


 , Feng Cao 1,*
, Feng Cao 1,*1 National Clinical Research Center for Geriatric Diseases, the Second Medical Center, Chinese PLA General Hospital, 100853 Beijing, China
Abstract
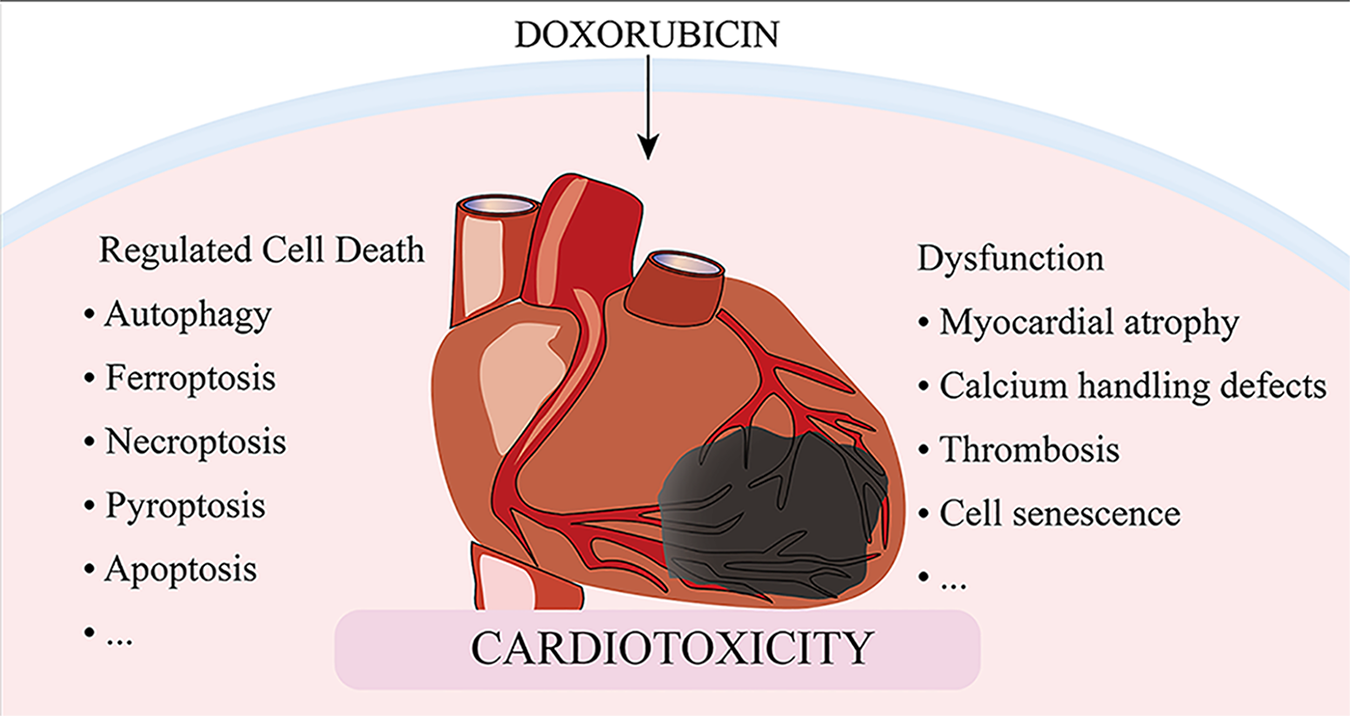
Despite recent advances in cancer therapy, anthracycline-based combination therapy remains the standardized first-line strategy and has been found to have effective antitumor actions. Anthracyclines are extremely cardiotoxic, which limits the use of these powerful chemotherapeutic agents. Although numerous studies have been conducted on the cardiotoxicity of anthracyclines, the precise mechanisms by which doxorubicin causes cardiomyocyte death and myocardial dysfunction remain incompletely understood. This review highlights recent updates in mechanisms and therapies involved in doxorubicin-induced cardiomyocyte death, including autophagy, ferroptosis, necroptosis, pyroptosis, and apoptosis, as well as mechanisms of cardiovascular dysfunction resulting in myocardial atrophy, defects in calcium handling, thrombosis, and cell senescence. We sought to uncover potential therapeutic approaches to manage anthracycline cardiotoxicity via manipulation of crucial targets involved in doxorubicin-induced cardiomyocyte death and dysfunction.
Graphical Abstract
Keywords
- anthracycline
- mechanism
- cardiotoxicity
- cell death
- dysfunction
The advent of chemotherapy drugs has greatly improved the survival rate of
cancer patients. However, a large number of surviving cancer patients suffer from
cardiac abnormalities induced by antitumor therapy [1]. Cardiovascular toxicity
is one of the most prevalent and potentially fatal adverse effects of antitumor
therapies, such as chemotherapy drugs [2]. Anthracyclines, a class of
chemotherapy drugs, are routinely used to treat a variety of tumors, such as
breast cancer, acute leukemia, sarcomas, Hodgkin’s and non-Hodgkin’s lymphomas,
and other hematologic and solid tumors [3]. As a typical representative of
anthracyclines, doxorubicin (DOX) has been extensively studied, and the incidence
of DOX-induced cardiotoxicity (DOXIC) increases with the cumulative dose [4, 5].
DOXIC may be a continuous process that begins with a decline in the function of
cardiomyocytes exemplified by decreased Ca
Regulated cell death (RCD) is implicated in general processes such as organogenesis and tissue remodeling, removal of redundant structures or cells, and regulation of cell numbers [13]. RCD can also be triggered by pathological stress when the adaptive processes that respond to stress fail. Five high-profile forms of RCD, including apoptosis, autophagy, necroptosis, ferroptosis, and pyroptosis, have been implicated in the pathogenesis of DOXIC [14]. In this review, we discuss the classical mechanisms of RCD and the specific pathways of cell death induced by DOX, to determine potential therapeutic targets to minimize the detrimental effects of DOX (Fig. 1).
 Fig. 1.
Fig. 1.Proposed mechanisms involved in DOX-induced cardiomyocyte
regulated cell death. Dox, doxorubicin; Alas1, aminolevulinate synthase 1; Bax,
Bcl-2-associated X protein; Bnip3, Bcl-2/adenovirus E1B interacting protein 3;
CamkII, calmodulin kinase II; CDK2, cyclin-dependent kinase 2; CSE, cysteine
gamma-lyase; GPX4, glutathione peroxidase 4; GSDMD, gasdermin D; GSDME, gasdermin
E; HMOX1, heme oxygenase-1; Mitol, E3 ubiquitin-protein ligase; MLKL, mixed
lineage kinase domain like pseudokinase; mTOR, mammalian target of rapamycin;
NLRP3, nod-like receptor protein 3; TFEB, transcription factor EB; TINCR,
terminal differentiation-induced non-coding RNA; GSH, glutathione; TfR,
transferrin receptor; Nrf2, nuclear factor erythroid 2-related factor 2; Caspase
1, cysteinyl aspartate specific proteinase 1; IGF2BP1, insulin
like growth factor 2 mRNA binding protein 1; RIPK3, receptor-interacting
serine-threonine kinase 3; mPTP, mitochondrial permeability
transition pore; Beclin1, Bcl-2-interacting protein 1; ULK1, unc-51 like
autophagy activating kinase 1; AMPK, adenosine 5
The most extensively studied type of cell death related to DOXIC is cell apoptosis, which is morphologically manifested as cell atrophy, increased cytoplasmic density, loss of mitochondrial membrane potential and permeability variation. As a result of these actions, complete apoptotic bodies are formed, which are absorbed and degraded by neighboring cells [15]. According to various mechanisms, apoptosis is divided into intrinsic apoptosis and extrinsic apoptosis. Intrinsic pathways, also known as mitochondrial or Bcl-2 (B-cell lymphoma 2)-mediated pathways, are activated by BH3 proteins (Bim, BMF, BID, PUMA, BIK, HRK, BAD, and NOXA) to initiate apoptosis in the face of intracellular stress caused by various physical and chemical factors [16, 17]. Downstream apoptotic factors released by mitochondria, such as cytochrome c and Smac (second mitochondria-derived activator of caspase) protein, also known as DIABLO, activate Bcl-2-associated X protein (Bax) which facilitates caspase cascade activation, which eventually results in protein cleavage and cell death [18]. In extrinsic apoptosis, the combination and activation of members of the tumor necrosis factor (TNF) receptor superfamily, the Fas/Fas ligand (FasL) system, and members of the death receptor (DR) family (DR3, DR4, and DR5) with corresponding ligands, results in the intracellular lethal signaling complex, which in turn activates caspase-8 and its effectors [19].
DNA damage in DOX-treated cardiomyocytes is thought to be the primary effect of
DOXIC. Apoptosis is a secondary response to DNA damage. Topoisomerase-II is
reported to be the direct target of DOX, which results in DNA double-strand
breaks and opens endogenous apoptotic pathways [20]. Bcl-2 antagonist killer 1 (Bak) and Bax, members of the Bcl-2 family, oligomerize in response to DNA damage, creating
a pore in the mitochondrial outer membrane that causes the release of cytochrome
C and the activation of caspases, resulting in apoptosis [21]. The insulin-like
growth factor 1 (IGF1) signaling pathway is a well-recognized cell survival
signal and exogenous IGF1 is capable of rescuing cardiomyocytes from apoptosis
triggered by DOX [22]. Endogenous hydrogen sulfide (H
As a key mechanism of intracellular degradation, autophagy, also named type II cell death, transports cellular constituents from the cytoplasm to the lysosome, where the cargos are broken down by double-membrane autophagosomes [27]. Autophagy functions as a major cytoprotective process by preserving cellular homeostasis and recycling cytoplasmic constituents. However, studies suggest that autophagy is a primary form of cell death and implicates autophagic cell death in the pathological process of DOX-induced cardiomyocyte death [28]. Anthracyclines have been found to induce dysregulated autophagy, which leads to an excessive amount of cardiomyocyte death [29].
The process of cell autophagy is initiated by AMP-activated protein kinase (AMPK) activation and mammalian target of rapamycin (mTOR) inhibition, both of which have been shown to induce unc-51 like autophagy activating kinase 1 (ULK1) activation. ULK1 promotes autophagy by phosphorylation and positive regulation of the autophagy-related protein Beclin-1 (BECN1), the signaling hub in the context of autophagy [30]. Bcl-2, an early gatekeeper of autophagy control, also inhibits BECN1 [31]. Current studies on the effect of anthracyclines on autophagy have reported conflicting results. Some studies have reported that anthracyclines induce autophagy [28], while others have reported that anthracyclines inhibit autophagy [32, 33]. Similarly, studies on the effects of genetic or pharmacological suppression of autophagy have yielded mixed results, with some indicating protective effects [34, 35], while others demonstrate that sustained reduction of autophagy cannot maintain protection against DOXIC [36, 37].
Recent studies have shown that these contradictory results may be due to the different effects of DOX on autophagy at different phases. DOX initially promotes autophagy but later prevents it. Thus, DOX leads to an accumulation of autophagosomes and autolysosomes that have not yet been destroyed, increasing the damage to cardiac cells, and ultimately resulting in cell death. DOX at low concentrations induces autophagy by up-regulating the expression of BECN1 [38]. In an adult DOXIC zebrafish model, a biphasic response in autophagy was observed: activation in the early stage and suppression in the later phase that is accompanied by cardiac functional decline [39]. Moreover, overexpression of autophagy related 7 proteins, a rate-limiting autophagy regulator, leads to therapeutic effects in the late phase but deleterious effects in the early phase of adult DOXIC [39]. In a more recent study using in vitro rat myoblast H9c2 cell culture model, DOX was revealed to block the progression of autophagy, particularly the fusion of autophagosomes with lysosomes. This decrease in the formation of autolysosomes and contributes to the accumulation of dysfunctional mitochondria and subsequent increase in cytotoxic ROS, eventually resulting in increased myocardial cell death [33]. The transcription factor E-box binding protein (TFEB) controls lysosomal signaling and function [40, 41]. DOX inhibits the expression of TFEB in cardiomyocytes. The inhibition of TFEB leads to decreased macrophage protein expression, inhibition of autophagy flux, impairment of lysosome cathepsin B activity, and activation of cell death [42]. However, recovery and/or activation of TFEB in DOX treated cardiomyocytes prevents DOX-induced cathepsin B activity inhibition, reduces DOX-mediated ROS overproduction, weakens the activation of caspase-3, and improves the cell’s viability [42]. These studies reveal that DOX hinders the process of autophagosome destruction, resulting in the accumulation of autophagosomes and the consequent overproduction of ROS and subsequent cell death.
A recently identified intracellular iron-dependent form of cell death process called ferroptosis is distinguished by intracellular iron accumulation and lipid peroxidation [43]. Recent studies have shown that ferroptosis, a novel form of regulated cell death mediated by iron-dependent lipid peroxidation, also plays a key role in DOX-induced cardiomyocytes death.
The main contributor to cardiac damage in DOX is mitochondrial-dependent
ferroptosis. By forming the DOX-Fe
Cell necroptosis is a programmed form of necrosis mediated by DRs, or inflammatory cell death. Receptor interacting protein kinase-1 and the -3/mixed lineage kinase domain-like protein (receptor-interacting protein [RIP]1/RIP3/mixed lineage kinase domain like pseudokinase [MLKL]) pathway is activated during canonical necroptosis [54, 55]. Specific DRs, including but not limited to TNF-R1, Fas receptors, and Toll-like receptors (TLR), are also responsible for sensing a variety of stimuli that cause necroptosis [56, 57].
Recent studies have shown substantial interplay between DOX-induced necroptosis
and other death modes of cardiomyocytes. DOX was shown to inhibit autophagy flux
[42]. The inhibition of autophagy flux leads to RIP1-RIP3 interaction and
activation of necroptosis in cardiomyocytes, which in turn leads to cell death
[58]. mTOR complex 1 (mTORC1) inhibition enhances autophagy and protects
cardiomyocytes against necroptosis via a TFEB-dependent mechanism [59].
DOX also activates necroptosis through a noncanonical RIP3/calmodulin-dependent
protein kinase II (CamkII) pathway. DOX treatment increases the amount of
receptor-interacting protein 3 (RIP3) in the myocardium. RIP3 binds to and
phosphorylates Ca
Pyroptosis is a highly inflammatory form of lytic programmed cell death [64]. The main morphological characteristics of pyroptosis include membrane perforation, cell swelling, leakage of cellular content, chromatin condensation and DNA fragmentation. Unlike apoptosis, the nucleus is mostly intact in pyroptosis. Pyroptosis has been found to be involved in many cardiovascular disorders including DOXIC [65]. The process of pyroptosis is usually incited by the formation of a large supramolecular complex termed the inflammasome as a result of various stimuli [2]. The inflammasome activates a different set of caspases from those associated with apoptosis, for example, caspase-1/4/5 in humans and caspase-11 in mice. The activated caspase-1 in turn cleaves gasdermin D (GSDMD) into the N-terminal domain. The GSDMD-N-terminal domain then forms pores in the plasma membrane, resulting in pyroptosis.
The involvement of the canonical nod-like receptor protein 3 (NLRP3) inflammasome pathway-mediated pyroptosis in the pathogenesis of DOXIC has been reported in several studies. The NLRP3/caspase-1/GSDMD axis was shown to contribute to DOXIC pathogenesis. DOX-induced dynamin-related protein 1-mediated mitochondrial fission and the ensuing NLRP3 inflammasome activation and pyroptosis could be rescued by NADPH oxidase 1 (NOX1) and NADPH oxidase 4 (NOX4) silence [66]. There is also evidence suggesting that DOX induces pyroptosis of cardiomyocytes in a GSDMD-dependent manner by directly binding to GSDMD and promoting GSDMD-N-mediated pyroptosis [67]. Similarly, DOX also causes pyroptosis of cardiomyocytes through the caspase-3/gasdermin E (GSDME) axis [68]. Human GSDME-positive SH-SY5Y and MeWo cells as well as mouse HL-1 cardiomyocytes exhibited GSDME-dependent pyroptosis upon activation of caspase-3 by chemotherapy drugs including DOX. Gsdme-/- mice were resistant to DOX-induced tissue damage [68]. Bcl-2/adenovirus E1B interacting protein 3 (Bnip3) is a pro-apoptotic protein. A previous study revealed the important role of Bnip3 in activating the caspase-3/GSDME axis. Cardiomyocyte pyroptosis is activated by the Bnip3/caspase-3/GSDME pathway following DOX treatment, suggesting that Bnip3 is a novel regulator of cell pyroptosis in DOXIC [69]. GSDMD is also involved in DOX-induced Bnip3-mediated mitochondrial damage and perforation in cardiomyocytes [67]. Apart from the extensively studied NLRP3 inflammasome, the nod-like receptor (NLR) family protein 1 NLRP1 inflammasome is also reported to be involved in DOX-induced pyroptosis. The E3 ubiquitin ligase TRIM25 has been reported to promote NLRP1 ubiquitination and to reduce NLRP1 stability, thereby inhibiting NLRP1-mediated pyroptosis in DOX-treated cardiomyocytes [70].
At present, some pharmacologic agents have been shown to inhibit pyroptosis in
animal experiments. MCC950 (a small molecule compound) is an NLRP3 inflammasome inhibitor, which can inhibit NLRP3-mediated pyroptosis and alleviate myocardial injury induced by DOX [71].
Curcumin inhibited DOX-induced pyroptosis of cardiomyocytes in a PI3K (phosphoinositide 3-kinase)/Akt (protein kinase B pathway)/mTOR dependent manner [72]. Calycoflavone (CAL), astragalus’ primary active component, reduces the pyroptosis of cardiomyocytes attributed to DOX, by preventing the
activation of the NLRP3 inflammasome [73]. By reducing the expression of
NOX1/2/3, TLR2/4, and the nuclear factor kappa-light-chain-enhancer of activated
B cells (NF-
Some non-coding RNAs have also been revealed to play essential roles in DOX-related pyroptosis. Terminal differentiation-induced noncoding RNA has recently been reported to direct insulin-like growth factor 2 binding protein 1 to increase the stability and expression of NLRP3, thereby inducing pyroptosis in DOX-treated cardiomyocytes [77].
Apart from drastic cell death, DOX also induces relatively mild cardiomyocytes
dysfunction that exacerbates its cardiotoxicity. In addition to the classical
manifestations of myocardial cell dysfunction caused by oxidative stress and
mitochondrial dysfunction, there are also some recently focused pathological
processes including myocardial atrophy, Ca
 Fig. 2.
Fig. 2.Proposed mechanisms involved in Dox-induced cardiac
dysfunction. Dox, doxorubicin; CaMKII, calmodulin kinase II; CDK2,
cyclin-dependent kinase 2; FOXO1, fork head box O1; FOXO3, fork head box O3; mTOR,
mammalian target of rapamycin; MURF1, muscle RING finger 1; NOX2, NADPH oxidase 2; PARP, poly ADP
ribose polymerase; PPAR
Myocardial atrophy is a significant factor in the decrease of left ventricular mass following anthracycline therapy, and cardiac atrophy is a common factor of DOX-induced cardiac dysfunction [78]. The loss of left ventricular myocardial cell mass increases with the increased level of myocardial cell damage [79]. There is evidence demonstrating that loss of myocardial mass rather than cardiomyocyte death is the major contributor to acute DOX cardiotoxicity [80]. Cardiomyocyte atrophy was viewed as an underestimated contributor to DOXIC [78].
Multiple pathways are involved in anthracycline-induced myocardial atrophy. Muscle ring finger-1 (MuRF1), a striated muscle-specific ubiquitin ligase, is required for DOX-induced cardiac atrophy in mice. MuRF1-deficient mice were protected from cardiac atrophy after DOX exposure with no loss in systolic function [81]. Fork head box O1 (FOXO1) is highly expressed in adult heart tissue and is essential for cardiac development [82, 83]. Through the transcriptional activation of atrophy-related target genes such as atrogin-1 and MuRF1, FOXO transcription factors regulate skeletal muscle atrophy in addition to apoptosis [84]. Increased FOXO1 levels in the heart result in atrophy of cardiomyocytes and loss of cardiac mass [85, 86, 87]. Depletion of FOXO1 protects against DOX-induced myocardial weight loss and decreased cardiac function [88]. NADPH oxidase 2 (NOX2) is another master regulator of myocardial atrophy. Mice lacking NOX2 display resistance to DOX-induced cardiac atrophy, which is associated with mitigated NADPH oxidase activity, oxidative/nitrosative stress, and inflammatory cell infiltration [89]. Myocardial atrophy caused by DOX is also reported to be under the control of the transient receptor potential canonical 3 (TRPC3)-NOX2 complex. The amplification of NOX2-mediated ROS signaling in cardiomyocytes can be inhibited by blocking TRPC3, thereby avoiding DOX-induced cardiac atrophy [90]. DOX is also reported to contribute to myocardial atrophy by suppressing the cell growing mTOR pathway [91]. This inhibitory effect has been demonstrated to be dependent on P53 activation, resulting in acute cardiac insufficiency and myocardial atrophy [80].
DOX cardiotoxicity is associated with dysregulation of Ca
Preventing Ca
The second leading cause of death in cancer patients is thromboembolic disease [111, 112]. Chemotherapy agents associated with DOX are known to cause thrombotic complications in cancer patients [113].
DOX promotes thrombosis through a variety of pathways. DOX has a direct effect
on platelets. DOX-induced venous thrombosis is strongly dependent on platelet
activation. Aspirin and clopidogrel, the two most potent antiplatelet
medications, have demonstrated considerable antithrombotic activity in
DOX-exacerbated stasis-induced thrombosis [114]. The procoagulant activity of
platelets increased by DOX can be related to the increased platelet exposure to
phosphatidylserine (PS) and PS-bearing microparticles, in which intracellular
Ca
DOX-induced vascular toxicity also contributes to DOX-induced venous thrombosis.
DOX administration was found to be correlated with smaller blood vessels (
Cardiomyocyte senescence is assumed to be one of the main contributing factors to the onset of DOXIC [119, 120]. Cardiomyocytes treated with DOX have a senile-like phenotype and exhibit aberrant troponin phosphorylation patterns, which may cause ineffective ventricular contraction [121]. Anthracycline-induced senescent cell accumulation is associated with long-term deterioration of cardiovascular homeostasis [3]. To date, most reports have demonstrated the pro-aging effects of anthracyclines on various types of cardiac cells.
DOX induces the increase of senescence phenotype as evidenced by increased
P16INK4a (official gene symbol CDKN2A, cyclin dependent kinase inhibitor 2A)
expression in cardiomyocytes [120]. Furthermore, studies have shown that
different DOX doses directly lead to different outcomes of cardiomyocytes, with
low dose inducing cardiomyocyte senescence while high dose inducing apoptosis
[122]. While low-dose DOX can cause downregulation of both telomeric repeat
binding factor 2 (TRF2) and telomeric repeat binding factor 1 (TRF1), increased
In addition to cardiomyocyte senescence, DOX also induces the senescence of other types of cardiac cells. DOX induces a DNA damage response in human cardiac progenitor cells, which results in telomere shortening and senescence [132]. Transient DOX treatment contributes to the onset of long-term senescence associated with a reduction in vascular endothelial growth factor receptor 2 levels in endothelial cells [133]. DOX-induced senescence has also been reported in human endothelial progenitor cells and vascular smooth muscle cells [134, 135].
There have been many studies on pharmacologic agents to retard cardiac
senescence induced by anthracyclines. Experimental evidence suggests that
anthracycline-induced cardiomyocyte senescence is eliminated by testosterone, and
that testosterone preconditioning prevents DOX-induced downregulation of TRF2 and
activation of p53 in H9C2 and neonatal mouse cardiomyocytes, resulting in
decreased expression of the aging markers senescence-associated
DOX results in cardiotoxicity, due to multiple forms of cell death and
myocardial dysfunction. DOXIC is an important public health problem. Though some
clinical trials have shown that non-drug interventions such as exercise, healthy
lifestyle, control of risk factors and treatment of comorbidities, as well as
pharmacologic interventions including
Conceptualization, writing, and editing, TW, YM, SG, WZ, DH and FC; supervision, DH and FC; provided financial support, DH and FC. All authors have read and agreed to the published version of the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by the National Natural Science Foundation of China (No. 82100372), the National Key Research and Development Program of China (No. 2022YFA1104700), China Postdoctoral Science Foundation (BX20200154 and 2021MD703962), Beijing Nova Program (Z211100002121048) and Beijing Natural Science Foundation (7232159).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.