



1 Key Lab of Medical Electrophysiology of Ministry of Education and Medical Electrophysiological Key Lab of Sichuan Province, Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease, Institute of Cardiovascular Research, Southwest Medical University, 646000 Luzhou, Sichuan, China
Abstract
Hypertension, a common cardiovascular disease, is primarily characterized by vascular remodeling. Recent extensive research has led to significant progress in understanding its mechanisms. Traditionally, vascular remodeling has been described as a unidirectional process in which blood vessels undergo adaptive remodeling or maladaptive remodeling. Adaptive remodeling involves an increase in vessel diameter in response to increased blood flow, while maladaptive remodeling refers to the narrowing or thickening of blood vessels in response to pathological conditions. However, recent research has revealed that vascular remodeling is much more complex. It is now understood that vascular remodeling is a dynamic interplay between various cellular and molecular events. This interplay process involves different cell types, including endothelial cells, smooth muscle cells, and immune cells, as well as their interactions with the extracellular matrix. Through these interactions, blood vessels undergo intricate and dynamic changes in structure and function in response to various stimuli. Moreover, vascular remodeling involves various factors and mechanisms such as the renin-angiotensin-aldosterone system (RAS), oxidative stress, inflammation, the extracellular matrix (ECM), sympathetic nervous system (SNS) and mechanical stress that impact the arterial wall. These factors may lead to vascular and circulatory system diseases and are primary causes of long-term increases in systemic vascular resistance in hypertensive patients. Additionally, the presence of stem cells in adventitia, media, and intima of blood vessels plays a crucial role in vascular remodeling and disease development. In the future, research will focus on examining the underlying mechanisms contributing to hypertensive vascular remodeling to develop potential solutions for hypertension treatment. This review provides us with a fresh perspective on hypertension and vascular remodeling, undoubtedly sparking further research efforts aimed at uncovering more potent treatments and enhanced preventive and control measures for this disease.
Graphical Abstract
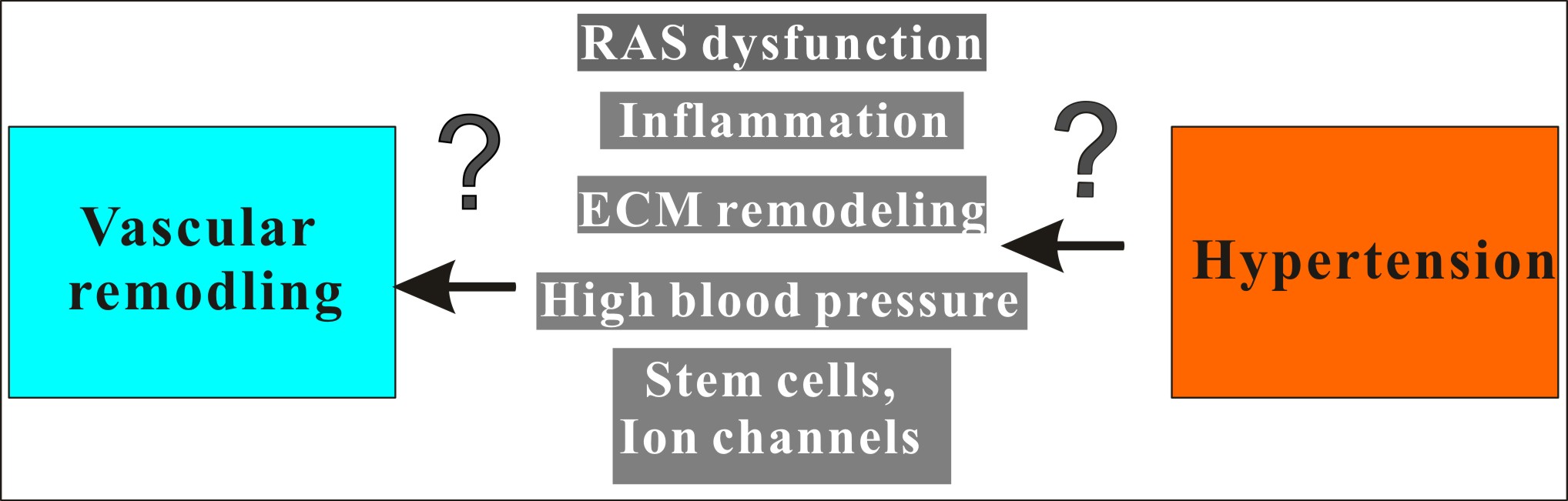
Keywords
- hypertension
- vascular remodeling
- vascular extracellular matrix
- renin-angiotensin system
- ion channel
- vascular resident stem cells
Hypertension is a significant risk factor for numerous cardiovascular, renal, and neurovascular diseases, with the frequency and mortality rates on the rise. Coronary heart disease, heart failure, atrial fibrillation, aortic valvular disease, sudden cardiac death, and abdominal aortic aneurysms, among others, have been linked to hypertension [1, 2]. Therefore, it is crucial to uncover novel mechanisms of hypertension and identify relatively effective treatment methods.
The arterial vessel wall is a complex structure and comprised of three distinct layers: the tunica externa, the tunica media and the tunica intima [3]. During the process of angiogenesis, the vessel wall closely monitors changes in its environment, integrating intercellular communication signals that ultimately affect the structure and function of blood vessels via the local production of mediators. Vascular remodeling is a crucial and positive process involving significant structural and functional changes. This process is governed by at least four cellular processes, including growth, death, migration, and synthesis or degradation of the extracellular matrix. Vascular remodeling is heavily influenced by the interaction among local growth factors, vasoactive substances, and hemodynamic stimulation, and it represents a dynamic response process directed towards hemodynamic conditions in the long term. However, it may also lead to vascular diseases and promote issues within the circulatory system [4].
Vascular structure changes can be broadly categorized as hypertrophic or non-hypertrophic remodeling. Hypertrophic remodeling involves the enlargement and proliferation of vascular smooth muscle cells (VSMCs), while non-hypertrophic remodeling involves a rearrangement of VSMCs. Remodeling can also be further categorized as inward, outward [5], or compensatory, depending on changes in the diameter of the blood vessel [6, 7]. Structural remodeling, first described by Folkow [8], is caused by various factors, such as the sympathetic nervous system (SNS) [9], renin-angiotensin-aldosterone system (RAS), extracellular matrix (ECM), endothelium-mediated mechanisms, mitochondrial dysfunction, and genetic factors. Vascular functional remodeling refers to the adaptive changes that occur in blood vessels in response to various physiological and pathological stimuli. This process involves a series of cellular and molecular events that aim to modify the structure and function of blood vessels to meet the changing demands of the body. One important aspect of vascular functional remodeling is the changes in cell growth, proliferation, and migration. These processes involve alterations in the number and size of VSMCs, endothelial cells (ECs), and fibroblasts within blood vessel walls. In addition, the regulation of vascular tone is another important aspect of vascular functional remodeling, in which the sympathetic nervous system (SNS) plays a crucial role. There exists a significant correlation between the SNS and vascular remodeling. The SNS has a vital function in promoting vascular remodeling by controlling vascular contraction and intimal thickening. However, in certain disease conditions, the excessive activation of the SNS can lead to abnormal vascular remodeling, thereby increasing the susceptibility to various cardiovascular diseases [10, 11, 12]. Changes in ion channel remodeling in vascular cells are also particularly important in this process. It is important to note that normal blood pressure is crucial in the development and differentiation of tissues and organs, achieved through shear and tensile forces. However, high blood pressure can result in abnormal biomechanical forces that cause vascular growth and development, vascular remodeling, and arterial stiffness. A recent study by Rasna Sabharwal in 2022 [13] explored the intrinsic structural plasticity of cerebral arterioles both during and after hypertension. It was discovered that during hypertension, arteriolar wall thickness, diameter, wall cavity ratio, and biostiffness undergo rapid changes that return to normal levels once blood pressure levels are reduced. However, inward remodeling occurs gradually and does not return to normal levels. Nevertheless, there is hope for improved disease outcomes in patients with hypertension [14]. Liu et al. [15] carried out a study to investigate the effects of long-term administration of losartan, aspirin, and atorvastatin on vascular remodeling in young spontaneously hypertensive rats (SHR). The results showed that these drugs not only have antihypertensive, anti-inflammatory, and lipid-lowering properties, but also improve vascular remodeling.
In summary, hypertension is a complex disease involving multiple pathophysiological mechanisms. By studying and exploring factors related to vascular remodeling, a better understanding of hypertension can be achieved. This article presents an extensive overview of the latest advancements in the field of ECM, RAS, and inflammation. Additionally, we explore the pivotal role played by ion channels present in blood vessels and stem cells in hypertension-induced vascular remodeling. Due to limitations in space, this review does not cover the influence of sympathetic nerves on vascular remodeling.
Vascular remodeling involves a multitude of factors and mechanisms, including
inflammation, cytokines, RAS, and mechanical stress experienced by the arterial
walls. Inflammation plays a critical role in vascular remodeling by promoting
endothelial dysfunction, VSMC proliferation, and ECM deposition [16, 17]. Elevated
levels of proinflammatory cytokines, such as interleukin-6 (IL-6) and tumor
necrosis factor-alpha (TNF-
 Fig. 1.
Fig. 1.Key factors that contribute to vascular remodeling. The main factors and important mechanisms of vascular remodeling are comprehensively summarized in the figure. RAS, renin-angiotensin system; ROS, reactive oxygen species; ECM, extracellular matrix; NADPH oxidase, nicotinamide adenine dinucleotide phosphate oxidase; Nox, the catalytic subunit of the NADPH oxidases; ECs, endothelial cells; VSMCs, vascular smooth muscle cells; SMCs, smooth muscle cells; CD, leukocyte differentiation antigen; Sca-1, stem cell antigen-1; c-kit, stem cell growth factor receptor.
ECM is a complex network composed of various proteins that closely interact with ECs and serve important biological functions [21]. Key proteins, such as collagen, fibronectin, and MMPs, play critical roles in maintaining blood vessel structure, promoting growth factor activity, and contributing to physiological and pathological processes associated with inflammation and immune response. Angiogenesis, the formation of new blood vessels, requires the degradation of the vascular basement membrane and the remodeling of the ECM to facilitate EC migration. The ECM not only provides mechanical stability but also controls vascular cell behavior and is central to vascular function and homeostasis. ECM remodeling, including generation, degradation, and changes in arterial tissue, is a hallmark of vascular remodeling in hypertension. Extracellular protease activity and receptor cleavage are associated with hypertension-related cell dysfunction. MMPs and cathepsins play crucial roles in the remodeling of ECM, making them pivotal regulators in various biological processes. Notably, their involvement in maintaining the integrity of vascular architecture and fine-tuning growth factor signaling has garnered significant interest among researchers and scientists.
MMPs are a group of enzymes that rely on Zn
Cathepsin serves as a crucial enzyme in reshaping the ECM. Its primary function is to facilitate the breakdown and regeneration of the matrix, thereby preserving the structure and functionality of tissues. As such, cathepsin has a significant impact on physiological and pathological processes, including cell migration, tissue repair, and cell activity. In the vascular system, Cathepsins belongs to the protease family and regulates the ECM under physiological and pathological conditions [33].
The MEROPS database lists over 700 human proteases, with 11 of them being cysteine cathepsins, including cathepsin B, C, F, H, K, L, O, S, V, W, and X. Cysteine cathepsins primarily contribute to the development of cardiovascular diseases, given their ability to degrade components of the ECM, particularly elastin. Cathepsin K, S, and V are capable of degrading elastin, which plays a crucial role in maintaining vascular integrity. Excessive elastin degradation, often associated with atherosclerosis, aneurysm, and chronic kidney disease [34, 35], can lead to a weakened vascular structure and rupture. It has been confirmed that overexpression of cathepsin S induces pulmonary artery remodeling [34]. Studies in the cardiovascular field have highlighted that vascular cells associated with atherosclerotic lesions overexpress cathepsin S without corresponding changes in cystatin C expression. This suggests that the balance between cathepsin and its inhibitors has shifted, facilitating cardiovascular wall remodeling. Further research indicates that the upregulation of cathepsin S in vascular disease can impede the integrity of the elastic layer and the basal layer of the intima. Microvessel growth is negatively impacted by cathepsin S inactivation, but does not impact vascular endothelial growth factor (VEGF) and basic fibroblast growth factor expressions. Overall, cathepsin S promotes ECM degradation. In addition to cathepsin S, cathepsin K is also considered an essential marker of vascular remodeling. It inhibits vascular smooth muscle cell proliferation and thus the vascular remodeling process [36]. It has been demonstrated that patients with endothelial dysfunction in chronic kidney disease have shown significant increase in the expression level of cathepsin D [37].
The Cathepsin L genes encode two different cathepsin proteins, namely cathepsin L and Cathepsin V. In a study by Lu et al. [38], it was found that vascular remodeling induced by Ang II as well as hypertension were mediated by mechanisms relating to cathepsin L/V-MEK/ERK. This confirmed that cathepsin proteins and related factors such as cystatin C and mitogen-activated protein kinase phosphorylation were up-regulated in mesenteric arteries and serum in hypertensive patients. Furthermore, cathepsin proteins are known to play a key role in the degradation and transfer of ECM. In an experiment involving the knockout of cathepsin proteins, Pan et al. [39] found a significant increase in collagen deposition in the medial aorta layer of cathepsin proteins knockout mice, while telomerase activity analysis suggested changes in vascular aging in cathepsin proteins knockout mice. In addition, a decrease in cathepsin proteins was observed in aging ECs [40]. Enhanced expression of A2, a member of the aldehyde dehydrogenase 1 family, was also reported. Activation of the AKT/ERK1/2-P21 pathway was found to promote cellular senescence, which may play a critical role in vascular senescence. These findings indicate that cathepsin proteins hold great potential as a therapeutic target for treating EC senescence. Yu et al. [41] revealed that elevated levels of cathepsin proteins in plasma were closely linked to coronary artery disease, providing evidence that circulating cathepsin proteins could be a promising biomarker for this condition. Furthermore, research on human atherosclerotic lesions and narrowed aortic valves has shown that, apart from cathepsin K and S, there is a notable increase in both mRNA and protein expression levels of cathepsin V [42]. Additionally, previous research has highlighted that the degradation of elastic fibers by cathepsin V, K, and S aids in the development of plaque-associated vascular calcification in VSMC [43]. As a result, cathepsin plays a crucial role in the process of vascular remodeling.
The RAS system, vascular remodeling, and hypertension are all interconnected through a complex network, with Ang II playing a significant regulatory role. Ang II is a multifunctional hormone that is produced by the RAS system. In the kidney, renin is released by the ECs of the glomerulus, which break down plasma angiotensinogen to generate a peptide precursor called angiotensin I. This is then transformed into Ang II by angiotensin-converting enzyme (ACE). Ang II plays a pivotal role in regulating hormonal responses in the body, such as the release of adrenaline and noradrenaline from the hypothalamus and adrenal medulla, which regulate water-sodium balance and the activity of the sympathetic nervous system. When Ang II binds to the Ang II type1/AT1 receptor (AT1R), it triggers an interaction between AT1R and heterotrimeric G proteins. This then leads to a cascade of second messenger signaling, involving key molecules such as inositol trisphosphate, diacylglycerol, arachidonic acid, and ROS. As a result, downstream effectors, including phospholipases C, A, and D, are activated. Furthermore, Ang II can stimulate inflammatory reactions, cell proliferation and differentiation, and contribute to various physiological and pathological processes such as cardiac hypertrophy and hypertension. Therefore, abnormal regulation of Ang II can affect the progression and treatment of hypertension, and other cardiovascular diseases.
One of the primary ways that Ang II contributes to vascular remodeling is by increasing oxidative stress [19]. The resulting oxidative stress may trigger the activation of MMPs, which can cause the deterioration of ECM and subsequently lead to vascular remodeling. Ang II increases oxidative stress by activating Nox to produce ROS [44]. Ang II is thought to enhance the generation of ROS by upregulating the expression and catalytic activity of the Nox family proteins [45]. The abundance of proteins of Nox family isoforms in VSMC will determine the physiological function of Ang [46]. Nox2 and Nox4 are primarily found in endothelial cells and cardiomyocytes, indicating their close association with cardiovascular diseases [47]. This aligns with the notion that the expression of Nox subunits in the vascular wall plays a crucial role in the microvascular remodeling observed in individuals with arterial hypertension. For example, Sortilin is a member of the vacuolar protein sorting 10 protein (VPS10P) receptor family. There is evidence suggesting that sortilin plays a key role in the dysregulation of sphingolipid metabolism and oxidative stress associated with human hypertension. Studies have found increased plasma acid sphingomyelinase (ASMase) activity, as well as elevated levels of plasma sortilin, sphingosine-1-phosphate receptor (S1P), and soluble Nox2-derived peptide (sNox2-dp) in hypertensive patients, with a more pronounced increase in those with uncontrolled blood pressure. Sortilin induces dysfunction of the mesenteric arterial endothelium through the activation of the Nox2 isoform, and this dysfunction can be prevented by lowering ASMase or sphingosine kinase 1 [48] Nox2 induces vascular oxidative stress by directly generating superoxide, whereas Nox4 primarily relies on the swift conversion of superoxide into hydrogen peroxide (H2O2) through dismutation [49].
Another main way through which Ang II contributes to vascular remodeling is by promoting inflammation in the vascular wall [19]. This is achieved by activating various pro-inflammatory cytokines and chemokines, leading to the recruitment of leukocytes and activation of resident immune cells. This, in turn, further drives vascular remodeling, as discussed in Section 3.2. Ang appears to be central and is able to activate and/or induce multiple factors involved in vascular fibrosis, to stimulate collagen fiber hyperplasia and ECM deposition by inducing the expression of connective tissue, growth factors, inflammatory factors, aldosterone and ET-1 [50, 51]. Consequently, this leads to the accelerated growth and development of VSMCs, ultimately promoting vascular remodeling. Additionally, Ang II triggers integrins, adhesion molecules, cytokines, and fibrosis, ultimately promoting inflammation and remodeling in the vascular system. Endothelial remodeling due to Ang II can induce both growth and apoptosis in cells, leading to a reduction in the outer diameter of blood vessels. However, it is important to note that the apoptosis is limited only to the periphery [20, 52]. A recent study has demonstrated the crucial role of AT1R expression in VSMCs in vascular remodeling within a mouse model of hypertension induced by Ang II infusion [53]. The underlying mechanism by which Ang II promotes vascular fibrosis and arterial stiffness should be highlighted. This process involves the interaction between adventitial fibroblasts, VSMC, immune cells, and ECM with inflammatory mediators and associated signal transduction pathways [20].
Stem cells are a type of highly potential cell that have unlimited self-renewal ability and multi-directional differentiation potential. Recent studies have demonstrated that vascular resident stem cells, which are present in the tunica intima, tunica media and tunica externa, play an essential role in cellular regeneration. The composition and distribution of vascular resident stem cells are key factors in the complex structure of arteries. The innermost layer, also referred to as the endothelial cell layer, contains a small population of endothelial progenitor cells (EPC) [54, 55], as well as notable stem cells such as Sca-1+ and CD34+. Meanwhile, the middle layer serves as a thick layer composed of VSMCs with a small population of stem cells such as Sca-1+ stem cells. Finally, the outer layer comprises a layer of connective tissue, and is home to a diverse group of cells, including fibroblasts, resident inflammatory cells, peri-vascular endothelial cells, adrenergic nerves, as well as stem cells (like pluripotent stem cells or bone marrow mesenchymal stem cells), and progenitor cells (including cells with differentiation potential for macrophages, endothelial cells, smooth muscle, and hematopoietic cells like Sca-1+, CD34+, c-Kit, and Flk-1, among others). These blood vessel stem/progenitor cells are spread throughout the entire structure of the vessel wall and are vital in the development of vascular diseases, as well as considered as the cell source for blood vessel remodeling.
A recent genetic cell lineage tracing study [56] showed that c-kit+ cells can be transplanted into blood vessels in animals and differentiate into VSMCs. Experiments on cell culture in vitro also support that these cells have stem/progenitor properties. Meanwhile, other studies [3] have confirmed that c-kit+ stem cells differentiate into vascular cells in small blood vessels. Another study using genetic lineage tracing technology in lung maintenance and repair processes has confirmed that c-kit+ cells was involved in the formation of pulmonary vascular endothelium [57]. Related studies have shown that CD146+ cells constitute the majority of embryonic aortic VSMCs, and rapid regeneration of the smooth muscle layer is crucial for successful repair of vascular injury. Jiang et al. [58] conducted single-cell RNA sequencing (sRNA-seq) on mouse femurs and found that the percentage of CD34+ expression showed a notable increase in the femoral arteries where lesions were detected. A series of data showed that most lumen and microvascular CD31+ endothelial cells were derived from non-bone marrow CD34+ cells to respond to vascular injury, while bone marrow CD34+ cells mainly caused an increase in CD45+ white blood cells, which may also be an important factor in neo-intima formation. Structural atherosclerosis, a well-established cardiovascular risk factor, is dependent on hematopoietic stem cells, specifically CD34+ cells. However, it is noteworthy that there is a negative association between the number of circulating CD34+ cells and cardiovascular disease. In a recent epidemiological study comprising Japanese men aged 60 to 69 years attending annual health examinations, Shimizu identified [59] that functional atherosclerosis differs significantly from structural atherosclerosis in terms of endothelial repair activity. Aggressive endothelial repair may result in an increase in both functional and structural atherosclerosis. The depletion of CD34+ cells leads to endothelial repair defects, which further exacerbates functional atherosclerosis instead of structural atherosclerosis. Therefore, the absence of structural atherosclerosis does not always indicate a favorable condition for the endothelium. Tang et al.’s [60] genetic lineage tracing and single-cell RNA sequencing found that after severe endothelial damage, Sca-1+ VSMCs migrate to the middle layer and generate new VSMCs, which have a greater tendency to expand compared to existing VSMCs, and are more effective in subsequent expansion than prior existing smooth muscle. Their study determined that Sca-1+ PDGFRa+ cells are a source of new smooth muscle healing after severe cross-cut artery injury. Research has shown that upon acute vascular injury, Sca1+ cells differentiate into myofibroblasts and are embedded in perivascular collagen and ECM. They found that the chromatin remodeler, Smarca4/Brg1, facilitates AdvSca1-SM myofibroblast differentiation [61].
Ion channels play a key role in various aspects of cell function, including pulse excitation and propagation, proliferation, migration, and apoptosis. The role of ion channels in vascular remodeling will be discussed in detail in the following Section 3.3. For stem cells, the importance of ion channels in cell proliferation, migration, and differentiation is increasingly recognized [62]. Research has shown that various membrane ion channels and pump proteins can effectively transport ions across lipid bilayers, establish membrane potential (Vm), and generate sustained signals that are more persistent than excitatory cell action potentials. This self-regulating bioelectric signal not only reflects the state of the cell, but also controls various characteristics of progenitor cells [63, 64]. Previous studies have suggested that the regulation of Vm can serve as an upstream factor influencing stem cell differentiation and can impact downstream processes, ultimately influencing the expression of genes related to progenitor cell maturation [65]. However, research on the relationship between stem cells and vascular remodeling is still limited. Further research will help elucidate the functional role of ion channels in stem cells and their role in vascular remodeling.
Hypertension is a chronic disease that is widely prevalent and characterized by
elevated arterial blood pressure. It is defined by systolic and/or diastolic
pressure readings
Prolonged high blood pressure can lead to vascular remodeling, including changes in the structure of blood vessels such as thickening of vessel walls and reduced distensibility. It also triggers abnormal growth, differentiation, migration, synthesis, and secretion of vascular tissue cells. Furthermore, disruptions in ion channel function may interfere with the normal regulation of blood vessel dilation and constriction. After suffering vascular injury, the vessel wall structure may undergo significant modifications, with intimal regeneration being a critical component in the healing process. Hypertension patients’ increased systemic vascular resistance is likely due to vascular remodeling being the leading cause. Changes to the blood vessels due to hypertension are complex and closely related to the fluctuations in hormone and vasoactive substance levels. Humphrey et al. [73] emphasize the importance of exploring the mechanism of hypertension-related vascular remodeling from a mechanical homeostatic perspective. Elevated blood pressure is the root cause of a series of arterial responses triggered by phenotypic changes in primary vascular wall cells and differential gene expression, leading to vascular remodeling. Aortic maladjustment is a condition characterized by fibrosis of the outer membrane, which greatly reduces its biomechanical function, resulting in impaired end-organ perfusion. This leads to an increase in the incidence and mortality of related diseases. The predominant components found in the walls of blood vessels are collagen and elastin, and the stiffness of arteries is determined by their presence [74]. Excessive deposition of collagen can damage the walls of blood vessels, leading to increased stiffness. The hypertrophy, sclerosis, and apoptosis of VSMCs contribute to the thickening of the inner layer of blood vessels and the stiffness of arteries. Studies have demonstrated that the increased stiffness of vascular ECM during hypertension activates stroma-binding receptor integrins and associated intracellular signaling pathways, including phosphatidylinositol 3-kinase/protein kinase B, beta-catenin, and RhoA/Rho-associated protein kinases. As a result, VSMCs undergo contraction, proliferation, and migration [75]. Elastin, a component of the ECM, also plays a crucial role in regulating VSMC behavior and the transition of their phenotype to a contractile state [76].
Recent research has highlighted the crucial role of an activated inflammatory
response and immune system in the development and progression of hypertension
[17] as well as its association with various complications. In individuals
diagnosed with high blood pressure, researchers have observed increased levels of
inflammatory biomarkers, suggesting the potential involvement of the immune
system in the development of hypertension. Furthermore, inappropriate immune
activation may contribute to high blood pressure by impacting different organs
like the microvasculature, kidney, and nervous system. It is widely known that an
imbalanced and overactive immune system can cause inflammation, which is
characterized by a surge of proinflammatory cytokines. High blood pressure
triggers the body’s natural inflammatory response, which leads to the
accumulation of white blood cells and the release of inflammatory factors. These
factors can build up under the lining of blood vessels, resulting in further
thickening of the walls and the formation of plaques [17]. Although inflammation
serves as a critical response of the body to foreign agents and promotes healing,
an excessive presence of inflammation can be detrimental. Inflammation and
endothelial dysfunction are intimately connected and contribute to every stage of
injury and thrombotic complications. According to a study by Adachi et
al. [77], following vascular guidewire injury, the expression of inflammatory
cytokines like TNF-
Traditionally, it was thought that essential hypertension was caused by changes
in hemodynamics. However, numerous studies have unequivocally confirmed that
inflammatory cytokines also hold a crucial role in promoting the progression of
hypertension through their impact on blood vessels and kidney function. One such
cytokine is IL-1
TNF-
In addition to cytokines, integrins also play a significant role in
inflammation. Budatha et al. [89] conducted a study on integrin
Ion channels are a crucial component of the cell membrane, comprising
specialized proteins that are expressed on the surface. These channels, including
Ca
Vascular tone is regulated by a variety of factors, including neural and humoral
stimuli. VSMCs integrate these signals and adjust the contractile state of VSMCs
by modulating intracellular calcium levels ([Ca
In addition to VSMCs, ion channels present in other cells of blood vessels also
play crucial roles in maintaining proper vascular function. For instance, it was
found that hypertension-induced vascular damage is caused by an increased
production of ROS and altered Ca
ECs release various vasodilating factors, including nitric oxide and
prostacyclin, in response to stimulatory and shear stress. Additionally, vascular
ECs are known to regulate VSMC contractility through the production of
endothelium-dependent hyperpolarization (EDH). The opening of small and
intermediate conductance calcium-activated potassium channels (SK
Piezo1 and Piezo2, two recently discovered mechanosensitive channels, combine
fine force sensing with regulated Ca
Fig. 2 illustrates the critical factors and innovative mechanisms involved in vascular remodeling in hypertension.
 Fig. 2.
Fig. 2.The key factors and crucial mechanisms of vascular remodeling under hypertension. The abnormal activity of major factors in hypertension leads to severe vascular remodeling, further exacerbating vascular injury and hypertension. RAS, renin-angiotensin system; ECM, extracellular matrix; ECs, endothelial cells; VSMCs, vascular smooth muscle cells.
Hypertension is a widespread cardiovascular disease that has a considerable impact on human health. While the pathogenesis of this disease is intricate, it is clear that it is intimately linked to vascular remodeling. Researchers are currently exploring different mechanisms that contribute to vascular remodeling in hypertension, such as changes in the ECM, inflammatory mechanisms, stem cell involvement, and ion channel involvement. However, the specific interactions between vascular structure and the resulting functional changes that contribute to the pathogenic effects of hypertension need further investigation. It is widely acknowledged that vascular structure and function modifications can exacerbate the effects of hypertension, and the underlying mechanisms are gradually being uncovered. These research findings offer valuable insights into the identification of new biomarkers, innovative therapies, and targets for treatment of treatment of hypertension and other cardiovascular diseases. In this review, we summarize the latest research progress of RAS, inflammation, ECM, and stem cells in vascular remodeling. We also discuss the latest research on ion channels found in vascular cells and stem cells. A comprehensive understanding of hypertension and vascular remodeling will undoubtedly fuel more research, leading to the discovery of more effective treatment methods and improved measures for disease prevention and control. Therefore, we hope that this summary of current knowledge will serve as a significant stimulus for future research in the field.
XYZ has made invaluable contributions in the area of data acquisition and reference collection. She has played a highly active role in drafting the manuscript, conducting thorough content reviews, and approving the final version for publication. Moreover, she has willingly assumed full responsibility for all aspects of the work. YY, on the other hand, has taken charge of organizing the article’s structure. She also has been involved in drafting the manuscript and reviewing it critically for important intellectual content, granted final approval for publication, and completed the necessary revisions prior to manuscript submission. She agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy.
Not applicable.
Not applicable.
This work was supported by the National Natural Science Foundations of China (grant numbers 31972909, 82070502 and 32171099) and the Sichuan Science and Technology Program (grant numbers 2022YFS0607, 23NSFSC1456 and 2021YJ0213).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.