
 , Guangping Li 1,*
, Guangping Li 1,*1 Tianjin Key Laboratory of Ionic-Molecular Function of Cardiovascular Disease, Department of Cardiology, Tianjin Institute of Cardiology, The Second Hospital of Tianjin Medical University, 300211 Tianjin, China
2 Department of Health Sciences, School of Nursing and Health Studies, Hong Kong Metropolitan University, 518057 Hong Kong, China
†These authors contributed equally.
Abstract
Background: Lung cancer is one of the major cause of death globally.
Crizotinib is a first-line drug used in treating non-small-cell lung cancer
(NSCLC). However, the pathophysiological mechanisms underlying its cardiotoxicity
are unknown. This study investigated the mechanisms of crizotinib-induced
cardiotoxicity and explored whether this toxicity can be prevented by the
angiotensin receptor/neprilysin inhibitor sacubitril/valsartan. Methods:
Male C57BL/6 mice were randomly divided into three groups: control, crizotinib
(40 mg
Keywords
- cardiotoxicity
- crizotinb
- sacubitril/valsartan
- Myh7
Lung cancer is one of the major cause of deaths globally. Non-small cell lung cancer (NSCLC) contributes to 80–85% of all lung cancer cases [1, 2, 3]. Rearrangements in the genes encoding for anaplastic lymphoma kinase (ALK) and v-ros UR2 sarcoma virus oncogene homolog 1 (ROS1) are observed in 2–7% and 1–2% of NSCLC samples, respectively [4, 5]. Crizotinib, an adenosine-triphosphate (ATP)-competitive small molecule inhibitor, was the first oral ALK inhibitor approved by the Food and Drug Administration (FDA) in August 2011 for the treatment of NSCLC to inhibit the receptor tyrosine kinases ALK, ROS1, and mesenchymal-epithelial transition (MET) [5, 6, 7, 8, 9]. Crizotinib can also be used to treat multiple myeloma [10, 11, 12] and, although crizotinib is prone to drug resistance with repeated use, it remains a promising option for the treatment of NSCLCs [13]. For ALK-positive NSCLC, crizotinib is more effective and better tolerated than chemotherapy [14, 15]. In NSCLC with ROS1 rearrangement, crizotinib can be used as first-line treatment [5, 6, 7, 8]; however, cardiotoxicity caused by different anti-cancer drugs has long been recognized [11]. Previous studies showed that crizotinib induces various cardiotoxicities such as bradycardia, QT prolongation, ventricular rhythm, and ventricular fibrillation [16, 17, 18]. Among these side effects, the most common reports are that crizotinib prolongs the QT interval and reduces heart rate [19, 20, 21]. Thus, patients receiving crizotinib should receive close and regular monitoring of both the QT interval and heart rate [20]. Early identification of the cardiotoxicities associated with crizotinib is conducive to rational drug use; however, the specific mechanism(s) underlying crizotinib cardiotoxicity remain unclear.
To avoid, or reduce, cardiotoxicity associated with anti-cancer drugs, the administration of cardioprotective agents is critical. For example, prophylactic administration of a renin-angiotensin system (RAS) antagonist partially attenuates the cardiotoxic effects of doxorubicin in a chronic mouse model of chemotherapy-induced cardiac insufficiency [22]. Left ventricular ejection fraction (LVEF) was increased, and troponin I (TnI) was decreased, during a 6-month follow-up period of anthracycline treatment combined with carvedilol, suggesting a protective effect for carvedilol against myocardial injury [23]. The angiotensin receptor/neprilysin inhibitor sacubitril/valsartan is also used to treat heart failure and hypertension [24, 25, 26]. Previous studies using either animal models or human clinical trials showed that sacubitril/valsartan reversed cardiac remodeling, modulated heart failure biomarkers, reduced arrhythmias, improved renal function, improved the quality of life, and reduced mortality and/or the risk of hospitalization [27, 28, 29, 30]. Sacubitril/valsartan has also demonstrated utility in the treatment of cancer therapy-related cardiac dysfunction [31, 32] and with findings of improved cardiac function and cardiac-related symptoms [33]. In a study that combined sacubitril/valsartan and doxorubicin, sacubitril/valsartan was found to attenuate doxorubicin-induced apoptosis and endoplasmic reticulum stress in cultured H9C2 cardiomyocytes [34]. Similar findings were observed in a doxorubicin-induced rat cardiotoxicity model that examined biochemical markers, contractile function, endoplasmic reticulum stress, and attenuated doxorubicin-induced apoptosis in rat heart [35]. However, whether sacubitril/valsartan can reduce the cardiotoxicity induced by crizotinib, as well as the molecular nature of crizotinib induced cardiotoxicity remains unclear. Herein, we sought to investigate the effects of crizotinib on cardiotoxicity and determine whether sacubitril/valsartan can ameliorate crizotinib-induced cardiotoxicity.
This study was approved by the Laboratory Animal Ethical Committee of Chinese
Academy Medical Sciences Institute of Radiation Medicine. A total of 36 male
C57BL/6 mice were divided randomly into three groups: control (CON group),
crizotinib (CRI group) and crizotinib + sacubitril/valsartan group (CRI + SV
group). Mice in the crizotinib group were administered 40
mg
Conscious animals were pre-warmed in a warm-up chamber at 36 °C–37 °C for 15 mins and their systolic, diastolic and mean arterial blood pressure (SBP, DBP and MBP, respectively) were recorded by using tail sleeve plethysmography (BP98AL, Softron, Tokyo, Japan).
After weighing mice, chest hair was removed with a hair removal cream, followed by anesthesia with 2% isoflurane. Transthoracic echocardiography was performed using Imaging System (Vevo 2100, VisualSonics, Toronto, Canada). Data collected included left atrial diameter (LAD), left ventricular diameter at systolic and diastolic period (LVIDs and LVIDd, respectively), left ventricular anterior wall thickness at systolic and diastolic period (LVAWs and LVAWd, respectively), left ventricular posterior wall thickness at systolic and diastolic period (LVPWs and LVPWd, respectively), interventricular septum thickness at systolic and diastolic period (IVSs and IVSd, respectively), pulmonary artery acceleration time (PAT), left ventricular fractional shortening (FS), and left ventricular ejection fraction (EF).
Using mice anesthetized with 1.5% tribromoethanol (0.02 mL/g; WXBD3759V, Sigma, St. Louis, Missouri, USA), and supported by a tracheal intubation ventilator, mouse chests were surgically opened with full exposure to the heart. Following this, the pericardium was removed, epicardial conduction velocity (CV), absolute inhomogeneity and inhomogeneity index was recorded and analyzed using the Electrical Mapping System (EMS64-USB-1003, MappingLab, Oxford, UK) and EMapScope 4.0 (MappingLab, Oxford, UK), as detailed previously [36].
A programmed electrical stimulation protocol was performed using electrodes on
the epicardial surface of the right ventricle (RV) and left ventricle (LV). The
stimulation was performed at eight beats (120 ms, 8
RNA extraction, transcriptome sequencing, and data analysis was performed by OE Biotech Co., Ltd. (Shanghai, China). In brief, total myocardial tissue RNA from myocardial in control (n = 3) and crizotinib groups (n = 3) was isolated by the mirVana™ miRNA ISOlation Kit (AM1561, Ambion, Austin, TX, USA). Following this, synthesis, purification and adapter ligation of cDNA was carried out. DNA libraries were created using TruSeq Stranded mRNA LTSample Prep Kit (NR604-02, Illumina, San Diego, CA, USA). The quality of libraries was assessed using an Agilent 2100 Bioanalyzer (2100, Agilent, Santa Clara, CA, USA). DNA libraries were sequenced using an Illumina sequencing platform (Nova6000, Illumina, San Diego, CA, USA).
p value
Mouse ventricular tissue was perfused with 10% neutral buffered formalin for 72 h at room temperature. These tissues were then dehydrated with an ethanol at different concentrations, followed by xylene and finally paraffin embedding and storage at –20 °C overnight. Embedded tissue was cut into 4 µm thick sections and hematoxylin and eosin (HE) (20220211, Solarbio, Beijing, China) staining was conducted to observe whether there are any changes in the arrangement or size of the cardiomyocyte nuclei. Masson Tricolor Staining (20220214, Solarbio, Beijing, China) was used to observe whether myocardial tissue was fibrotic.
RNA extraction (0000458714, Promega, Beijing, China) followed by reverse
transcription of RNA into cDNA was conducted using a reverse transcription kit
(X0222, Tiangen, Beijing, China). Subsequently, RT-qPCR was conducted using SYBR
green (P31221, TransGen, Beijing, China) and a Quant Gene 9600 System (9600,
Bioer Technology, Hangzhou, China). The 2
| Gene name | Primer sequence | |
| Myh7 | forward | GACAGGAAGAACCTACTGCG |
| reverse | GAACTTGGACAGGTTGGTGT | |
| Ciart | forward | AGTGAAGAAGCTGCATACCG |
| reverse | CAGCTCCCGTAGTACCAAAG | |
| Ngp | forward | GAGGCCCTTCGACAACTAAG |
| reverse | TTCTGACTAGAAGGCGGAGT | |
| Lcn2 | forward | TGACAACTGAATGGGTGGTG |
| reverse | GATGCTCCTTGGTATGGTGG | |
| Ptgds | forward | CTCCTTCTGCCCAGTTTTCC |
| reverse | AATCCCAAGAGACCCAGGAG |
Myh7, myosin, heavy polypeptide 7, cardiac muscle, beta; Ciart, circadian associated repressor of transcription; Ngp, neutrophilic granule protein; Lcn2, lipocalin 2; Ptgds, prostaglandin D2 synthase.
Total tissue protein was isolated by RIPA buffer (01408/15322, Cwbio, Beijing,
China) and PMSF protease inhibitor (01392/06122, Cwbio, Beijing, China). Protein
concentrations were measured using a bicinchoninic acid (BCA) Protein Concentration Assay Kit. The
protein samples were separated on an 8% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel and subsequently
transferred to a polyvinylidene fluoride (PVDF) membrane. The PVDF membrane was
blocked with 5% milk, subsequent incubation with primary
The data analysis was carried out using Origin 6.0 (OriginLab, Northampton,
MA, USA) and SPSS 17.0 (SPSS Inc., Chicago, IL, USA) software.
The results are presented are expressed as mean
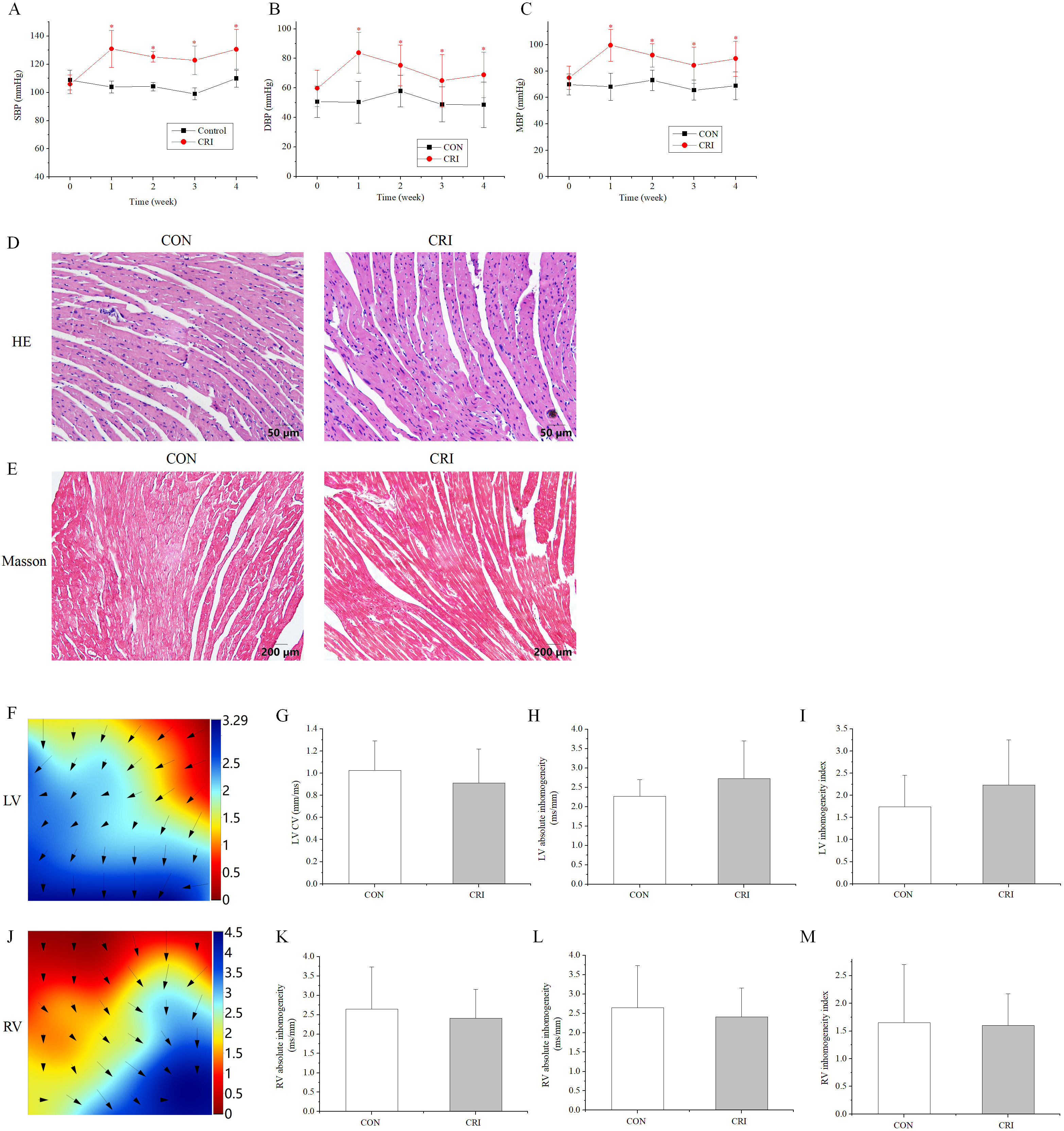
To study the cardiotoxicity caused by crizotinib, the blood pressure (BP) of mice after crizotinib administration was measured. The SBP, DBP and MBP were significantly higher following crizotinib treatment for 1 week (n = 10, SBP, p = 0.000; DBP, p = 0.000; MBP, p = 0.000), and remained significantly elevated at 4 weeks after treatment (n = 10, SBP, p = 0.000; DBP, p = 0.009; MBP, p = 0.001) when compared to the control group (Fig. 1A–C). HE and Masson staining was performed on ventricular muscle tissue obtained from mice 4 weeks after crizotinib use. No significant changes in myocardial tissue structure nor significant myocardial fibrosis was observed in control and crizotinib group mice (n = 5) (Fig. 1D,E). We also recorded the characteristics of epicardial electrical conduction of mice using mapping. Fig. 1F is a representative epicardial electrical mapping of LV. The calculated CV (n = 5, p = 0.553), absolute inhomogeneity (n = 5, p = 0.365), and inhomogeneity index (n = 5, p = 0.404) of LV were not significantly different between the control and crizotinib groups (Fig. 1G–I). A representative epicardial electrical mapping of RV is shown in Fig. 1J. The calculated CV (n = 5, p = 0.532), absolute inhomogeneity (n = 5, p = 0.702), and inhomogeneity index (n = 5, p = 0.926) of RV was also found to not change significantly in control and crizotinib groups (Fig. 1K–M).
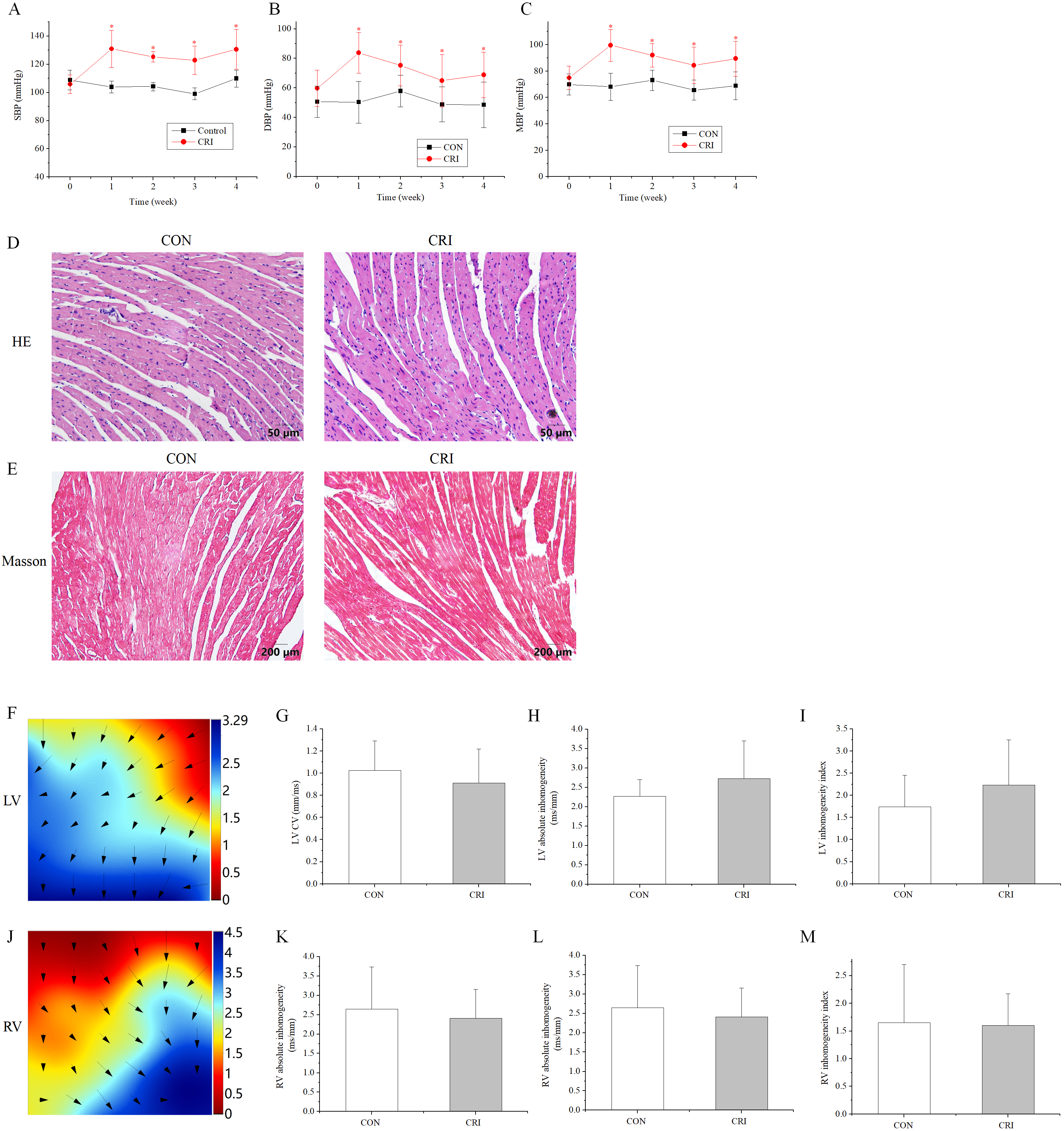 Fig. 1.
Fig. 1.Effect of crizotinib on BP, myocardial pathology, and electrical
conduction characteristics in control and crizotinib group mice. (A) Effects of
crizotinib on SBP. (B) Effects of crizotinib on DBP. (C) Effects of crizotinib on
MBP. (D) Typical sample of HE staining in control and crizotinib group. (E)
Typical sample of Masson staining in control and crizotinib group. (F)
Representative epicardial electrical mapping of recorded LV. (G) CV of LV. (H)
Absolute inhomogeneity of LV. (I) Inhomogeneity index of LV. (J) Representative
epicardial electrical mapping recording of RV. (K) CV of RV. (L) Absolute
inhomogeneity of RV. (M) Inhomogeneity index of RV. *p
To screen for alterations in gene expression following crizotinib treatment of
myocardial tissue, we performed transcriptomic analysis on control and crizotinib
group mice. Three mouse myocardial tissue samples in each group were analyzed.
p value
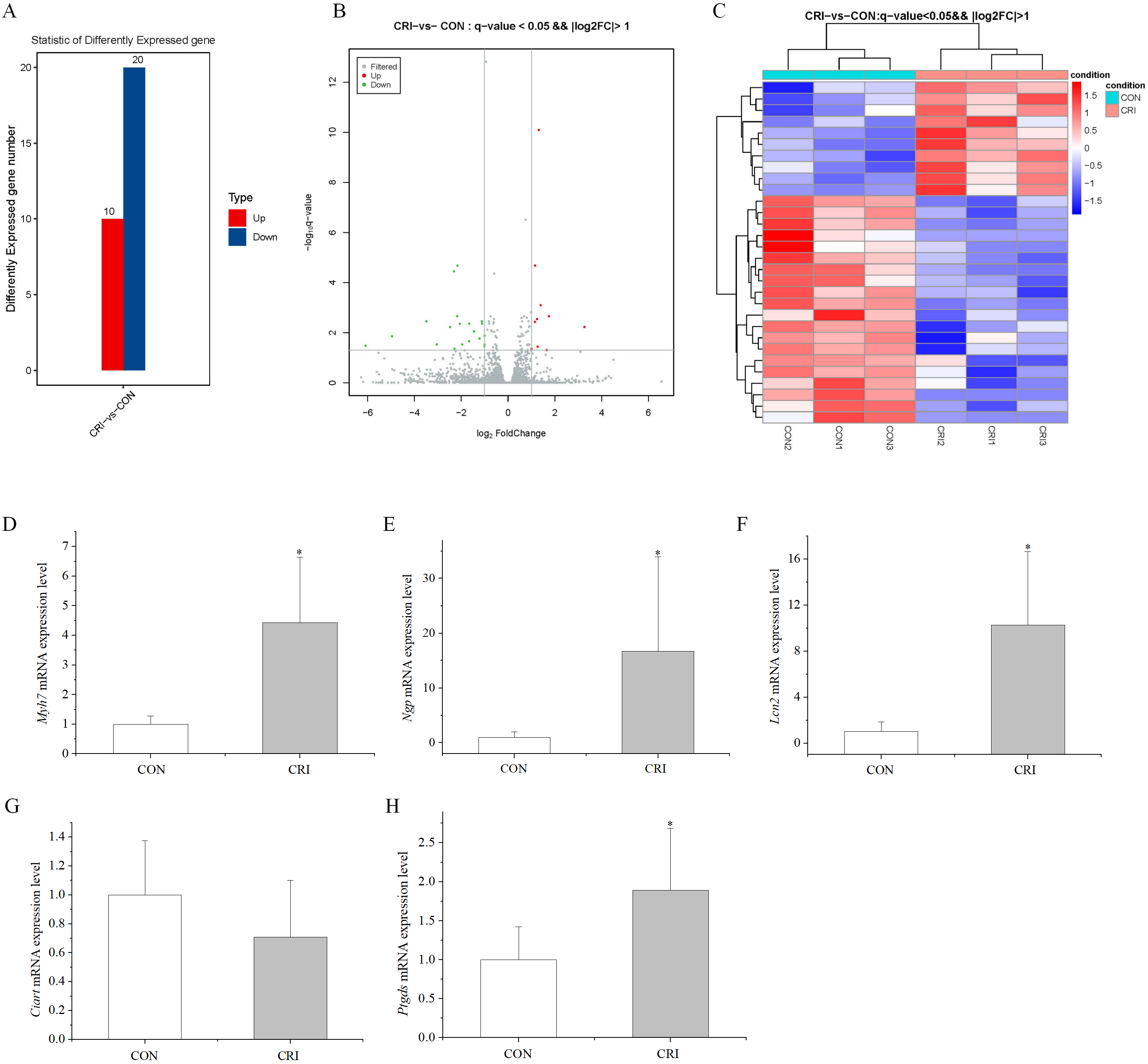
Among the DEGs, we were most interested in highly expressed genes after crizotinib exposure. This led us to select the top five differentially up-regulated genes for validation using RT-PCR (primer sequences given in Table 1). These genes included Myh7 (myosin, heavy polypeptide 7, cardiac muscle, beta), Ciart (circadian associated repressor of transcription), Ngp (neutrophilic granule protein), Lcn2 (lipocalin 2), and Ptgds (prostaglandin D2 synthase). RT-PCR results showed that 4 of these 5 genes displayed significantly increased expression in crizotinib mice (n = 5, Myh7, p = 0.007; Ngp, p = 0.015; Lcn2, p = 0.011; Ciart, p = 0.245; Ptgds, p = 0.016), which was in good agreement with the results of transcriptomic analysis except for Ciart (Fig. 2D–H).
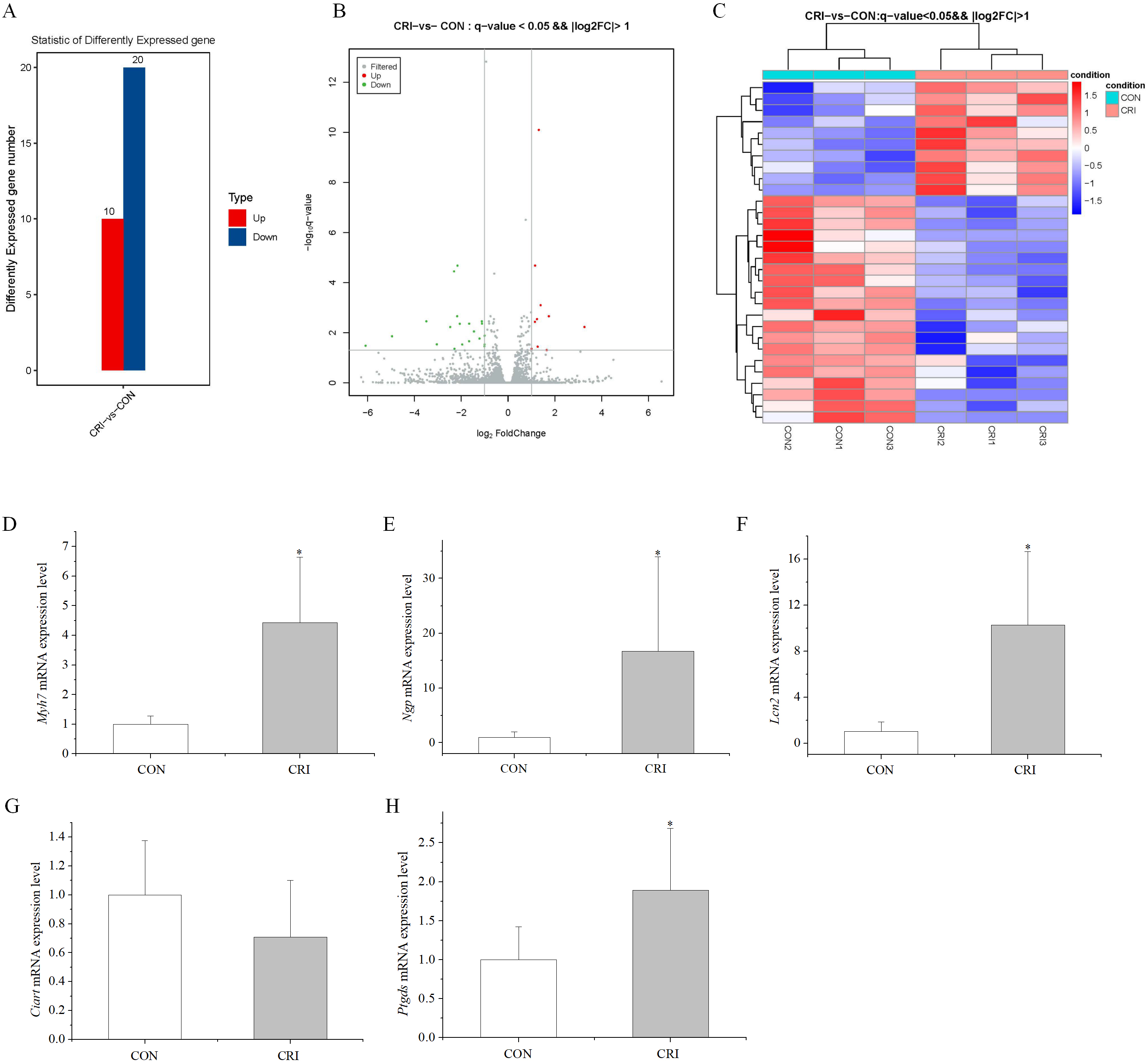 Fig. 2.
Fig. 2.Characteristics of DEGs. (A) 30 DEGs
identified in the crizotinib group, including 10 up-regulated and 20
down-regulated genes (the ordinate is differently expressed up-regulated and
down-regulated gene number). (B) Volcano map of DEGs (the abscissa is log
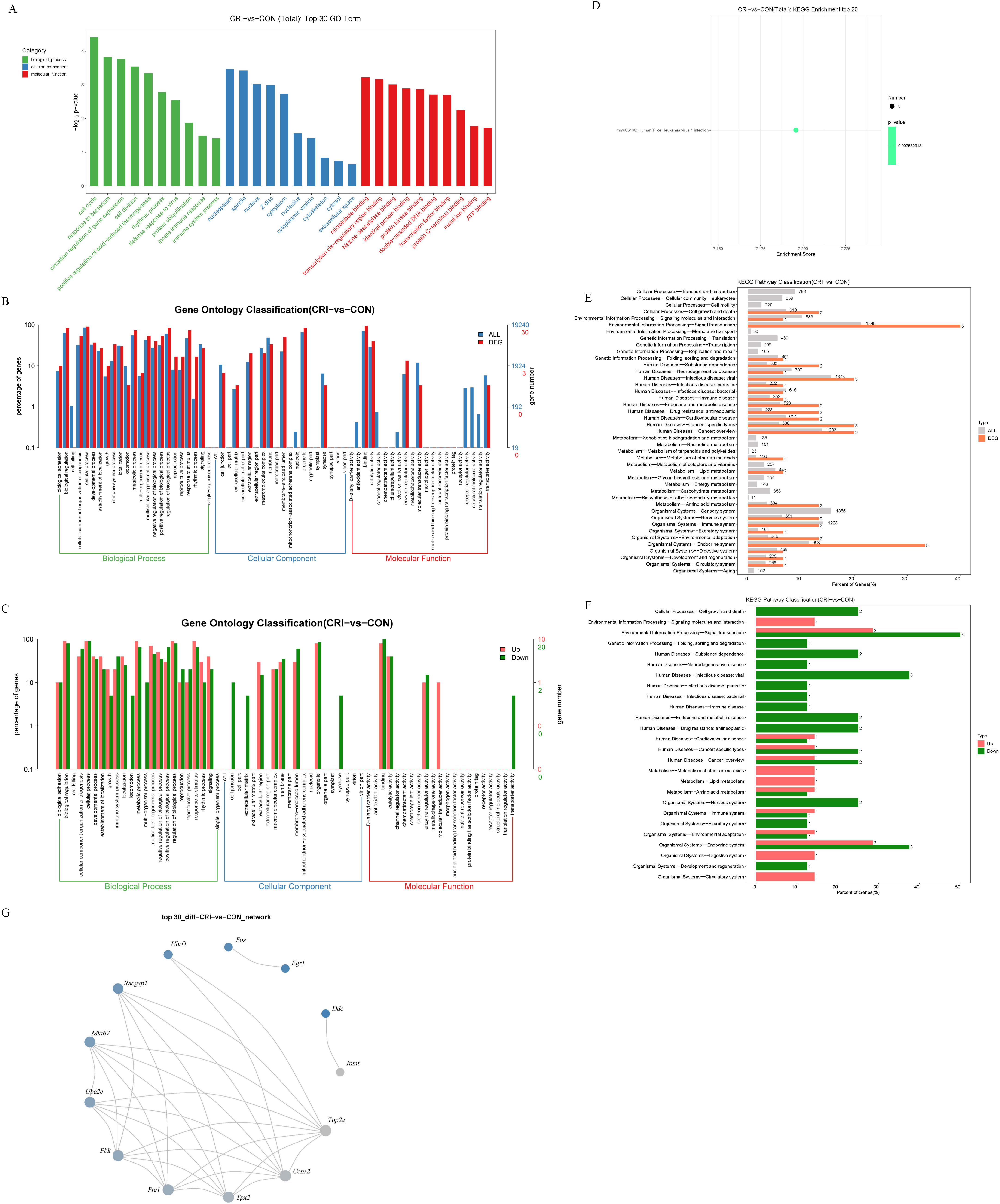
Following identification of DEGs, we next analyzed these genes using GO/KEGG to understand their functions. These genes were grouped into categories according to their characteristics in “biological process”, “cellular component”, and “molecular function”. The top 3 GO terms for “biological process” were “cell cycle”, “response to bacterium”, and “circadian regulation of gene expression”. The top 3 GO terms for “cellular component” were “nucleoplasm”, “spindle”, and “nucleus”. The top 3 GO terms for “molecular function” were “microtubule binding”, “transcription cis-regulatory region binding”, and “histone deacetylase binding” (Fig. 3A). According to the assigned characteristics of “biological process”, “cell composition” and “molecular function” of these genes, their level 2 function was graded. The functional distribution of all DEGs at GO Level 2 is shown in Fig. 3B. The functional distributions of differentially up-regulated and down-regulated genes at GO Level 2 is shown in Fig. 3C.
KEGG analysis was performed on the identified DEGs to systematically analyze the their regulatory role. KEGG enrichment of the top 20 identified genes is shown in Fig. 3D, the roles only include the term “human T-cell leukemia virus 1 infection”. Further, the distributions of all genes and DEGs at KEGG Level 2 is displayed in Fig. 3E. The distributions of up and down-regulated DEGs at KEGG Level 2 are shown in Fig. 3F. Finally, we investigated the interaction relationship between DEGs using the STRING database (Fig. 3G). This analysis of gene interactions showed that one gene interacts directly or indirectly with another or more other genes.
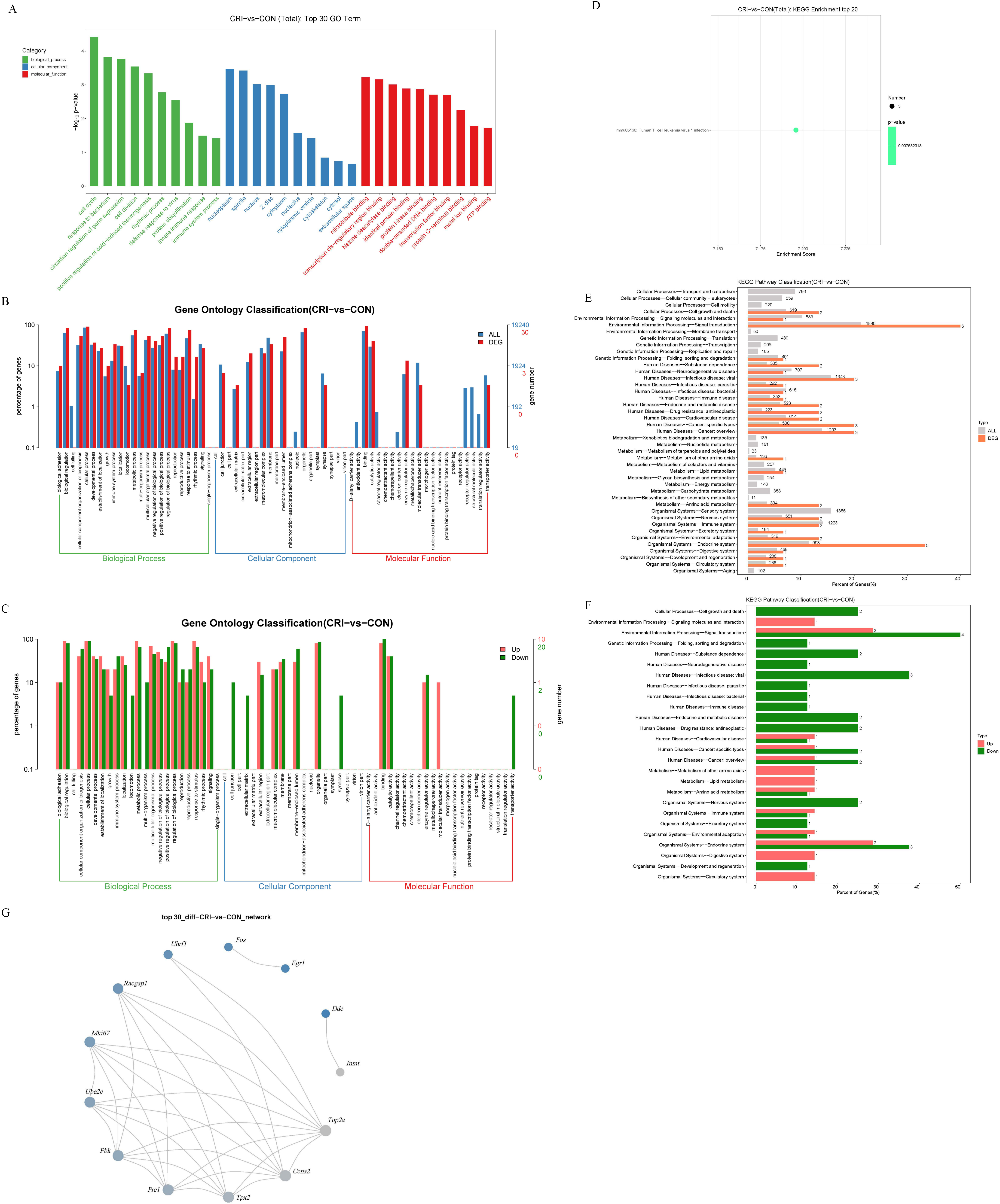 Fig. 3.
Fig. 3.GO and KEGG analysis of DEGs. (A) Top 30 GO terms. The abscissa
is the GO term, the ordinate is –log
Among the DEGs, we paid close attention to those related to human cardiovascular diseases. We screened 7 items in KEGG enrichment by classification_level1 ‘human disease’ or ‘organismal Systems’ and classification_level2 ‘cardiovascular diseases’ or ‘circulatory system’ (Table 2). This analysis indicated that Myh7 is not only highly expressed, but also involved in multiple processes in KEGG enrichment. Myh7 is closely related to a variety of cardiomyopathies, myocardial contraction, and adrenergic signaling in cardiomyocytes. Therefore, Myh7 may be a potential gene target associated with crizotinib-induced cardiotoxicity.
| ID | Term | Classification_level1 | Classification_level2 | p values | GeneID |
| mmu05416 | Viral myocarditis | Human diseases | Cardiovascular disease | 0.132 | Myh7 |
| mmu04260 | Cardiac muscle contraction | Organismal systems | Circulatory system | 0.135 | Myh7 |
| mmu05410 | Hypertrophic cardiomyopathy | Human diseases | Cardiovascular disease | 0.147 | Myh7 |
| mmu05414 | Dilated cardiomyopathy | Human diseases | Cardiovascular disease | 0.152 | Myh7 |
| mmu04261 | Adrenergic signaling in cardiomyocytes | Organismal systems | Circulatory system | 0.234 | Myh7 |
| mmu05418 | Fluid shear stress and atherosclerosis | Human diseases | Cardiovascular disease | 0.227 | Fos |
| mmu05417 | Lipid and atherosclerosis | Human diseases | Cardiovascular disease | 0.316 | Fos |
KEGG, Kyoto Encyclopedia of Genes and Genomes; Myh7, myosin, heavy polypeptide 7, cardiac muscle, beta; Fos, FBJ osteosarcoma oncogene.
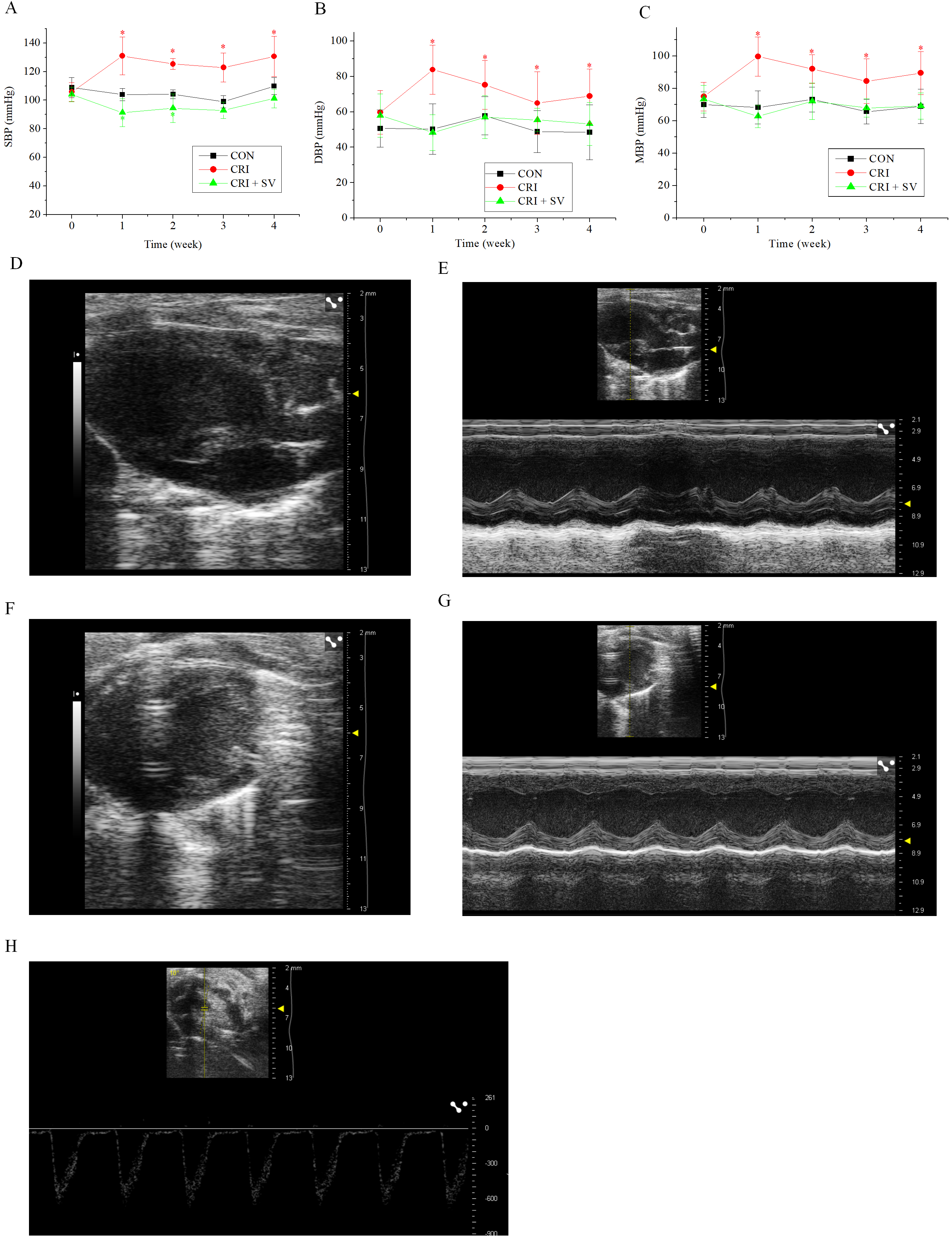
In view of the above results, crizotinib cardiotoxicity appears to be principally manifested as an increase in BP. Sacubitril/valsartan is a commonly used drug for BP reduction, thus we added an additional animal group to our study composed of crizotinib combined with sacubitril/valsartan, this was termed the crizotinib + sacubitril/valsartan group. The SBP, DBP and MBP line charts of the three experimental groups are shown in Fig. 4A–C. It can be seen that that the rise of SBP, DBP and MBP was caused by crizotinib at different timepoints (n = 10). However, data gathered indicate that sacubitril/valsartan given in combination with crizotinib can reduce elevated BP at 4 weeks when compared with the control group, SBP, p = 0.054; DBP, p = 0.473; MBP, p = 0.967.
To observe the effect of the crizotinib + sacubitril/valsartan combination on cardiac function we recorded the echocardiography of control, crizotinib, and crizotinib + sacubitril/valsartan groups. Fig. 4D–H shows typical images of the parasternal LV long-axis view. Also shown are B-type and M-type echocardiograms of long and short-axis views, and doppler pulse wave of pulmonary valve flow. Echocardiographic parameters of these three groups are shown in Table 3. The results indicated that there was no significant difference in IVSd, LVPWs, LVPWd, LVIDs, LVIDd, LVAWs, LVAWd, FS, EF or LAD among the three groups (n = 10). However, we found that the PAT of crizotinib group increased and the IVSs decreased when compared with the control group (n = 10). However, these two values returned to the level of the control group after crizotinib was combined with sacubitril/valsartan (n = 10).
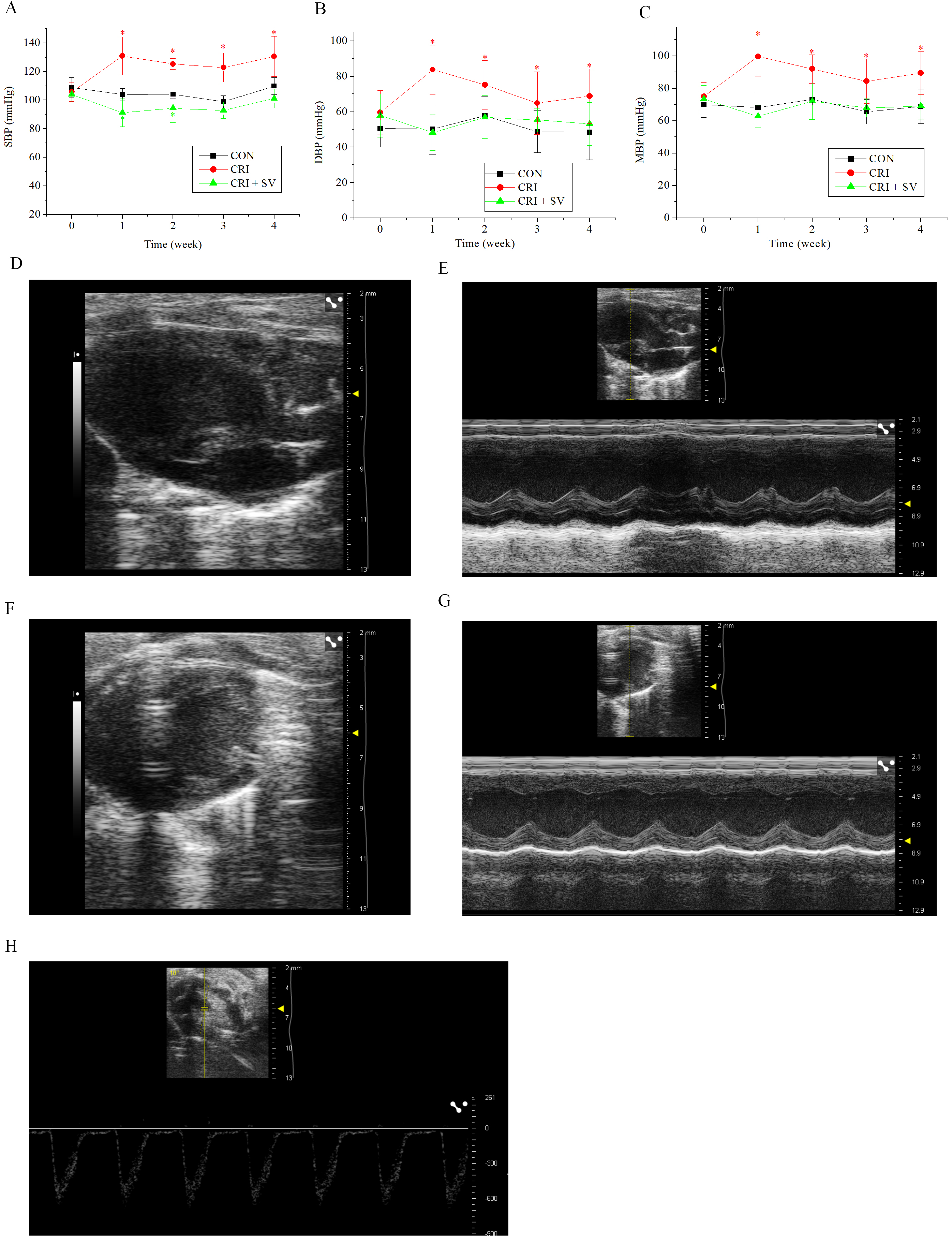 Fig. 4.
Fig. 4.Effect of crizotinib and sacubitril/valsartan on BP and cardiac
function in control, crizotinib, and crizotinib + sacubitril/valsartan mouse
groups. (A) Effects of crizotinib and sacubitril/valsartan on SBP. (B) Effects
of crizotinib and sacubitril/valsartan on DBP. (C) Effects of crizotinib and
sacubitril/valsartan on MBP. (D,E) B- and M-type echocardiogram long-axis
view of the parasternal LV. (F,G) B- and M-type echocardiogram short-axis
view. (H) Doppler pulse wave of pulmonary valve flow. *p
| CON (n = 10) | CRI (n = 10) | CRI + SV (n = 10) | p values | |
| PAT (ms) | 15.97 |
20.39 |
15.53 |
0.000 |
| IVS;s (mm) | 1.37 |
1.09 |
1.32 |
0.045 |
| IVS;d (mm) | 0.83 |
0.72 |
0.78 |
0.198 |
| LVPW;s (mm) | 1.41 |
1.38 |
1.42 |
0.933 |
| LVPW;d (mm) | 0.96 |
0.92 |
0.94 |
0.894 |
| LVID;s (mm) | 1.94 |
2.04 |
2.03 |
0.847 |
| LVID;d (mm) | 3.30 |
3.26 |
3.30 |
0.910 |
| LVAW;s (mm) | 1.35 |
1.33 |
1.29 |
0.793 |
| LVAW;d (mm) | 0.80 |
0.90 |
0.77 |
0.113 |
| FS (%) | 41.55 |
37.72 |
39.00 |
0.642 |
| EF (%) | 71.53 |
68.47 |
70.25 |
0.806 |
| LAD (mm) | 2.34 |
2.39 |
2.38 |
0.954 |
*p
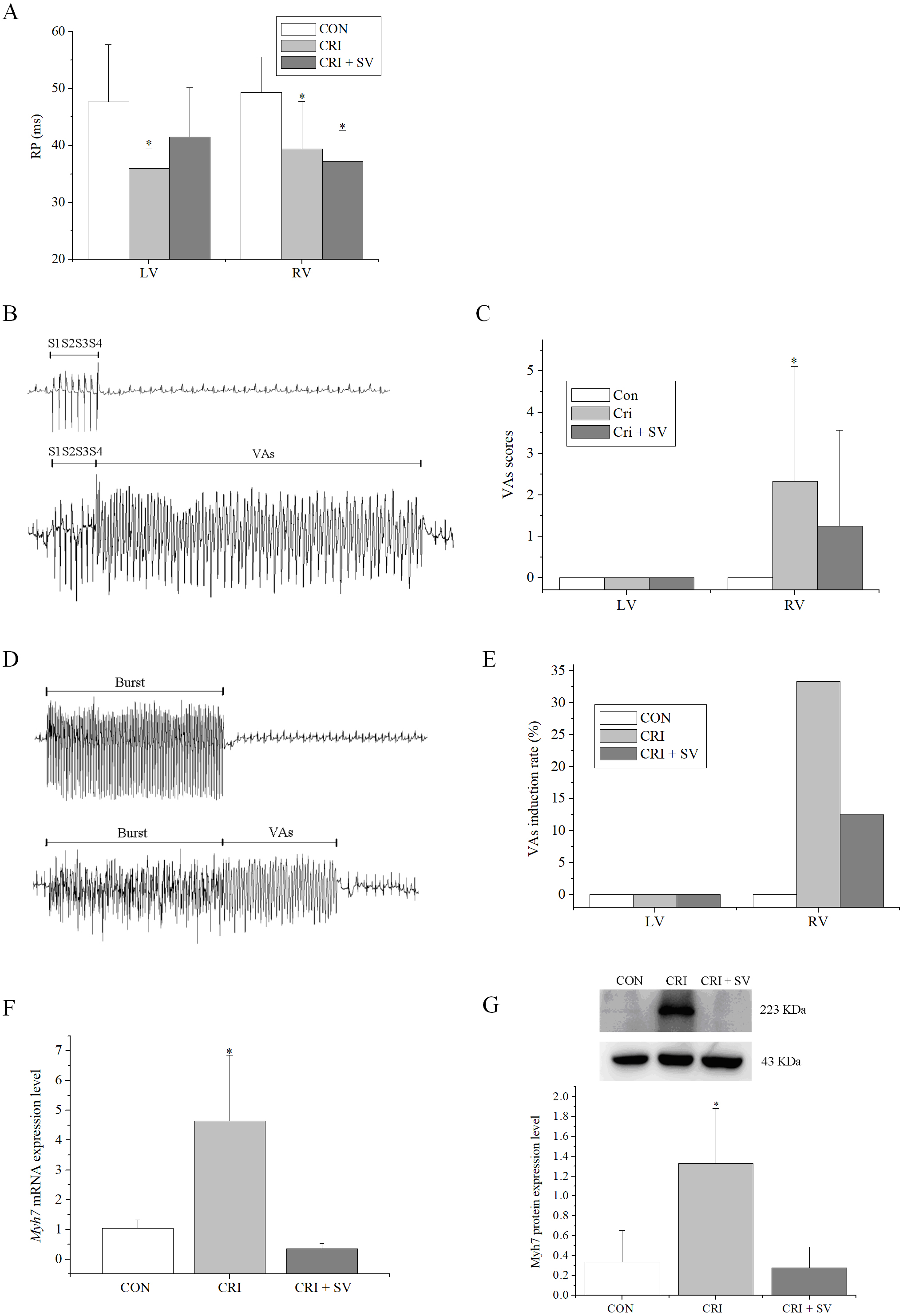
The effects of combination crizotinib and sacubitril/valsartan on cardiac electrophysiology in vivo were explored. Analysis of ECGs showed that the crizotinib group displayed faster heart rates, shorter RR intervals, and longer QTc compared to the control group (Table 4). Moreover, these abnormalities were restored in the crizotinib + sacubitril/valsartan group. A programmed electrical stimulation protocol was performed by stimulating the epicardial surface of the LV and RV. The RPs of LV and RV in the control (n = 7), crizotinib (n = 9), and crizotinib + sacubitril/valsartan (n = 8) groups are shown in Fig. 5A. A typical example of VAs occurring after 8 S1 stimulation followed by one to three extra stimuli (S2, S3, and S4) is shown in Fig. 5B. The VAs scores of the three groups were calculated and are shown in Fig. 5C and a typical example of VAs induced by burst stimulation is shown in Fig. 5D. The effects of crizotinib and sacubitril/valsartan on VAs induction rate after burst stimulation in the three groups are given in Fig. 5E. These data showed that crizotinib reduced the RPs of the LV and RV (LV, p = 0.006; RV, p = 0.010), increased the VAs score (p = 0.045), and increased the induction rate in the RV. Most of these abnormalities were prevented in the crizotinib + sacubitril/valsartan group when compared with controls (LV RPs, p = 0.130; VAs score of RV, p = 0.280). The exception to this was the RV RPs (p = 0.003).
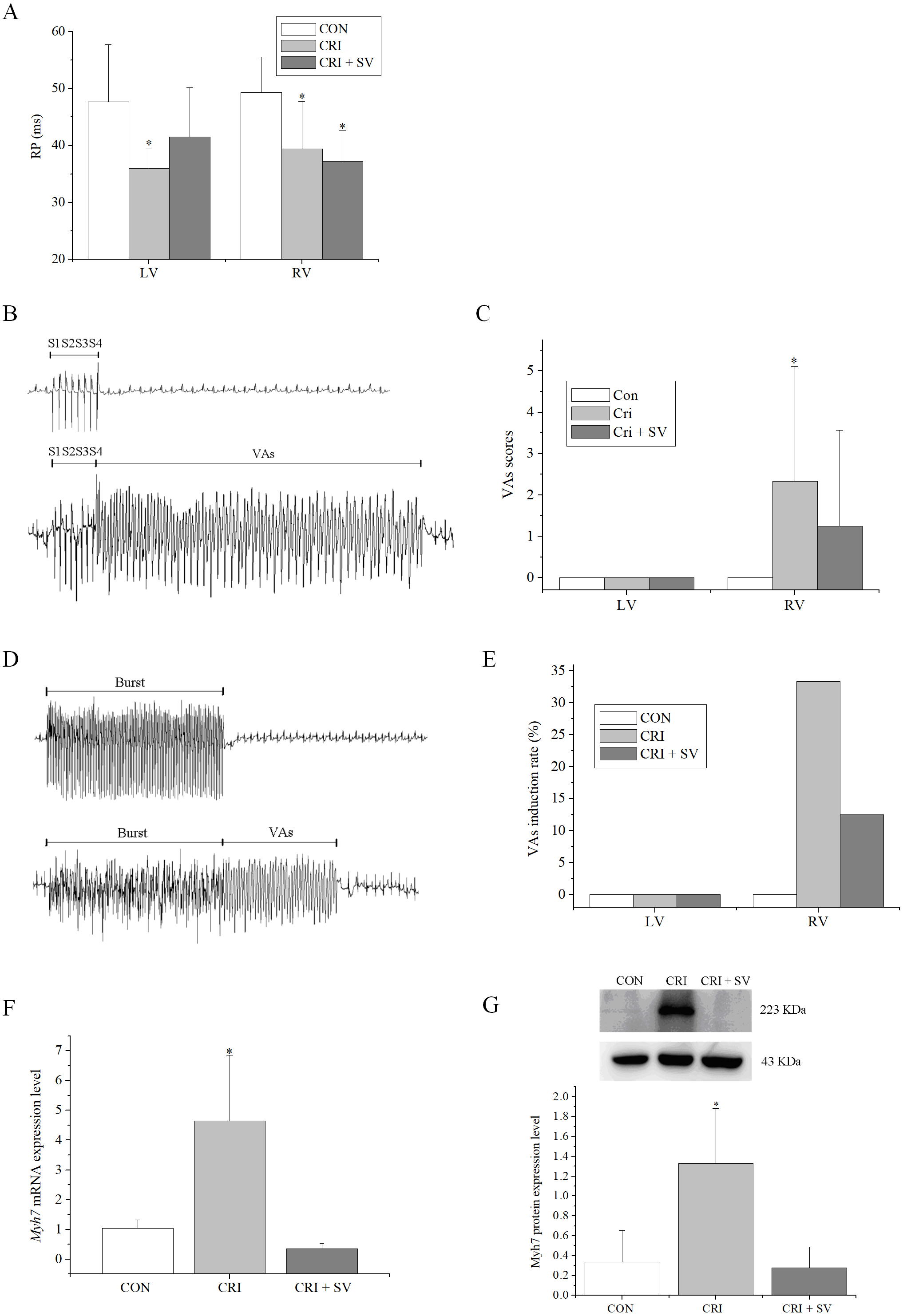 Fig. 5.
Fig. 5.Effects of crizotinib and sacubitril/valsartan on ventricular
electrophysiology in control, crizotinib, and crizotinib + sacubitril/valsartan
groups. (A) Effects of crizotinib and sacubitril/valsartan on ventricular RP.
(B) Typical examples of VAs occurring after 8 S1 stimulation followed by one to
three extra stimuli (S2, S3, and S4). (C) Effect of crizotinib and
sacubitril/valsartan on VAs score. (D) Typical examples of VAs occurring after
burst stimulation. (E) Effect of crizotinib and sacubitril/valsartan on VAs
induction rate after burst stimulation. (F) Effects of crizotinib and
sacubitril/valsartan on Myh7 mRNA expression. (G) Effects of crizotinib
and sacubitril/valsartan on Myh7 protein expression. *p
| CON (n = 10) | CRI (n = 15) | CRI + SV (n = 10) | p values | |
| BW (g) | 25.18 |
23.27 |
23.74 |
0.197 |
| HR (bpm) | 389.70 |
459.13 |
401.80 |
0.053 |
| RR interval (ms) | 160.20 |
133.73 |
153.20 |
0.046 |
| PR interval (ms) | 44.70 |
36.00 |
46.40 |
0.030 |
| QT interval (ms) | 74.90 |
81.73 |
64.40 |
0.014 |
| QTc (s) | 0.19 |
0.22 |
0.16 |
0.000 |
*p
Finally, changes in the Myh7 expression levels by crizotinib with or without sacubitril/valsartan were determined by western blot and RT-PCR (Fig. 5F,G). Crizotinib increased both the mRNA and protein abundance of Myh7 in the myocardium (n = 5, mRNA, p = 0.001; protein, p = 0.000). This effect was blunted by the use of crizotinib combined with sacubitril/valsartan when compared to controls (n = 5, mRNA: p = 0.414; protein: p = 0.803).
In this study we reported the cardiotoxic side effects associated with crizotinib exposure, including increased BP and prolonged QTc intervals. These were associated with increased right VAs scores and induction rates, and increased myocardial expression of Myh7 which is the most frequently mutated gene in hypertrophic cardiomyopathy. Most of these abnormalities were limited by co-treatment with sacubitril/valsartan.
Lung cancer is one of the major contributor to deaths globally, with NSCLC accounting for a large proportion of the tumor burden [1, 2, 3]. Crizotinib is approved for the treatment of NSCLC cases in which rearrangements in the genes encoding for ALK, ROS1 and MET are found [5, 7, 8, 9, 38]. Approximately 2–7% and 1–2% of NSCLC samples show rearrangements in ALK and ROS1, respectively [5]. For ALK inhibition in NSCLC, crizotinib is more effective and better tolerated than chemotherapy [39, 40]. However, patients treated with crizotinib develop drug resistance, requiring the use of second-generation ALK inhibitors to overcome crizotinib resistance. ROS1 rearrangement defines a second molecular subgroup of NSCLC for which crizotinib is highly active [7], and crizotinib displays marked antitumor activity in patients with advanced NSCLC with ROS1 rearrangement. In NSCLC with ROS1 rearrangement, crizotinib can be used as first-line treatment [5, 6, 7, 8]. Currently, only crizotinib is used for Anaplastic Large Cell Lymphoma (ALCL), belonging to the first generation of this drug class [4]. In crizotinib phase I studies, 94 percent of patients displayed some degree of tumor shrinkage during the study. In a phase 3, open-label trial comparing the differences between crizotinib and chemotherapy, the median progression-free survival of crizotinib was significantly higher than that of chemotherapy [7]. Previous studies have confirmed that crizotinib is superior to pemetrexed cisplatin or carboplatin, and is associated with a reduction in the main symptoms associated with lung cancer including cough, pain, and dyspnea [6].
Cardiotoxicity caused by anti-cancer drugs, including hypertension, arrhythmias, QTc interval prolongation, and left ventricular systolic dysfunction have long been a focus of attention [11]. Previous studies have demonstrated crizotinib-related cardiotoxicities of QT prolongation, bradycardia, ventricular fibrillation, and ventricular tachycardia [16]. In NSCLC, crizotinib was found to cause adverse cardiovascular side effects such as bradycardia, QT interval prolongation, ventricular rhythm, ventricular fibrillation, and pericarditis [17]. Another study reported QT interval prolongation, mild motion wall abnormalities in the left anterior wall and chamber door, small amounts of pericardial effusion, and even transient ventricular tachycardia and ventricular fibrillation [41]. In our mouse study, crizotinib exposure led to side effects such as increased BP, prolonged QTc, and inducible ventricular arrhythmias. We also observed a significant prolongation in PAT in the crizotinib group, suggesting that increased pulmonary artery pressure may cause right ventricular dysfunction in mice [42], whereas long QT interval is mainly associated with impaired ventricular function and cardiac exhaustion [43].
Crizotinib can lead to increased caspase activation, cholesterol accumulation, and ion channel dysfunction [42]. Effective control of tumor growth can be achieved by dose-dependent inhibition of tyrosine phosphorylation of MET kinase and ALK [19]. After inhibition of 2-DIG-mediated glycolysis, crizotinib is inhibited by cell proliferation, migration, ATP production, mitochondrial transmembrane potential, or apoptosis signaling of mitochondria-associated cells. These findings suggest that crizotinib induces mitochondrial hypofunction and compensatory hyperoxic metabolism, without maintenance of adequate ATP levels. Moreover, the exchange pattern and inadequate supply of ATP may be an antitumor property of crizotinib [44]. Crizotinib is also a MET inhibitor, and MET has been implicated in cardiovascular remodeling after tissue injury as well as regulating mRNA levels of Glut4 and Ppars [45]. Further, the inhibition of potassium channels encoded by human ether-a-go-go (hERG)-related genes can lead to delayed repolarization, prolonged QT intervals, and life-threatening polymorphic ventricular tachycardia or Torsades de Pointes [46].
To further elucidate the mechanisms responsible for crizotinib-induced cardiotoxicity and to identify the genes that underlie its pathological effects, transcriptome sequencing of cardiac muscle tissue was used and this approach identified 10 up-regulated and 20 down-regulated genes in response to crizotinib exposure. Using GO and KEGG analysis of these DEGs, we selected terms directly related to cardiovascular disease and the circulatory system. Myh7 is both highly expressed and involved in multiple processes in KEGG enrichment, including a variety of cardiomyopathy, myocardial contraction, and adrenergic signaling in cardiomyocytes. Therefore, Myh7 may be a potential gene target of crizotinib-induced cardiotoxicity.
The Myh7 gene encodes the beta myosin heavy chain subunit of cardiac myosin
(beta-MHC). Changes in myosin expression can affect the contractile capacity of
cardiomyocytes and lead to abnormal myocardial structure and/or function.
Modification of myosin may affect the mechanical function of the myocardium and
are therefore considered to be linked to myocardial dysfunction leading to heart
failure [47]. To date, 186 and 73
Sacubitril/valsartan is used clinically in hypertension and heart failure. It can reverse left ventricular hypertrophy and delays left ventricular remodeling. These effects reduce the risks of cardiovascular death or hospitalization, improve symptoms, in-hospital outcomes and mortality in patients with heart failure [49, 50]. Clinical practice guidelines classify sacubitril/valsartan as a Class I recommendation as an alternative to angiotensin converting enzyme inhibitor [51] and that sacubitril/valsartan is associated with a reduced incidence of VAs in heart failure with reduced ejection fraction (HFrEF) [52, 53]. In patients with nonischemic DCM, the use of sacubitril/valsartan can also improve ventricular function and clinical outcomes [54]. In regards to possible cardiac protection in cancer patients receiving anti-cancer therapies, the international guidelines for sacubitril/valsartan are less clear. However, an increasing body of evidence has reported the benefits of sacubitril/valsartan [24, 27] on, for example, doxorubicin-related cardiotoxicity [34]. Specifically, sacubitril/valsartan can limit doxorubicin-induced apoptosis and endoplasmic reticulum stress in cultured H9C2 cardiomyocytes and can improve biochemical markers, contractile function, and endoplasmic reticulum stress in a rat doxorubicin-induced cardiotoxicity model [35]. Indeed, the potential benefits of sacubitril/valsartan in patients with cancer treatment-related cardiac insufficiency are increasingly recognized [28] and results shown in this study indicate that sacubitril/valsartan can significantly reduce the cardiotoxicity caused by crizotinib.
Crizotinib induced a range of cardiotoxic side effects in a mouse model, and that increased expression of Myh7 represents a biomarker for this cardiotoxicity. These cardiovascular abnormalities can be largely prevented by the co-administration of sacubitril/valsartan.
NSCLC, Non-small cell lung cancer; ALK, Anesthenic lymphoma kinase; LV, Left ventricular; RV, Right ventricular; VAs, Ventricular arrhythmias; VRP, Ventricular refractory period; BW, Body weight; HR, Heart rate; BP, Blood pressure; RP, Refractory period; QTc, QT interval correction; GO, Gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEGs, Differentially expressed genes; FC, FoldChange; RAS, Renin-angiotensin system; DCM, Dilated cardiomyopathy; PAT, Pulmonary artery acceleration time; LAD, Left atrial diameter; FS, Fractional shortening; LVIDs, Left ventricular diameter at systolic period; LVIDd, Left ventricular diameter at diastolic period; LVAWs, Left ventricular anterior wall thickness at systolic period; LVAWd, Left ventricular anterior wall thickness at diastolic period; LVPWs, Left ventricular posterior wall thickness at systolic period; LVPWd, Left ventricular posterior wall thickness at diastolic period; IVSs, Interventricular septum thickness at systolic period; IVSd, Interventricular septum thickness at diastolic period; SBP, Systolic arterial blood pressure; DBP, Diastolic arterial blood pressure; MBP, Mean arterial blood pressure; HE, hematoxylin and eosin; PVDF, Polyvinylidene fluoride; CV, Conduction velocity; AI, Absolute inhomogeneity; II, Inhomogeneity index; ATP, Adenosine-triphosphate; RT-PCR, Real time-polymerase chain reaction; BCA, Bicinchoninic acid; FDA, Food and Drug Administration; SDS-PAGE, Sodium dodecyl sulfate polyacrylamide gel electrophoresis; LSD, Least significant difference; EF, Ejection fraction; ECG, Electrocardiogram.
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
GL and TL designed the research study. LC and JD performed the research, analyzed the data and wrote the manuscript. GT provided help and advice on interpretation of data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was approved by the Animal Ethical and Welfare Committee of Chinese Academy Medical Sciences Institute of Radiation Medicine (Ethics approval number: IRM-DWLL-2021200).
Thanks to all the people who helped us in the experiment completion and manuscript writing. Thanks to all the peer reviewers for their valuable comments and suggestions.
This work was supported by grants from National Natural Science Foundation of China (NO. 82100342), the Tianjin Natural Science Foundation (16JCQNJC12000), China Postdoctoral Science Foundation (2016M601274), Key Laboratory of Scientific Research Foundation of the Second Hospital of Tianjin Medical University (2019ZDSYS14), and Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-029A).
The authors declare no conflict of interest. Tong Liu and Gary Tse are serving as Guest Editors of this journal. We declare that Tong Liu and Gary Tse had no involvement in the peer review of this article and have no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Jerome L. Fleg.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.