
- Academic Editor
†These authors contributed equally.
Cardiovascular diseases are the first cause of death worldwide, with a heavy social and economic impact. They include a wide range of pathological conditions, among which cardiac fibrosis represents a common pathogenetic hallmark. The fibrotic process is driven by cardiac mesenchymal stromal cells, namely fibroblasts, which become activated, proliferate, and differentiate into myofibroblasts in response to several stimuli, in the end secreting extracellular matrix proteins, and mediating cardiac tissue remodelling and stiffening. A specific therapy for the exclusive treatment of cardiac fibrosis is still lacking. Given the growing quest for reducing the burden of cardiovascular diseases, there is increasing interest in the search for new effective anti-fibrotic therapies. In this review, we will briefly summarize the limited pharmacological therapies known to act, at least in part, against cardiac fibrosis. Then we will present novel potential active molecules, molecular targets, and biotechnological approaches emerged in the last decade, as possible future therapeutic strategies for cardiac fibrosis, with a specific focus on targeting fibroblast activation and function.
Heart disease is a leading cause of death worldwide and its prevalence is expected to increase in the next decades [1, 2, 3], representing a heavy social and economic burden globally. Cardiac fibrosis is one of the key underlying pathogenetic mechanisms of many chronic cardiac diseases, and its therapeutic management remains an unmet clinical need [4].
The fibrotic process develops either acutely as a circumscribed scar due for example to acute ischemia and cell death, or in the interstitium as a continuum positive feedback of multiple attempts for repair reactions. In this case, it evolves into a chronic degenerative/inflammatory response with an increasing number of mesenchymal stromal cells turning into activated cardiac fibroblasts (CFs). Under physiological conditions, most stromal cells (including CFs) are static, but when an injury occurs, CFs become activated, proliferate, and then secrete a large amount of collagen fibers, leading to extra-cellular matrix (ECM) resorption and deposition [5]. These phenomena will progressively increase myocardial stiffness, hamper myocardial conduction, reduce the efficiency of the cardiac muscle contraction, all of which will ultimately lead to heart failure [6, 7].
At present, there is no specific treatment targeting exclusively myocardial fibrosis. The increasing quest for novel effective drugs against fibrosis and remodelling respond to international research priorities for the development of precision and personalized medicine approaches to chronic non-communicable diseases in the aging population, which could reduce the cardiovascular disease burden effectively. Indeed, novel efficient anti-fibrotic drugs may reduce the cost of surgical-clinical interventions and of standard life-long medication, as well as the incidence and average time of hospitalization.
In this review, we will provide a brief overview of the current medications known to interfere, at least in part, with the mechanisms of cardiac mesenchymal and fibroblast activation and function. Then we will present a selection of novel potentially active molecules, whose mechanism of action is not necessarily fully elucidated yet, together with a review of novel possible molecular targets in CF activation. We will also present a selection of epigenetic pathways and non-coding RNAs known to interfere directly with fibroblast differentiation. Finally, we will present some examples of biological and biotechnological approaches (e.g., direct reprogramming, T-cell immunotherapy) that have been recently described against cardiac fibrosis.
Cardiac fibrosis development is directly associated with the activation of the
renin-angiotensin-aldosterone system (RAAS) [8]. The progression of cardiac
dysfunction reduces renal perfusion, so the juxtaglomerular apparatus starts to
release renin [9]. The consequence of this release is an increment of the
angiotensin II level produced by the angiotensin-converting enzyme (ACE). For a
long time, angiotensin II has been considered the principal promoter of cardiac
fibrosis [10]. In fact, angiotensin II directly drives numerous myofibroblast
functions such as, cell proliferation and migration, transforming growth factor beta (TGF-
Aldosterone is a steroid hormone produced by adrenal cortex under stimulation of angiotensin II [20]. It is a transcriptional regulator and is responsible for upregulating pro-fibrotic genes. Aldosterone and angiotensin II work together by activating the mitogen-activated protein kinase (MAPK) pathway in cardiomyocytes [21, 22], as well as in CFs [23, 24]. Interestingly, aldosterone can exert its profibrotic effect also when angiotensin II is inactivated, suggesting its independent role in promoting fibrosis [25]. There are two drugs—spironolactone and eplerenone—which have revealed potential antifibrotic effects clinically. They are receptor antagonists of the mineralocorticoid family, blocking aldosterone signaling. Zannad et al. [26] verified the serum level reduction of fibrosis markers and collagen synthesis after spironolactone treatment. More data on their specific effect on CFs is needed, though.
Pharmacological approaches for the treatment of cardiac fibrosis include
targeting the adrenergic receptor system, as well. This neurohormonal pathway
contributes to the maintenance of cardiovascular homeostasis through the release
of norepinephrine, a neurotransmitter that binds to both type
Other types of
Besides
Many molecules have been described in the literature as able to directly act on cardiac stromal cells and fibroblasts, reducing fibrotic activation and features. In some cases, the signalling pathways involved have been defined, although for many of them detailed insights on signal transduction and molecular mechanisms are still lacking or incomplete (Table 1, Ref. [47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62]). Nonetheless, they represent an important starting point for further validation and translational studies.
| Molecule | Target | Mechanism of action | Model system | References |
| Vinpocetine | PDE1 | Activation of NF- |
In vitro | [47] |
| In vivo | ||||
| IC86340 | PDE1A | Reduced fibroblast activation, ECM deposition, and expression of pro-fibrotic genes. | In vitro | [48] |
| In vivo | ||||
| TP-10 | PDE10A | Reduced cardiac remodeling and fibrosis. | In vivo | [49] |
| Dapagliflozin | SGLT2 | AMPK |
In vitro | [59] |
| In vivo | ||||
| Berberine | IGF-1R | Downregulation of IGF-1R activation, reducing MMP2, MMP9, |
In vitro | [50] |
| Naringenin | ERK, JNK, PI3K/Akt, SMAD3 | Inhibition of TGF- |
In vitro | [51, 52] |
| Bufalin and lycorine | Unclear/multiple | Suppression of collagen I and matrix components in activated fibroblasts. | In vitro | [58] |
| Piperlongumine | Unclear/multiple | Reduced expression and transcriptional activity of KLF4 in fibroblasts. | In vivo | [53] |
| Reduced phosphorylation of Akt and preserved levels of FoxO1 in cardiomyocytes. | ||||
| Curcumin | Unclear/multiple | Activation of SIRT1 with reduced MMP2 and 9, collagen I and III levels in fibroblasts. Reduced IL-18 secretion and TGF- |
In vivo | [54, 55, 56] |
| Trehalose | Autophagy | Reduced expression of markers of activated fibroblasts, and reduced fibrotic and inflammatory paracrine signaling. Increased pro-angiogenic paracrine support. | In vitro | [57] |
| MCC950 | NLRP3 complex | Blocking of TGF- |
In vivo | [60] |
| pUR4 | Fibronectin polymerization | Reduced cell proliferation and migration, and ECM deposition. Reduced remodeling and fibrosis. | In vitro | [61] |
| In vivo | ||||
| Verteporfin | YAP/TAZ-TEADs complex | Downregulation of the genes induced by TGF- |
In vitro | [62] |
| In vivo |
PDE, phosphodiesterase; SGLT2, sodium-glucose cotransporter-2; IGF-1R, insulin-like growth factor 1 receptor; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase; PI3K/Akt, phosphatidylinositol3-kinase/protein kinase B; SMAD, small mothers against decapentaplegic; NLRP3, NOD-like receptor family pyrin domain containing 3; YAP/TAZ, Yes-associated protein/transcriptional coactivator with PDZ-binding motif; TEADs, TEA domain transcription factors; ECM. extra-cellular matrix; TGF-
Some active molecules occur naturally in food and could be used in clinics due to the wide range of pharmacological effects.
Vinpocetine is derived from a natural plant alkaloid and is used as a dietary
supplement. It appears to act through an IkappaB kinase (IKK)-dependent pathway, attenuating CF
activation and ECM synthesis, independently from small mothers against decapentaplegic (SMAD) phosphorylation or
activation [47, 63]. Moreover, both in vitro and in vivo, it
targets cyclic nucleotide phosphodiesterase 1 (PDE1), whose expression is
up-regulated in the heart under RAAS and beta-adrenergic stimulation [48, 64].
Vinpocetine could also act through IKK with anti-inflammatory effects because of
its role in nuclear factor kappa B (NF-
Concerning cyclic nucleotide PDEs as promising targets for the modulation of the
fibrotic process, the PDE1A form has been identified as a key regulator of
fibroblast activation and ECM remodelling in the heart [48]. This isoform was
found to be specifically enhanced in activated myofibroblasts both in
vitro upon angiotensin II and TGF-
Berberine is derived from a natural plant alkaloid able of targeting the
insulin-like growth factor I receptor (IGF-1R). IGF-1 is an important mediator of
cardiac hypertrophy, but its continuous expression in the myocardium contributes
to pathological remodelling and interstitial fibrosis [70]. The IGF-1R is
upregulated in CFs isolated from diabetic hearts, characterized by overexpression
of matrix metalloproteinase matrix metallopeptidase-2 (MMP-2) and matrix metallopeptidase-9 (MMP-9),
Naringenin, a flavonoid present in citrus fruits, exerts a cardioprotective
effect by suppressing extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and phosphatidylinositol3-kinase/protein kinase B (PI3K/Akt) signalling pathways. In
vitro, naringenin caused the inhibition of TGF-
Piperlongumine is another alkaloid that is naturally isolated from the long
pepper piper, and it has been proposed as a potential natural compound for the
clinical treatment of cardiac hypertrophy and fibrosis. Angiotensin-induced
expression of profibrotic proteins in neonatal CFs was significantly decreased by
piperlongumine treatment, reducing the expression of Krüppel-like factor 4
(KLF4), and its recruitment to the promoter regions of the pro-fibrotic cytokines
TGF-
Curcumin is a spice-derived pigment, widely studied as an anti-cancer,
anti-oxidant, and anti-inflammatory agent [73, 74]. Recently, it was investigated
also as a modulator of cardiac fibrosis. Hsu and colleagues [75] reported
reduction of TGF-
A recent study reported the ability of curcumin to also modulate autophagy. In this case, it inhibited mammalian target of rapamycin (mTOR) signalling thus enhancing the autophagic process. Consequently, all pro-fibrotic markers such as procollagen I and III in rat hearts treated with isoprenaline were reversed by curcumin administration [56]. However, whether this in vivo effect was due to direct or indirect mechanisms has not been defined yet.
The disaccharide trehalose—derived from myocytes—is known to be a natural activator of autophagy. It has shown protective effects on the heart [79], and can specifically enhance autophagy also in resident cardiac mesenchymal stromal cells [80] promoting anti-fibrotic responses. Autophagy boosting in these cells can reduce the expression of markers of activated fibroblasts, and preserve a less pro-fibrotic and pro-inflammatory paracrine function. When cells are exposed to metabolic stress, which is a main mediator of myocardial fibrosis, trehalose treatment also increased pro-angiogenic effects of cardiac mesenchymal stromal cells [57]. This effect has been also confirmed in human cells derived from diabetic patients, where the enhancement of autophagy by trehalose, combined with oxidative stress reduction by nitrogen oxides (NOX)-inhibitors, may contribute to reducing fibrotic activation and specification of primitive stromal cells [81].
A recent study by Schimmel et al. [58] has identified fifteen substances with antiproliferative effects in human CFs by high-throughput natural compound library screening. The validation by multiple in vitro fibrosis assays and selection by stringent algorithms have identified the steroid bufalin and the alkaloid lycorine as potentially effective antifibrotic molecules. Both were assayed for suppression of collagen I and matrix components production in activated fibroblasts, and the most significant effects were mediated by bufalin [58].
Dapagliflozin is a sodium-glucose Cotransporter-2 (SGLT2) inhibitor. SGLT2 is
involved in the pathogenetic mechanisms of diabetes by increasing the risk of
cardiovascular disorders, such as MI, stroke, and heart failure [82]. The
development of diabetic cardiomyopathy is caused by functional and metabolic
changes that occur in the myocardium at multiple levels [57]. In the last years,
studies have shown how SGLT2 inhibitors exhibit cardiovascular safety and
benefits [83]. Dapagliflozin blocks glucose resorption in the proximal tubule of
the kidney and promotes glucosuria [84, 85, 86], but it also mediates antifibrotic
effects in an AMPK
MCC950 is a selective inhibitor of the inflammasome complex NOD-like receptor family pyrin domain containing 3 (NLRP3), that has been
proposed to reverse transverse aortic constriction (TAC)-induced cardiac
remodelling. It is known that the prevention of myocardial remodelling is
possible by suppressing the expression or activation of NLRP3 before TAC surgery
[89]. Zhao et al. [60] have reported that NLRP3 activation promotes
pathological cardiac remodelling due to pressure overload in a mouse model,
whereas MCC950 treatment significantly attenuated fibrosis. Specifically, it
reduced
A new strategy proposed to prevent the development of cardiac fibrosis is the intervention on ECM protein polymerization. Specifically, the small peptide pUR4, a fibronectin polymerization inhibitor, has been tested in mouse models of ischemia/reperfusion (I/R) injury to attenuate pathological remodelling, tissue fibrosis, and cardiac dysfunction. In vitro, pUR4 administration to murine and human myofibroblasts led to a reduction of cell proliferation and migration, as well as ECM protein deposition, thus ameliorating pathological features [61].
A recent study aimed to demonstrate how pharmacological interference with
mechano-sensing cues could oppose stromal cell activation and myofibroblast
differentiation. Indeed, mechano-sensing signalling due to myocardial stiffening
plays a key role in remodelling progression. In particular, the shuttling of the
main Hippo transcriptional component, the YAP (Yes-associated protein)/TAZ
(transcriptional coactivator with PDZ-binding motif) complex, exerts homeostatic
control of cardiac matrix, and a specific function in myocardial remodelling
after injury [91, 92]. Garoffolo et al. [62] tested treatment with
Verteporfin, a drug known to prevent the association of the YAP/TAZ complex with
their cognate transcription factors TEA domain transcription factors (TEADs), on human fibroblasts activation and
differentiation. The results revealed prevention of the fibrotic process by
significant downregulation of more than half of the genes induced by
TGF-
It is worth mentioning that the expanding knowledge on the mechanism of action
of commonly prescribed drugs may represent a starting point as repurposing
strategy towards cardiac fibrosis. Statins are the most common drugs used to
inhibit cholesterol biosynthesis. Recently, several studies have demonstrated
their capability of exerting other beneficial effects on cardiovascular diseases
[93, 94]. Specifically, atorvastatin was studied as a potential drug to ameliorate
cardiac fibrosis. It was used to treat CFs in vitro and achieved
reduction of Heat Shock Protein 47 (HSP47) levels. Indeed, HSP47 is a collagen
specific molecular chaperon, normally induced by TGF-
Besides the discovery and testing of novel molecules with one or more known targets, in the last decades many studies have focused on the identification of new potential molecular targets for cardiac fibrosis (Fig. 1), yet in the absence of known modulators.
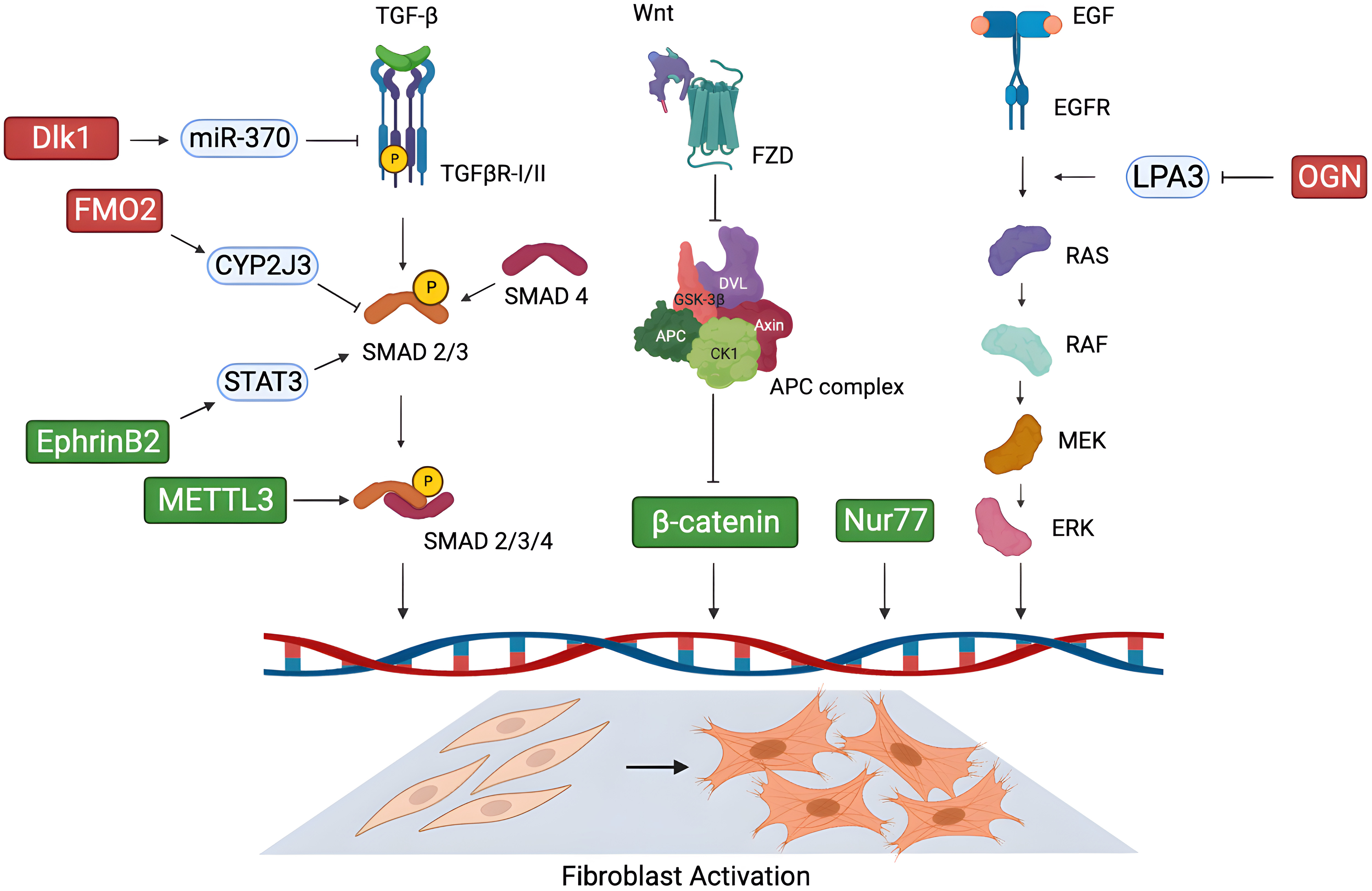 Fig. 1.
Fig. 1.Novel possible molecular targets for cardiac fibrosis. New
potential targets proposed in the literature in recent years for the treatment of
cardiac fibrosis, and their mechanism of action in modulating cardiac fibroblast
activation through key signalling pathways. (Green: pro-fibrotic factors; red:
anti-fibrotic factors). Figure was created with the Biorender Software
(https://www.biorender.com/). Dlk1, delta-like homolog 1; FMO2, flavin-containing monooxygenase 2; CYP2J3, cytochrome P450
superfamily 2J3; STAT3, signal transducer and activator of transcription 3; EphrinB2, erythropoietin-producing
hepatoma interactor B2; METTL3, methyltransferase-like 3; TGF-
Osteoglycin (OGN) is a small leucine-rich proteoglycan secreted in the ECM, whose role in the fibrotic process is still debated. OGN was found to be overexpressed in the heart of patients with hypertensive [96] and ischemic disease [97]. In 2018, Zuo et al. [98] identified OGN as a negative regulator of cardiac fibrosis in response to chronic hypertension-induced injury. Angiotensin II infusion in OGN deficient (OGN-/-) mice revealed increased interstitial fibrosis and diastolic dysfunction. Accordingly, in vitro angiotensin II treatment on OGN-/- CFs led to enhanced cell proliferation and migration via activation of the epidermal growth factor receptor (EGFR) signalling, while OGN overexpression had the opposite effect. Specifically, the authors demonstrated that in physiological conditions OGN attenuates cardiac fibrosis binding to lysophosphatidic acid 3 (LPA3), thus preventing the activation of the Rho-dependent EGFR/ERK signalling, which induces fibroblast proliferation and migration [98].
Another interesting modulator of cardiac fibrosis is the nuclear receptor Nur77.
It is involved in stress responses in different cell types [99, 100, 101], and it seems
to play a dual role in the fibrotic process in heart tissue. A study by
Medzikovic et al. [102] highlighted how Nur77 promoted CF
differentiation into myofibroblasts. Conversely, its knockdown led to a reduction
of myofibroblast marker expression and ECM proteins production, as well as
decreased proliferation and wound healing capacity in rat cells stimulated with
isoproterenol or TGF-
Among the factors that take part in the fibrotic process, it is worth mentioning
fine regulation of fibrosis-related gene expression carried out by the
N6-methyladenosine transferase methyltransferase-like 3 (METTL3) at the post-transcriptional level. METTL3
is the catalytic subunit of the methyltransferase complex which mediates mRNA
methylation [103, 104, 105]. Recently, Li et al. [106] discovered the role of
METTL3 as a promoter of CF activation and ECM remodelling in the process of
MI-induced fibrosis. Specifically, METTL3 exerted its pro-fibrotic properties via
tuning the m6A modification of mRNAs encoding collagens and factors of the
TGF
In 2019, delta-like homolog 1 (Dlk1), a member of the epidermal growth factor
(EGF)-like family involved in the control of cell differentiation [107], has been
identified as a key anti-fibrotic factor in the heart. In detail, authors found
that Dlk1 negatively regulated fibroblast-to-myofibroblast differentiation
through the activation of miR-370, which in turn inhibited the TGF-
Another factor whose pro-fibrotic activity has been recently uncovered is erythropoietin-producing hepatoma interactor B2 (EphrinB2), a cell surface transmembrane ligand ubiquitously expressed in mammals, mediating organ-specific regulation of the fibrotic process [109, 110]. Increased expression of EphrinB2 has been detected in human and mouse hearts with severe cardiac fibrosis. In vivo silencing of EphrinB2 led to the attenuation of cardiac fibrosis and improvement of cardiac function, while its overexpression in CFs mediated myofibroblast differentiation and activation in vitro. In detail, EphrinB2 exerted its pro-fibrotic effects mainly via activating the signal transducer and activator of transcription 3 (STAT3) signalling in CFs, and promoting the interaction between STAT3 and SMAD3, thus inducing the fibrotic response [111]. Given this evidence, EphrinB2 could be considered as another promising candidate target against cardiac fibrosis.
Recently, a previously unknown anti-fibrotic activity of flavin-containing
monooxygenase 2 (FMO2) has been characterized by Ni et al. [112]. FMO2 is
an enzyme with nicotinamide adenine dinucleotide hydrogen/flavin adenine dinucleotide (NADH/FAD)-dependent activity, whose expression was found
specifically enriched in CFs, but not in cardiomyocytes, and significantly
down-regulated upon cardiac injury in rats, mice, and non-human primates. FMO2
silencing in rat hearts induced spontaneous fibrosis and altered cardiac
function; in contrast, induction of FMO2 expression in infarcted rat and monkey
hearts attenuated pathological remodelling and cardiac dysfunction. Authors
demonstrated that FMO2 inhibited SMAD2/3 activation through an enzymatic
activity-independent way: mechanistically, FMO2 was found to bind to cytochrome
P450 superfamily 2J3 (CYP2J3), thus preventing the interaction of the latter with
SMURF2, an E3 ubiquitin ligase, which selectively mediates the
proteasome-dependent degradation of phosphorylated SMAD2/3 in the nucleus [113].
Thus, FMO2 inhibits TGF
The Wnt/
Vainio et al. [118] has explored the effects of monoclonal antibody
(mAb) therapy against connective tissue growth factor (CTGF), whose increased
expression has been correlated with fibrotic diseases of virtually every human
organ [119], including the heart [120]. CTGF-mAb administration in a mouse model
of MI resulted in reduced cardiac fibrosis and hypertrophy, as well as increased
survival and left ventricular function. Heart tissue also showed increased
expression of genes associated with developmental and repair processes, and
reduction of inflammatory and fibrotic genes. Mechanistically, CTGF-mAb treatment
on human CFs in vitro attenuated the expression and production of
A new interesting potential target for the treatment of cardiac fibrosis has
been identified by Schafer and colleagues [121]. In this study, RNA-sequencing
analysis of human CFs stimulated in vitro with TGF-
In the search for new potential therapeutic targets against cardiac fibrosis, the field of epigenetic approaches opens the possibility of fine tuning the activation and the progression of the fibrotic process. In the wide scenario of epigenetic regulators and non-coding RNAs, we briefly present examples of potential targets recently identified for therapeutic intervention in this pathological condition, focusing on those with proven specific activity in CFs.
In recent years, numerous studies have suggested that epigenetic regulation,
including histone methylation, has extensive roles in fibrosis [124]. Wang
et al. [125] studied how the lysine demethylase 5B (KDM5B), a histone
H3K4me2/me3 demethylase, can affect the pathophysiological events in cardiac
fibrosis. Initially, they demonstrated KDM5B upregulation in CFs in response to
pathological stress. Secondly, it was proposed the molecular mechanism through
which KDM5B exerts its action: it demethylates the activated H3K4me2/3
modification bounding the activating transcription factor 3 (Atf3) promoter,
inhibiting its expression. In the literature [126] Atf3 is considered an
antifibrotic regulator of cardiac fibrosis, and its suppression is correlated
with the activation of TGF-
Another fundamental epigenetic modulation is histone acetylation/deacetylation,
that enhances or opposes the accessibility of transcriptional factors,
respectively. It is known that small molecules acting as histone deacetylase
(HDAC) inhibitors play a role in the attenuation of cardiac remodelling and
fibrosis [127]. Nural-Guvener et al. [128] proposed a strategy through
selective class I-HDAC inhibition against CFs activation. In particular, they
treated cluster of differentiation 90-positive (CD90+) CFs in vitro with mocetinostat (an HDAC inhibitor),
showing reduction of collagen III and
In a previous study, it was demonstrated that a group of epigenetic reader
molecules called bromodomain and extra-terminal (BET) acetyl-lysine binding
proteins, play a crucial role in regulating pathological fibrosis [129]. In
particular, Stratton and Bagchi et al. [130] identified by
RNA-sequencing the bromodomain-containing protein 4 (BRD4) involvement in cardiac fibrosis promoting the activation
of quiescent CFs into Periostin (Postn)-positive cells. The authors validated the
pro-fibrotic role of BRD4 using the BET bromodomains inhibitor JQ1. Specifically,
cultured primary adult rat ventricular fibroblasts stimulated with TGF-
The pivotal regulatory role of non-coding RNAs (ncRNAs) in every biological process is well established and consolidated, and so it is for the process of cardiac fibrosis. In the last years, many ncRNAs, mainly microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and more recently also circular RNAs (circRNAs), have been discovered as fine tuners of cardiac fibrosis, paving the way to their therapeutic targeting. Indeed RNA-targeting therapeutics have a high potential for the treatment of several diseases.
LncRNAs contribute to cardiac diseases, including fibrosis. One of the first
lncRNAs reported to be dysregulated during chronic cardiac remodelling is the
Maternally Expressed Gene 3 (Meg3), which was found specifically expressed in CFs
and downregulated during late cardiac remodelling in mouse hearts after TAC
[131]. Meg3 was known to act as a transcriptional regulator of gene expression,
recruiting and guiding chromatin remodelling complexes to target sites [132, 133].
In 2017, it was identified as a regulator of the expression of the
fibrosis-related gene matrix metalloproteinase-2 (MMP2) in CFs through the
recruitment of p53, this latter induced by TGF-
In 2019 Hao et al. [134] identified the lncRNA AK137033, named Safe
(Sfrp2 antisense as fibrosis enhancer), specifically enriched in the nuclei of
CFs. Its expression resulted up-regulated upon both MI induction in mouse hearts
and TGF-
The Nuclear Enriched Abundant Transcript 1 (NEAT1) is a lncRNA involved in cancer and some fibrosis-related diseases. Its expression was found elevated in patients with heart failure. Neat1 acts as a pro-fibrotic lncRNA, by inducing Smad7 promoter methylation through recruitment of enhancer of zeste homologue 2 (EZH2). Silencing of NEAT11 significantly alleviated the progression of cardiac fibrosis and dysfunction in a mouse model of heart failure, while overexpression of Neat1 had the opposite effect. This suggests that NEAT11 could be a good candidate for targeted antisense therapy [135].
The scaffold attachment factor B interacting lncRNA (SAIL) was shown to be reduced in cardiac fibrotic tissue and activated CFs. In vitro overexpression of SAIL decreased proliferation and collagen production of CFs, and could alleviate cardiac fibrosis in a mouse model of cardiac fibrosis due to MI. SAIL blocks the access of the scaffold attachment factor B (SAFB) to RNA pol II and reduces the transcription of fibrosis-related genes. The human conserved fragment of SAIL (hSAIL) could also suppress the proliferation and collagen production of human CFs [136].
The lncRNA taurine upregulation gene 1 (TUG1) was found to be overexpressed after MI, and this correlated with reduced levels of miiR-133b in rat models of myocardial fibrosis. Also, angiotensin-treated cardiac myofibroblasts revealed increased expression of TUG1. In addition, TUG1 knockdown prevented myofibroblast activation, while its overexpression increased cell proliferation and collagen production. Authors proved TUG1 could act as a sponge against miR133b in CFs, thus promoting the expression of CTGF, a target of miR133b. On the basis of this evidence, the axis TUG1/miR133b/CTGF could be a new potential target for the treatment of cardiac fibrosis [137].
Several miRNAs play a central role in the regulation of cardiac fibrosis. MiR-21 is highly expressed in cardiac cells, preferentially in non-myocytes, and it is known to be up-regulated and involved in the development of cardiac fibrosis, mainly promoting the activation of the ERK-MAPK pathway in CFs [138]. MiR-21 inhibition has been already proven effective for the prevention of cardiac fibrosis and dysfunction in small animal models of heart injury [139, 140]. Recently, Hinkel et al. [141] have tested the feasibility and anti-fibrotic efficacy of miR-21 silencing also in large animal models of heart failure, laying the foundation for potential applications in humans. In detail, the local, intracoronary delivery of locked nucleic acid (LNA)-antimiR-21 in pig hearts after I/R injury led to a reduction of adverse myocardial remodelling, associated with decreased fibroblast proliferation and macrophage infiltration, as well as improvement of cardiac function [141].
A critical miRNA in cardiac fibrosis is miR-125b, which has been proven to be
necessary and sufficient for fibroblast-to-myofibroblast transition. MiR-125b
expression was found to be enhanced in failing human hearts and in various animal
models of cardiac fibrosis, as well as in CFs stimulated with TGF-
Another regulator of fibroblast-to-myofibroblast transition was identified in
miR-146b-5p, which is overexpressed in the blood of patients with myocardial
ischemia, and in the heart of mice with MI. Interestingly, miR-146b-5p is
up-regulated in CFs, endothelial cells, and macrophages by hypoxia-induced NF-
In contrast, a miRNA with anti-fibrotic properties with a potential therapeutic
application is miR-101. This miRNA was found to be down-regulated in the
peri-infarct area of rats undergoing coronary artery ligation, as well as in CFs
stimulated with angiotensin II. On the contrary, in vitro overexpression
of miR-101 suppressed fibroblast proliferation and ECM remodelling protein
production, mainly targeting c-Fos, a downstream factor of the TGF-
Circular RNAs have been involved in the progression of both cancer and cardiovascular diseases. Given their peculiar secondary structure, with a covalent link bridging each end of the linear RNA which is converted into its circular form, circular RNAs are the most stable RNAs. This feature makes them optimal targets in RNA therapeutics.
In a rat model of post-ischemic myocardial fibrosis, circHNRNPH1 expression was
found increased specifically in CFs. The expression of circHNRNPH1 could restrict
the differentiation of CFs into myofibroblasts by de-repressing TGF-
Similarly, circNFIB (which is conserved un humans) was found to be decreased in
mouse cardiac samples post-MI, and its overexpression could attenuate CF
proliferation [146]. Its action is dependent on TGF-
The circHIPK3 expression markedly increased in CFs and heart tissues in a
mechanism mediated by TGF-
A recent study on patients with cardiac hypertrophy showed a significant decrease in yet another circRNA, that is the circYap isoform [148]. In a mouse model of hypertrophy, the expression of circYap was negatively correlated with the expression of fibrosis genes. The restoration of circYap via ectopic expression improved heart function and cardiac fibrosis in a pressure-overload mouse model of hypertrophy. CircYap expression could directly affect both survival and cell morphology of fibroblasts in vitro [148].
Finally, the circ_LAS1L was found to be down-regulated in acute MI patients and CFs in another clinical study. The circ_LAS1L functional role is to sponge miR-125b, thereby de-repressing this miRNA target genes involved in activation, proliferation, and migration of CFs [149].
The continuous advancements in medical and biotechnological research have led to the development of innovative approaches based on biological and biotechnological therapies designed to counteract cardiac fibrosis. They include the use of biological non-cellular products, in situ reprogramming strategies, and immunotherapy approaches.
Several adult cell populations have been studied as therapeutic and/or regenerative products for the injured heart, including selected resident cardiac mesenchymal populations. Cardiosphere-Derived Cells (CDCs) have been shown to have beneficial effects, mainly through a protective paracrine action [150, 151], mediated in part by the release of extracellular membrane vesicles (EMVs), such as exosomes [152]. Human and rat dermal fibroblasts were treated with cardiosphere-derived EMVs (CSp-EMV) and then injected into rat hearts subjected to left anterior descending artery ligation [153]. In vitro treated fibroblasts displayed a less fibrotic phenotype, with significantly reduced levels of fibroblast specific protein 1 (FSP1) and discoidin domain receptor 2 (DDR2), and showed increased secretion of pro-angiogenic factors, such as stromal-cell derived factor 1 (SDF-1) and vascular endothelial growth factor (VEGF). Moreover, they exerted pro-angiogenic and anti-apoptotic effects in vitro. The in vivo administration of exosome-primed fibroblasts attenuated adverse remodelling, reduced the infarct area, and enhanced cardiac function. Authors speculated this therapeutic activity could be due to the specific profile of miRNAs carried by the EMV, since a similar effect on cardiomyocytes was previously proven for exosomes from CDCs [152]. This is an interesting example of a biological product directly influencing the functional phenotype of fibroblasts, with a relevant therapeutic outcome.
Recently, also the curative effect of exosomes derived from cortical bone stem
cells (CBSCs) has been tested in murine models of I/R injury. CBSCs were already
proven to exert beneficial effects, in terms of decreased scar size and improved
cardiac function, when injected in mouse and pig models of MI [154]. The same
effects were recapitulated in I/R mouse hearts where CBSC-derived exosomes
(CBSC-dEXOs) were administered, highlighting their key role in mediating
cardio-protection [155]. Mechanistically, in vitro treatment of CFs with
CBSC-dEXOs decreased the expression of pro-fibrotic genes, such as collagen I,
collagen III, and MMP2, even to a greater extent than whole CBSC-conditioned
medium from which the exosomes were isolated. In addition, CBSC-dEXOs were also
proven to significantly reduce CF activation induced by TGF-
Cardiac regeneration aims at restoring functional cells that have been lost in the damaged heart. A therapeutic option to recover lost cardiomyocytes is the direct reprogramming of fibroblasts into cardiomyocytes through genetic tools or small molecules. This strategy could be also viewed as a way of simultaneously replacing potentially fibrotic cells with new parenchymal cells, thus representing also an anti-fibrotic approach. Several strategies have been proposed for the delivery of direct reprogramming factors in recent years, and we briefly summarize a selection based on artificial vectors/carriers.
Mónica P. A. Ferreira et al. [156], developed a functionalized
spermine-modified dextran-based (AcDXSp) nanoparticle that allows the release of
two molecules—CHIR99021 and SB431542—capable of inducing fibroblast
reprogramming. Further functionalization of the nanoparticles with atrial natriuretic peptide (ANP) allowed
tropism towards cardiac cells [157]. Specifically, AcDXSP nanoparticles were
incubated with non-myocytes, and the effect of the release of the active
molecules was confirmed. In fact, the administration of CHIR99021 significantly
enhanced
Poly (lactic-co-glycolic acid) (PLGA)-polyethyleneimine (PEI) nanocarriers
encapsulated with microRNAs represents a low-toxicity system to induce the direct
reprogramming of cardiac human fibroblasts into cardiomyocyte-like cells. These
nanoparticles were loaded with miR-1 and miR-133a, that initiate and drive muscle
development and differentiation [159, 160]. Direct fibroblasts reprogramming was
evaluated by the expression of markers such as cardiac troponins and
Qiaozi Wang et al. [162] have developed another non-viral system to reprogram CFs in situ, by coating neutrophil-mimicking membranes on mesoporous silicon nanoparticles (MSNs) with the FH peptide, and loading them with miR-1, -133, -208, and -499 (miR-Combo). Homing of the nanoparticle to the injured heart was achieved by the natural inflammation-homing capacity brought by neutrophil membranes, while the FH peptide’s high affinity to tenascin-C (TN-C), specifically expressed by CFs in the injured hearts, granted cell tropism. This study reported the intravenous injection of the nanoparticles loaded with the miR-combo in a mouse model of myocardial I/R, and evaluated the efficient in situ reprogramming of CFs into cardiomyocyte-like cells. Despite low rates of reprogramming, this strategy of delivering showed nonetheless an improvement in cardiac function, and attenuation of fibrosis in vivo [162].
Recently an innovative strategy based on chimeric antigen receptor T-cells (CAR-T) was proposed as an additional method to reduce cardiac fibrosis. The gene signature of CFs derived from healthy versus diseased human hearts were analysed, and fibroblast activation protein (FAP) was found as a specific marker of activated fibroblasts which is not expressed in cardiomyocytes. The study suggested that the adoptive transfer of antigen specific CD8+ T cells targeting the FAP protein can lead to partial removal of activated CFs in vivo, with significant reduction of cardiac fibrosis and restoration of function after injury in mice [163]. Joel G Rurik et al. [164] developed a therapeutic approach to generate transient CAR-T cells directly in vivo by delivering modified messenger RNA (mRNA) in T cell–targeted lipid nanoparticles (LNPs). Specifically, they injected CD5-targeted LNPs in a mouse model of heart failure, favouring the delivery of modified mRNA encoding for CAR in T lymphocytes, thus producing CAR-T cells in vivo. The transient CAR-T cells generated were specific for the FAP protein, so their action reduced the abundance of activated fibroblasts and consequently fibrosis, also restoring cardiac function after injury [164].
The quest for novel therapeutic strategies against cardiac fibrosis has taken multiple promising directions, that have provided encouraging preclinical data in recent years. A key obstacle in finding one good therapeutic option for treatment of myocardial fibrosis is represented by the complexity of the signalling network involved in the activation of repair mechanisms, which includes responses to surrounding stress and cell death signals, as well as the powerful crosstalk with the immune cells compartment. Therefore, any novel therapeutic approach will have to deal with, and possibly overcome, all the opposing signals in the microenvironment of the injured myocardium, making the quest quite complex. Moreover, different aetiologies and/or pathogenetic stimuli may be differentially sensitive to the same strategy, therefore future studies will have to define also the specifics of the population of potential target patients. Finally, given these many levels of complexity, future developments of therapies might work best with a synergistic approach, where multiple pathways are targeted by the same molecule or by combined treatments.
EF, CC, SM, FT performed bibliographic search and manuscript drafting. EF and CC prepared the figures. SM and FT prepared the table. VP, FP and IC participated conception and design, drafted the manuscript, and revised it critically for important intellectual content. All authors have read and agreed to the published version of the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
IC is supported by grants # RM12117A805ED2FD and # RG11916B85CDBF76 from Sapienza University of Rome, and by grant # RG1221816BC8E766 from the Italian Health Ministry. FP is supported by Grant # A0375-2020-36621 from Regione Lazio (POR-FESR 2014-2021). EF is supported by grant # AR12218167E8FA71 from Sapienza University of Rome.
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.