidence
is questionable because these data were not confirmed before by other
investigators over the last 10 years. Moreover, it was obtained data that
microthrombi is not involved in MVO [10, 11, 18]. Placebo-controlled studies in
patients with STEMI + PCI have been performed [10, 11, 18]. The control group
received a placebo, whilst alteplase was injected into coronary arteries of
patients of the treatment group [10, 11, 18]. The MVO area was measured by MRI
[18]. It was found that alteplase did not alter the MVO size measured by MRI [10, 11, 18]. Alteplase had no effect on infarct size but promoted the development of
intramyocardial hemorrhage in patients with TIMI flow grade
 Fig. 1.
Fig. 1.Microvascular obstruction. Gadolinium contrast-enhanced cardiac magnetic resonance (DE-CMR) imaging of the basal, middle and apical (a, b, c) short-axis slices. Hypointense areas correspond to no-reflow in the projection of the anterior-septum wall of the left ventricle (LV) of the heart (a, b, c) of the LV in the mode of delayed contrast (inversion recovery sequence). HAL, head anterior left; CA, contrast agent; RAH, right anterior head; FPR, foot posterior right; LPF, left posterior foot; BP, body position; ST, slice thickness.
Microembolization and microthrombi do not play a significant role in the pathogenesis of MVO.
It has been shown that the microvascular obstruction score is lower in patients
with STEMI and PCI who received aspirin than in patients without aspirin [46].
The MVO area was measured by MRI [45]. Patients with STEMI received heparin in
the pre-hospital stage following PCI [57]. No-reflow was detected
angiographically. Pretreatment with heparin contributed to a decrease in the
incidence of no-reflow (TIMI = 0–1) by 13% (p
A correlation between the incidence of MVO and ADP-induced platelet aggregation
in patients with STEMI and PCI was demonstrated [47]. The MVO area was measured
by MRI [47]. A correlation between the incidence of MVO and platelet-neutrophil
aggregation was also shown. The incidence of MVO and platelet-monocyte
aggregation is also correlated [47]. Consequently, platelets could be involved in
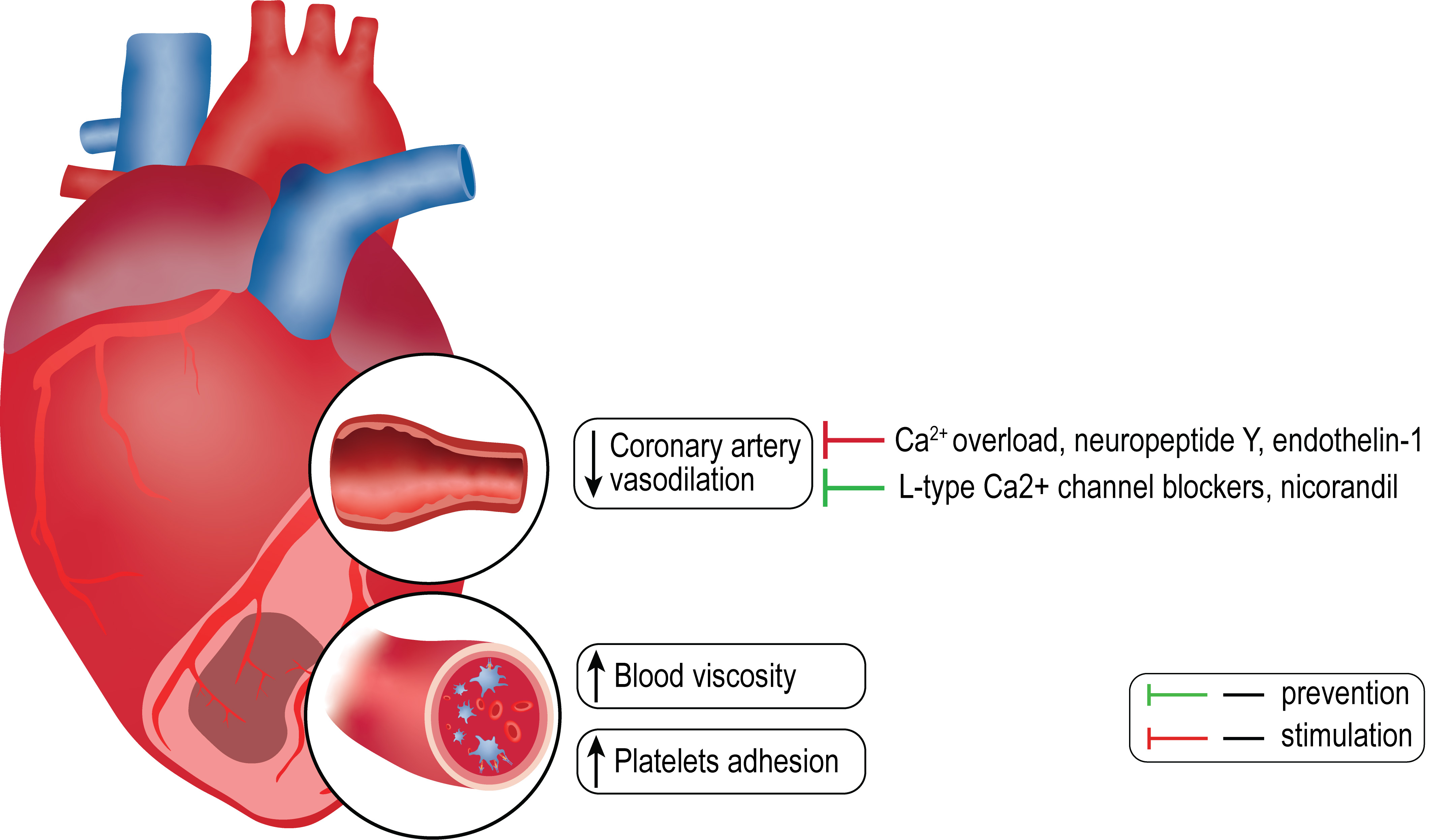
MVO (Fig. 2) by releasing a strong vasoconstrictor - thromboxane A2 [78]. Chronic
administration of aspirin, a non-selective inhibitor of thromboxane A2 synthesis,
resulted in a decrease in the serum concentration of thromboxane B2, a stable
metabolite of thromboxane A2 [78]. These findings demonstrate that thromboxane A2
could be involved in MVO. Microvascular obstruction was more frequently observed
in patients with STEMI + PCI and high platelet reactivity than in patients with
low platelet reactivity [79]. No-reflow was detected as ST-segment regression
 Fig. 2.
Fig. 2.Some hypothetical pathogenic factors of microvascular obstruction.
The role of disturbances of endothelial-dependent vasodilation in MVO remains
unclear because the standard endothelial-dependent vasodilator, acetylcholine,
was not used in the therapy of MVO in patients with AMI. However,
endothelium-independent vasodilators (L-type Ca
It was shown that acute coronary syndrome is associated with a rise in whole blood viscosity [85]. It was found that whole blood viscosity was higher in patients with STEMI + MVO than in patients with STEMI without MVO [37]. It could be suggested that whole blood viscosity could be involved in the development of MVO in patients with AMI.
It was reported that LV volume was increased in patients with AMI and MVO for
six months after AMI, but not in patients with AMI without MVO [38]. No-reflow
was detected angiographically [38]. It was demonstrated that infarct size and
severe microvascular obstruction were positively correlated with adverse
myocardial remodeling for six months after AMI [86]. Other investigators obtained
similar evidence of the involvement of MVO in the pathogenesis of adverse
post-infarction remodeling of the heart [63, 68, 74, 87]. Adverse left
ventricular remodelling occurred in 27% of patients 1 year after STEMI and PCI
[76]. Infarct size and MVO were predictors of adverse remodelling according to
MRI findings [76]. LV remodelling was defined as
However, Dregoesc et al. [89] could not find a relationship between post-infarction remodeling of the heart and MVO.
Thus, MVO promotes the development of adverse post-infarction remodeling of the heart.
It was reported that inflammation is involved in ischemia reperfusion (I/R) cardiac injury [90, 91].
It was shown that a high MVO score positively correlated with a rise in the
plasma concentration of C-reactive protein (CRP), plasma leukocytes, and peak
value of creatine kinase and negatively correlated with high LV ejection fraction
in patients with STEMI and PCI [46, 65]. The peak of CD14
Consequently, C-reactive protein and interleukins could be involved in the pathogenesis of microvascular obstruction.
The role of reactive oxygen species (ROS) in the pathogenesis of no-reflow has
not been studied before in patients with AMI. The L-type Ca
Intracoronary administration of sodium nitroprusside, a NO donor, improved the TIMI flow grade in patients with AMI and PCI [94, 95]. However, other investigators reported that intracoronary administration of sodium nitroprusside did not alter CBF in patients with AMI and PCI [25, 26, 50]. Amit et al. [25] evaluated no-reflow by ST-segment elevation resolution. Niccoli et al. [26] used TIMI grade. Nazir et al. [50] used MRI. These studies included larger groups of patients with AMI and PCI, therefore their results are more significant. Consequently, these data demonstrate that coronary artery spasm is not involved in the pathogenesis of MVO.
It has been reported that the concentration of endothelin-1 in coronary sinus plasma was 1.7 pmol/L in patients with stable angina and 3.0 pmol/L in patients with AMI [96]. The plasma concentration of endothelin-I in the aorta was higher in patients with AMI than in patients with angina [96]. Microvascular obstruction was evaluated in patients with STEMI and PCI (n = 128) by MRI [51]. The plasma endothelin-1 level on admission was associated with MVO and the mortality rate [51]. These data demonstrated that a rise in the concentration of endothelin-1 in blood could contribute to MVO development. Neuropeptide Y (NPY) is also a strong vasoconstrictor that is released from sympathetic terminals [97]. It was reported that intracoronary administration of NPY resulted in coronary artery spasm in volunteers [98]. The plasma NPY level was higher in patients with STEMI and no-reflow (TIMI = 0–2) than in patients without MVO [6]. In contrast, Herring et al. [52] did not find differences in TIMI flow score between patients with high NPY levels and patients with the low NPY levels in coronary sinus blood in patients with STEMI. However, the microcirculatory resistance was higher in patients with high NPY levels in coronary sinus compared to patients with a low concentration of NPY [52].
These data demonstrate that NPY could be involved in the formation of MVO. However, correlation analysis between NPY and endothelin-1 levels and the no-reflow area has not been performed, therefore further studies on the role of NPY and endothelin-1 in the pathogenesis of MVO are required.
It was reported that intravenous injection of arginine vasopressin resulted in coronary artery spasm and ST elevation in rats [99]. Vasopressin triggered a contractile response of isolated coronary arterioles isolated from the heart of patients undergoing cardiac surgery [100]. However, a role for vasopressin in the formation of MVO has not previously been studied.
Na
Intracoronary administration of epinephrine reportedly completely reverses
no-reflow in 9 of 12 patients with STEMI and PCI [27]. It should be noted that
this group was too small, thereby it is unclear whether
The role of endogenous epinephrine in the prevention of MVO in patients with AMI
and PCI remains unclear. Correlation analysis of the MVO size and treatment with
the
The angiotensin II receptor antagonists were not used before for the treatment of no-reflow, therefore a role for angiotensin II in MVO remains unclear.
Intracoronary infusion of adenosine decreased the incidence of MVO in patients with AMI and PCI [29, 30, 71]. These investigators detect MVO by angiography [29, 30, 71]. Consequently, adenosine could alleviate MVO.
It has been reported that diabetes contributes to I/R cardiac injury [72]. Hyperglycemia has been reported to be accompanied by MVO in patients with diabetes and AMI [16, 31, 53]. Iwakura et al. [31] used intracoronary myocardial contrast echocardiography to detect the no-reflow area. Jensen et al. [16] and Ota et al. [53] used MRI to measure the MVO area. We also found that a combination of MVO and intramyocardial hemorrhage is more common in patients with hyperglycemia and diabetes mellitus. We used MRI to measure the MVO area.
However, the MVO area can be identical in patients with diabetes and without diabetes [21]. This was shown using MRI [21]. Investigators did not analyze the interaction between type 2 diabetes, insulin-dependent diabetes, the incidence of MVO, and the MVO area. The molecular mechanism of aggravation of MVO by diabetes remains unclear.
Pinacidil, an ATP-sensitive K
There are MRI data to suggest that MVO is accompanied with interstitial edema
[10, 11, 18, 21, 50, 52, 54, 55, 56, 58, 102, 103, 104]. This pathology is observed in
50% of patients with STEMI and PCI [50]. Edema could induce extrinsic
compression of coronary arteries and trigger MVO. Chen et al. [104] found that
Myocardial Extracellular Volume Fraction was larger in patients with MVO and IMH
by MRI. Myocardial edema in patients with STEMI and MVO was greater than in
patients without MVO (p
Thus, microembolization and microthrombi do not play a significant role in the development of MVO. MVO promotes the development of adverse post-infarction remodeling of the heart. Platelets, increased blood viscosity, vasoconstriction, inflammation, NPY, endothelin-1, and myocardial edema could be involved in the pathogenesis of MVO. However, their role in the development of MVO requires further studies because data on their involvement in MVO formation are preliminary and need clarification. The diabetes-induced aggravation of MVO remains unclear.
Microvascular obstruction is often accompanied by intra-myocardial hemorrhage. A combination of MVO and IMH was found in 35–51% of patients with STEMI and PCI, where the hemorrhage area was about 3% of the LV mass [13, 19, 22, 56, 58, 66]. MVO and IMH areas were measured by MRI mass [13, 56, 66]. IMH without MVO was observed in 15% of patients with STEMI and PCI mass [66]. It was reported that the IMH area reached a maximum 24 h after the restoration of coronary perfusion and was about 4% of the left ventricle in pigs with coronary artery occlusion (CAO, 40 min) and reperfusion (24 h), while the maximum MVO area peaked in these pigs was 120 min after the restoration of coronary perfusion [102]. MVO and IMH areas were evaluated by MRI [102]. The largest area of IMH was identified in rats subjected to a 90-minute CAO followed by 48 hours of reperfusion [106]. The IMH area was measured in rats by MRI [106]. The onset of microvascular obstruction precedes the destruction of microvessels and the subsequent emergence of IMH. The presence of IMH was linked to poorer outcomes and the development of unfavorable ventricular remodeling. In patients with AMI, a larger IMH area was associated with longer ischemic durations and delayed reperfusion events [107]. Intra-myocardial hemorrhage was developed in pigs after a 40–120-min CAO and followed reperfusion [102]. According to Ma et al. [13] IMH did not develop before successful reperfusion of the heart and IMH size was correlated with infarct size and the MVO area. Investigators used CMR for the measurement of MVO and IMH areas. Intra-myocardial hemorrhage often develops in STEMI patients who have wider and deeper Q waves [108]. Anticoagulant and antiplatelet therapy could contribute to the occurrence of IMH in patients with AMI and PCI [108]. It was reported that the use of alteplase in patients with STEMI and PCI promotes an appearance of IMH [11]. A combination of MVO and IMH was observed in 36%–44% of patients with STEMI and PCI according to MRI data [13, 56, 66]. The pathogenesis of IMH remains unclear. It was reported that patients with IMH had higher CRP, inteleukin-6, fibrinogen, and neutrophils levels compared to patients without IMH [58, 88]. We found that the appearance of IMH is accompanied by an increase in the plasma CRP level was 13-fold in patients with STEMI and MVO. The role of inflammation in the development of IMH requires further investigation.
In summary, IMH is a common manifestation of AMI, especially in patients with STEMI. It is possible that inflammation is involved in the pathogenesis of IMH.
In our opinion, microvascular obstruction could be a target for the treatment of reperfusion cardiac injury. In recent years, much attention has been paid to dual antiplatelet therapy (DAPT, three possible combinations: aspirin and clopidogrel; aspirin and prasugrel; aspirin and ticagrelor) for the prevention of MVO [76, 109]. Some investigators performed DAPT in 97% of patients with STEMI and PCI [76].
The incidence of MVO in patients with STEMI and PCI receiving the P2Y
It has been reported above that intracoronary infusion of adenosine prevents the appearance of MVO in patients with AMI and PCI [30, 32, 71]. Nevertheless, Niccoli et al. [26] demonstrated that the intracoronary delivery of adenosine did not influence the occurrence of MVO in a cohort of patients (n = 160) with STEMI who underwent PCI.
Nazir et al. [50] also could not find an improvement in MVO in patients (n = 168) with STEMI and PCI. It should be noted that adenosine can aggravate ischemic/reperfusion cardiac injury in patients with AMI through triggering coronary steal [110]. Consequently, adenosine cannot be recommended for the treatment of AMI and MVO.
It was reported above that sodium nitroprusside, a NO donor, did not alter the
MVO area [25, 26, 50]. However, nicorandil, a NO donor and K
In dogs with intact coronary arteries and no myocardial hypoxia,
Consequently, epinephrine could be used for therapy of MVO, but it should be evaluated for its negative effects.
L-type Ca
Consequently, L-type Ca
These data demonstrated that clopidogrel, ticagrelor, and nicorandil reduce the incidence of MVO (Table 3). Epinephrine, verapamil, and nicardipine decrease the MVO area (Table 3).
| Drugs | Effects | Reference |
| Prasugrel | Incidence of MVO |
[54] |
| Ticagrelor | Incidence of MVO |
[54] |
| Tirofiban | MVO area |
[17] |
| DAPT | MVO area |
[109] |
| Adenosine | Incidence of MVO |
[30, 32, 71] |
| Adenosine | Incidence of MVO no effect | [26, 50] |
| Nitroprusside | MVO area no effect | [25, 26, 50] |
| Nicorandil | Incidence of MVO |
[8, 33, 101] |
| Epinephrine | MVO area |
[28, 29] |
| Verapamil | MVO area |
[24, 34, 35, 36, 39] |
| Nicardipine | MVO area |
[36, 41] |
Note. MVO, microvascular obstruction; AMI, acute myocardial infarction; DAPT, dual antiplatelet therapy.
Thus, MVO could be the result of an imbalance between vasodilation and vasoconstriction. Microvascular obstruction could be the result of coronary microvascular injury and, above all, the result of endothelial cell damage. It could be an inflammation injury of coronary microvessels. However, anti-inflammatory agents, for example, glucocorticoids have not been used before for treatment of MVO. Therefore, we cannot evaluate a role for inflammation in microvascular injury in patients with AMI and PCI.
Many questions are still waiting for an answer. It is unclear whether there is a
role for endothelial cell injury in the pathogenesis of MVO in patients with AMI
and reperfusion of the heart. There is no indisputable evidence of the
involvement of inflammation in the development of MVO. A role for ROS in the
pathogenesis of MVO is also yet to be studied. The role of necroptosis and
pyroptosis in the pathogenesis of MVO in patients with AMI and PCI is also not
studied. The role of thromboxane A2, vasopressin, angiotensin II, disturbances of
nitric oxide production and prostacyclin synthesis in the formation of MVO was
not studied before. The role of neuropeptide Y and endothelin-1 in the
development of MVO is required in further investigations. It is unclear of the
role of coronary artery spasm in the formation of MVO. It was reported that
sodium nitroprusside, a donor NO and endothelium-independent vasodilator, did not
improve CBF in patients with AMI and PCI. However, verapamil, an L-type
Ca
Platelets could be involved in the development of microvascular obstruction in
patients with AMI and PCI. Ca
LNM, EVV and NVN had the idea for the paper, reviewed and edited it critically for important intellectual content, FF, JMP, ERD were responsible for curating data, IAD performed visualization, VVR and AAB performed the literature search, GZS, AVM, BKK, AVK, AEG, SVD, JOS substantially contributed to the conception of the paper, wrote the manuscript, designed the figures and critically revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This article was supported by state assignment 122020300042-4.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.