
- Academic Editor
Cardiovascular disease is one of the major diseases threatening the health of Chinese residents, and the death rate has long been the highest on the disease spectrum in China. With the progress of population aging, the prevalence and mortality of cardiovascular diseases remain on the rise, and the current treatment effect on and prognosis of heart failure (HF) are not satisfactory. It is particularly important to explore the potential pathogenic mechanisms of HF and identify new therapeutic targets.
Studies have shown that the number of people suffering from cardiovascular
diseases in China is currently approximately 330 million, and 8.9 million are
heart failure patients [1]. Heart failure (abbreviated as HF) is a group of
syndromes in which various structural or functional heart diseases lead to
impaired ventricular filling and/or ejection function, and cardiac blood output
cannot meet the metabolic needs of body tissues, with clinical manifestations of
pulmonary and/or body circulation stasis and insufficient blood perfusion to
organs and tissues. According to the ejection fraction, HF with reduced ejection
fraction (HFrEF, left ventricular ejection fractions (LVEF)
The intestinal flora is a relatively complex ecosystem composed of a variety of intestinal microorganisms that reside in the human gut, they are large in number and diverse. Each microorganism has the capacity to produce hundreds of different known and unknown metabolites that act on the intestine itself [1]. Intestinal flora dysbiosis is a pathological lesion caused by the inhibition of sensitive intestinal bacteria due to age, diet, drug abuse, disease and other factors, following which uninhibited bacteria take the opportunity to multiply, thus causing flora dysbiosis and imbalance of intestinal flora metabolites, resulting in the disruption of normal physiological combinations. Studies have shown that intestinal flora dysbiosis is closely related to the development of HF, that metabolites of the intestinal flora, mainly short-chain fatty acids (SCFAs), trimethylamine-N-oxide (TMAO) and bile acid (BA), are involved in the development of HF through various metabolic pathways, and they affect the prognosis of HF [2]. Diet, drugs (antibiotics), probiotics and fecal microbiota transplantation (FMT) can alter the intestinal flora components and provide new therapeutic ideas for the treatment of HF.
Dietary interventions are the safest and most effective way to improve the intestinal flora. Marques et al. [3] administered a high-fiber diet or added acetate as a dietary intervention in mice using an HF model and observed that it led to an increase in SCFA levels, in turn resulting in blood pressure levels being effectively controlled and myocardial hypertrophy and myocardial fibrosis being reduced, thus improving cardiac function. The American College of Cardiology/American Heart Association guidelines formally adopted a strong recommendation, the Dietary Approaches to Stop Hypertension (DASH), which recommends a diet that is rich in fruits, vegetables, whole grains and low-fat dairy products, including meat, fish, poultry, nuts and legumes, and that limits sugary foods and beverages, red meat and legumes, and added fats. Several observational studies have been shown to reduce the incidence of HF [4, 5, 6]. The Mediterranean diet, mainly high in fruits, vegetables, legumes and whole grains and low in red/processed meats and refined carbohydrates, is beneficial in delaying cardiovascular disease (CVD) and HF. In fact, a systematic review and meta-analysis comparing randomized, controlled trials of the Mediterranean diet involving 10,950 people showed that the Mediterranean diet reduced the incidence of HF by 70% [7].
Antibiotic interventions, one of the most common and effective experimental
interventions to regulate the intestinal flora in clinical practice. A variety of
antibiotics have been shown to reduce the levels of inflammatory factors in the
body, such as interleukin-1
Probiotics are a group of intestinal physiological bacteria that live in mutually beneficial symbiosis with their hosts and contribute to their health [11]. Probiotics in clinical use include bacterial and fungal microorganisms, including Lactobacillus spp., Bifidobacterium spp. and Saccharomyces boulardii [12]. Probiotics can colonize the intestine, prevent the adherent colonization of pathogenic bacteria, regulate intestinal flora disorders, and improve the intestinal inflammatory response. It has been reported that treatment of rats with a drink containing Lactobacillus plantarum 299v 24 hours before coronary artery ligation reduced infarct size and improved left ventricular function [10, 13]. In another study, it was found that the administration of Lactobacillus rhamnosus GR-1 treatment significantly improved left ventricular hypertrophy and increased left ventricular ejection fraction in a rat model of acute myocardial infarction. In a randomized, double-blind, controlled study, Saccharomyces boulardii was beneficial in patients with HF, with short-term improvements in left ventricular ejection fraction and reductions in serum creatinine and inflammatory markers [14].
FMT is the transplantation of functional bacteria from healthy human feces into the patient’s gastrointestinal tract to improve the structure and composition of the intestinal flora, and it can reverse dysbiosis of the intestinal flora and re-establish its normal function in inflammatory bowel disease [15, 16], but the therapeutic role of FMT in other diseases is unclear. Standardization and optimization of FMT procedures are essential, including screening of suitable donors, development of noninvasive delivery methods (e.g., capsules), and identification of active ingredients to develop a rational and individualized therapeutic strategy. The ability of FMT to improve HF by restoring the diversity and function of the intestinal flora requires extensive experimental and clinical studies.
Chinese medicine interventions are rich in a variety of chemical components. In particular, Chinese medicine compounds not only contain alkaloids, polysaccharides, glycosides and other effective drug components, but they are also rich in vitamins, dietary fiber and other nutrients, with a variety of pharmacological effects. They can also provide intestinal nutrition, promote the repair of intestinal mucosal epithelial cells and the expression of tight junction proteins, regulate intestinal immunity, reduce intestinal inflammation, and facilitate the intestinal flora. Single Chinese medicines [17], Chinese medicine monomers and Chinese medicine compounds can take the intestinal flora as an effective target to play a pharmacological role in the prevention and treatment of chronic HF. For example, Bao Yuan Tang, which is a Proprietary Chinese Medicine, can improve the dysregulated intestinal flora in rats with isoproterenol-induced myocardial hypertrophy, thereby regulating the metabolism of short-chain fatty acids, bile acids, and amino acids involved in the intestinal flora so that the downstream pro-inflammatory, pro-oxidative, and pro-myocardial hypertrophy signaling pathways can be effectively inhibited, thus exerting its effects of lowering blood pressure and blood lipids, alleviating myocardial hypertrophy, and improving cardiac function [18].
A large body of evidence suggests that cytokines and chemokines are closely
associated with HF. The proinflammatory cytokines TNF-
In the early 1990s, studies showed significantly elevated levels of circulating
TNF-
There are 11 cytokine members and 10 receptors that make up the IL-1 family; of
these, the most well-studied pathogenic mechanisms in the cardiovascular system
are the IL-1
A number of cytokines have been implicated in the development of cardiovascular disease, including IL-11, leukemia inhibitory factor (LIF), cardiolipin-1 and tumor suppressor M. These are typical members of the gp130 family of cytokines, the most well-studied of which is IL-6. Tocilizumab significantly reduced circulating blood levels of N-terminal pro brain natriuretic peptide (NT-proBNP) in patients with rheumatoid arthritis without previous cardiovascular disease, indirectly demonstrating its protective effect in slowing the progression of HF [27]. In another trial, in patients with non-ST-segment elevation myocardial infarction (NSTEMI), a single effective dose of tocilizumab before coronary angiography was effective in suppressing not only systemic validation levels but also in reducing troponin T release, indirectly suggesting that suppression of inflammation is effective in protecting against myocardial necrosis. This experiment provides ample evidence of the protective effect of tocilizumab against ischemic myocardial injury in acute coronary syndromes [28]. The complexity of the effect of IL-6 on the inflammatory cascade is demonstrated by the opposite trend in serum C-reactive protein (CRP) and chemokine C-X-C motif chemokine ligand 10 and C-C motif chemokine ligand 4 (CXCL10 and CCL4) levels, which well illustrates the uncertainty of the classical and cross-signaling effects of cytokines in anti-IL-6 therapeutic agents. In this context, more studies and more definitive results are needed to confirm the use of IL-6 targeting for the treatment of HF, which remains a potential target for HF therapy and still holds good promise to be explored [29].
Chemotactic cytokines of 8–12 kDa, called chemokines, are responsible for regulating cellular localization as well as migration in development, homeostasis and inflammation in the body [30]. Of these, XC, CC, CXC and CX3C chemokines are the four most studied subfamilies. Among the inflammatory CC chemokines regarding HF, C-C motif chemokine ligand 4/major capsid protein-1 (CCL2/MCP-1) has the ability to inhibit cardiac remodeling and prevent persistent myocardial damage leading to ischemic necrosis, and has been suggested in numerous studies as a potential therapeutic target for HF [31, 32]. CCL2 in endothelial cells, vascular smooth muscle cells, monocytes and cardiomyocytes, whose expression is consistently and stably upregulated in animal models of HF with cardiac injury and cardiac remodeling, may be associated with Toll-like receptor (TLR)-mediated signaling activation, neurohumoral cascade responses or pro-inflammatory cytokine-mediated pathways [31, 33, 34]. As research continues to ascend, persistent high expression of CCL2 in HF animal experiments, with or without myocardial infarction, is closely associated with myocardial dysfunction, fibrosis, and ultimately cardiac remodeling. In contrast, in patients with HFrEF, peripheral blood CCL2 levels showed a significant positive correlation with heart failure symptoms and left heart systolic function [35]. In patients with advanced HF, peripheral blood CCL2 levels are also positively correlated with the occurrence of adverse cardiovascular events [36]. CCL2-driven pro-inflammatory signaling leads to a sustained increase in immune response in failing cardiomyocytes, resulting in further swelling, necrosis and thus an increased risk of death from heart failure; it has been reported that in animal models of myocardial infarction, blocking the CCL2/chemokine receptor 2 (CCR2) axis is effective in reduce infarct size and protect surviving myocardium [37, 38]. At the same time, CCL2 has a powerful fibrotic effect, causing dead myocardium to fill the gap left by myocardial death through fibrosis, which to some extent accelerates myocardial remodeling [39]. A number of researchers have also suggested that CCL2 has a direct myocardial damaging effect, rapidly leading to increased risk of death due to cardiac contractile dysfunction [40]. Therefore, effective inhibition of the CCL2/CCR2 axis is expected to be a target for the future treatment of HF.
Wnt is a cysteine-rich glycoprotein in the extracellular matrix (EMC) that plays
a key role in a variety of pathological processes including neurodegeneration,
osteoporosis, cancer, cardiac arrhythmias, and myocardial infarction. The Wnt
(wingless/integrated) signaling pathway is a fundamental signaling pathway that
regulates heart and vascular development and plays a critical role in the
development of Frizzled (Fzd) receptors, low density lipoprotein (LDL) receptor related protein 5/6 (LRP5/6) receptors and the downstream
signaling molecules glycogen synthase kinase 3
Myocardial hypertrophy is a compensatory response of the heart. Some findings
have confirmed that the Wnt/
Currently, some breakthrough research progress has been made in regulating the
activity of the Wnt signaling pathway by targeted degradation techniques. Using
targeted degradation technology to precisely modulate Wnt signaling pathway
activity, which in turn can be used to treat heart diseases, such as HF, Yeguang
Chen’s research team successfully synthesized axin-derived peptides based on
binding to
Other proteins in the Wnt signaling pathway can also be regulated by targeted degradation techniques. For example, Dsh proteins can regulate both classical and nonclassical Wnt signaling pathways, and to treat pathophysiological processes, such as cardiac hypertrophy, myocardial fibrosis, myocardial repair, and myocardial remodeling. In addition, the axin protein can be degraded by both the ubiquitin-proteasome pathway and the autophagy-lysosome pathway, and by designing specific PROTAC or autophagy-tethering compound (ATTEC) small molecule compounds, the function of the Wnt signaling pathway can also be precisely regulated (Fig. 1).
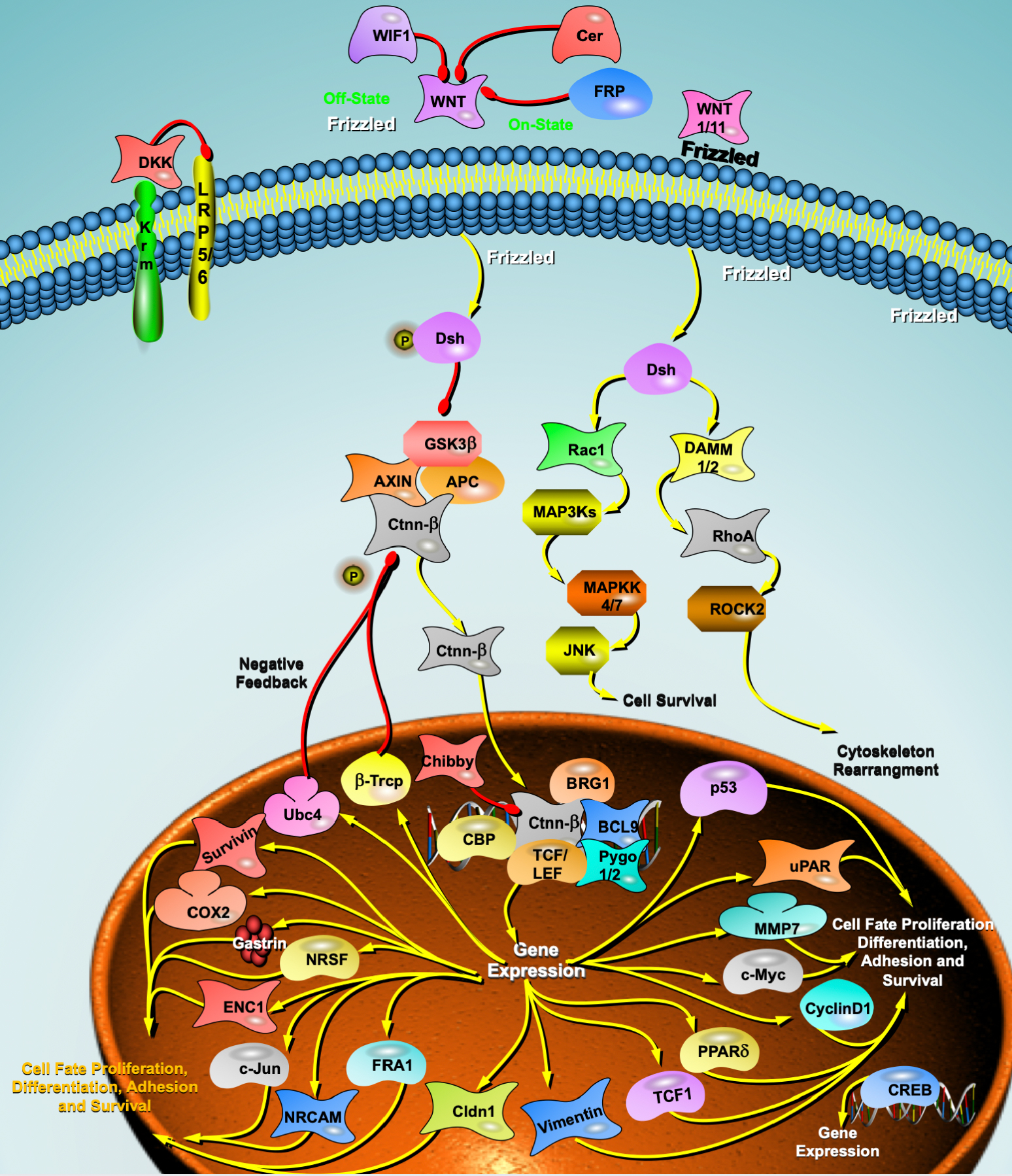 Fig. 1.
Fig. 1.Wnt signaling pathway and heart failure. DKK, Dickkopf; WIF1, WNT inhibitory factor 1;
BRG1, Brahma-related gene 1;
Krm, Kremen;
LRP5/6, lipoprotein receptor-related protein 5/6;
FRP, Frizzled receptor proteins;
Dsh, Dishevelled;
GSK3
Autophagy is the phagocytosis of eukaryotic cells by their own cytoplasmic proteins or organelles to achieve the metabolic needs of the cell itself and the renewal of some organelles through lysosomal degradation, the most important of which is mitochondrial autophagy in selective autophagy. Studies have shown that mitochondrial autophagy (mitophagy) is closely associated with HF [46, 47].
Mitochondrial autophagy is the process by which autophagic vesicles selectively wrap and transport damaged mitochondria to lysosomes for hydrolysis. This concept was first proposed by Lemasters in 2005 [48]. The current findings suggest two main types of mitochondrial autophagy mechanisms: ubiquitin dependent and non-ubiquitin dependent. Among them, ubiquitin-dependent mitochondrial autophagy includes PTEN-induced putative kinase 1 (PINK1)/parkin signaling pathway-mediated mitochondrial autophagy and non-parkin-dependent mitochondrial autophagy, while non-ubiquitin-dependent mitochondrial autophagy is directly mediated by mitochondrial autophagy receptors.
Song et al. [49] observed that parkin-mediated reduction in mitochondrial autophagy in cardiomyocytes from Mitofusin-2 (MFN2) gene mutant mice induced cardiac hypertrophy and HF. Chen et al. [50] found in a clinical study that PINK1 protein levels were significantly reduced and mitochondrial autophagy was inefficient in patients with advanced HF, whereas normal expression of PINK1 and parkin could attenuate myocardial cell injury, delay the progression of HF, and prolong patients’ lives.
It was found that the PINK1/parkin-mediated mitochondrial autophagy pathway is
a potential target for the treatment of HF. In an experimental study of safranin,
it was observed that parkin-mediated mitochondrial ubiquitination was
significantly inhibited after knockdown of the PTEN-induced putative kinase 1 (PINK1) gene, counteracting
the beneficial effects of safranin on HF, suggesting that safranin could promote
PINK1/parkin signaling pathway-mediated mitochondrial autophagy for the treatment
of HF [51]. Xiong et al. [52] found that overexpression of PINK1
promoted parkin translocation to the damaged mitochondrial outer membrane and
phosphorylation, maintained cardiomyocyte homeostasis, and attenuated myocardial
injury in a mouse model of angiotensin II-induced HF. In a tacrolimus (TAC)-induced mouse
model of HF, Wang et al. [53] showed that high expression of
AMP-activated protein kinase
Noncoding RNAs (NCRs) are RNAs that do not encode proteins. MicroRNAs (miRNAs)
are a class of single-stranded noncoding RNAs of approximately 22 nucleotides in
length, and mature miRNAs contribute to the posttranscriptional degradation of
target mRNAs by binding specifically to the 3
A growing number of studies have shown that microRNAs (miRNAs) play an important role in cardiovascular disease and are involved in several pathophysiological processes associated with HF, such as myocardial remodeling, cardiac hypertrophy, myocardial fibrosis, apoptosis, and hypoxia [55, 56]. An increasing number of miRNAs have exhibited a dysregulated and predominantly upregulated pattern in the later stages of end-stage HF [57]. The etiology of HF (e.g., ischemic, aortic stenosis, or idiopathic cardiomyopathy) is associated with differentially expressed miRNA patterns [58]. Thus, miRNAs play an active role in the development and progression of HF.
In the plasma of hypertrophic cardiomyopathy (HCM) patients diagnosed with no symptoms of HF, miR-29a, the
only miRNA associated with left ventricular hypertrophy and fibrosis, was found
to be significantly upregulated [59]. In addition, miRNAs can be used to
determine the prognosis of HF patients. Qiang et al. [60] examined
miRNAs in endothelial progenitor cells (circulating from monocytes) from 106 HF
patients and found that low levels of miR-126 were associated with cardiovascular
death in patients with ischemic HF, while high levels of miR-508a-5p were
associated with nonischemic HF patients. Additional studies have shown that
decreases in miR-18a-5p and miR-652-3p during hospitalization for HF predicted
180-day mortality [61]. One meta-analysis examining the expression of circulating
miRNAs and patient prognosis [62], which included four relevant articles
assessing 19 circulating miRNAs in 867 patients, showed that low expression of
miR-1, miR-423-5p, miR-126, miR-21, miR-23, miR-30d, miR-18a-5p, miR-16-5p,
miR-18b-5p, miR-36b-5p, miR-206a-3p, miR-2313-3a-3p, and miR-423-128 was
associated with significantly poorer overall survival in HF patients (p
It has also been shown that microRNAs can be used as biomarkers of response to HF treatment. For example, one animal study found [63] that plasma levels of miR-16, miR-20b, miR-93, miR-106b, miR-223 and miR-423-5p were elevated in rats with hypertension-induced HF compared to controls. Nie et al. [64] showed that miR-217 was highly expressed in the myocardial tissue of patients with chronic HF and exacerbated pressure load-induced cardiac dysfunction. These findings suggest that miR-217 plays an important role in myocardial hypertrophy and dysfunction and could be a therapeutic target for HF.
Long noncoding RNAs (lncRNAs) are a set of functional RNA molecules greater than 200 nucleotides in length that do not encode proteins and are generally present within longer coding genes, between coding genes, or in antisense to coding sequences [65]. LncRNAs are involved in cell differentiation, proliferation, apoptosis and autophagy, affecting the development and progression of various cardiovascular diseases, such as hypertension, atherosclerosis and HF. Liu et al. [66] showed that lncRNA H19 is highly expressed in the embryonic period, and its expression gradually decreases as individuals mature. The expression of lncRNA H19 decreases as individuals mature but increases again when vascular damage or cardiac insufficiency occurs. The results of another study observed a significant increase in H19 expression in patients with HF and in a mouse model of myocardial hypertrophy [67]. Han et al. [68] showed that high expression of H19 could be associated with disorders of lipid metabolism, and these findings suggest that the extent of HF might be understood by measuring H19 expression levels. In addition, studies have shown that the lncRNA long intergenic noncoding RNA predicting cardiac remodeling (LIPCAR) is upregulated early and downregulated later in the plasma of HF patients, and it has also been suggested that LIPCAR might be used as an HF biomarker [69]. Although the mechanisms of action of only a few lncRNAs have been elucidated, further systematic and comprehensive studies can be conducted to shed more light on the complex regulatory mechanisms and functional targets of lncRNAs, providing new ideas for the diagnosis and treatment of cardiovascular diseases and new targets for the development of new drugs.
The types of RNA modifications include N
Differentially m
Mathiyalagan et al. [72] found that obesity-associated protein (fat
mass and obesity-association protein, FTO) is a demethylase that plays an
important role in myocardial homeostasis and remodeling during cardiac
contraction. FTO overexpression attenuates the ischemia-induced elevation of
m
The renin angiotensin system (RAS) plays a very important role in regulating normal physiology and the mechanisms of cardiovascular disease development. Angiotensin converting enzyme (ACE) is an important key enzyme of the classical RAS pathway angiotension converting enzyme-angiotensin II-type 1 (ACE-Ang II-AT1), which converts angiotensin (Ang) I to Ang II, and the increase in Ang II is an important part of the RAS involved in the development of cardiovascular diseases. Another RAS pathway, ACE2-Ang (1-7) MAS, exerts action antagonistic to the classical RAS pathway, and the two pathways are functionally antagonistic to each other, giving the RAS a dual effect [73]. Activation of the ACE2/Ang-(1-7) pathway effectively delays the progression of HF, and changes in the balance between ACE/Ang II and ACE2/Ang (1-7) play an important pathophysiological role in HF failure. Ang (1-7) is a cardioprotective peptide in the RAS that counteracts the cardiotoxic effects of Ang II and has protective effects against pathological cardiac remodeling and HF [74]. The ACE2-Ang (1-7) pathway, known as the second metabolic axis of the RAS, plays a crucial role in cardiovascular disease. Ang (1-7) plays a cardioprotective role and has the potential to contribute to HF treatment.
Advanced glycation end products (AGEs) are compounds produced by the
nonenzymatic reaction of free amino groups in proteins, lipids, and nucleic acids
with reducing sugars (e.g., glucose, fructose, pentose, etc.), i.e., the Maillard
reaction, and they are classified as endogenous or exogenous according to their
sources [75]. The exogenous pathway from food is the main source of AGEs in
humans. Protein-bound AGEs have been most extensively studied, and AGEs affect
downstream HF-related signals, including the proto-oncogene protein p21 (ras),
stress-activated protein kinase (SAPK), the activator of transcription (STAT)
pathway, and several other pathways, by coupling to cell membrane proteins and
altering their structures or directly activating cell surface receptors [76, 77],
promotes ventricular remodeling and myocardial injury during heart failure, and
also participates in inflammation, oxidative stress and apoptosis of myocardial
tissue and autophagy of cardiomyocytes. Gao et al. [78] found
that Receptor of Advanced Glycation Endproducts (RAGE) activation in a mouse model caused
excessive autophagic activity through activation of the RAGE-NF-
The most important exogenous source of AGEs is food, and the way in which food
is cooked is the most important factor affecting the content of AGEs. The
formation of AGEs is accelerated in high-temperature, long and deeply dry cooking
methods (grilling, frying, deep-frying, etc.), so changes in cooking habits and
methods can help to reduce the accumulation of AGEs in the body [79]. Toprak
et al. [80] observed that the mechanism of the diastolic effect of
alagebrium (ALT-711) on carotid arteries in healthy animals is not only to reduce the
cross-linking of AGEs with collagen but that ALT-711 has the ability to improve
the uptake and release of Ca
Due to the difficulties in targeting drugs to receptors and intracellular pathways, many years ago, researchers proposed cardiac gene therapy as an alternative therapeutic approach, whereby cell-targeted delivery of exogenous genes (transgenes) would produce “therapeutic” proteins that could compensate for pathological downregulation or counteract harmful molecular processes. The selection of appropriate targets and the availability of effective gene vectors are the key requirements for the success of this therapeutic approach.
A study published by Leiden’s research team in 1990 provided a history of
cardiac gene therapy. In a landmark study, rat cardiomyocytes were transfected
in vivo by direct injection of plasmid DNA containing the
As research on cardiac gene therapy has continued, a number of directions have
been identified according to which good translational potential could exist. AC6
(adenylate cyclase) catalyzes the conversion of ATP into cyclic adenosine
monophosphate (cAMP), a molecule essential for cardiac function; S100 calcium binding protein A1 (S100A1) is a
protein that regulates sarcoplasmic reticulum Ca
Chinese medicine refers to natural drugs and their processed substitutes that are guided by the theory of Chinese medicine, have a unique theoretical system and application form, used to prevent and treat diseases and have rehabilitation and health care effects, mainly including plant drugs, animal drugs and mineral drugs [17]. Chinese medicine is divided into proprietary Chinese medicine and Chinese herbal medicine according to the processing undergone (Table 1). Chinese herbal medicines are rich in various chemical components, especially compounds that not only contain alkaloids, polysaccharides, glycosides and other effective drug components but are also rich in vitamins, dietary fiber and other nutrients, with a variety of pharmacological effects. The multitarget therapeutic effect of Chinese medicine can effectively avoid the adverse therapeutic effect caused by the defective therapeutic target and the weakened efficacy caused by the defective drug metabolism in the process of Western medicine treatment, and Chinese medicine can also effectively alleviate the clinical symptoms of patients with chronic HF, improve the prognosis of patients and improve quality of life. Chinese medicine monomers, single Chinese medicines and Chinese medicine compounds all have the characteristics of multichannel and multitarget therapy, which can regulate the body as a whole and affect the physiological functions of multiple systems of the body, so the targeted treatment of HF with Chinese medicine is also worthy of exploration [17, 86].
| Proprietary Chinese medicine | Herbal medicine |
| Baoyuan Tang, Danshen Yin, Shengmai San, Taohongsiwu Tang, Tinglidazaoxiefei Tang, Xuefuzhuyu Tang, Zhenwu Tang, Danqi Pill, Fufang danshen Dripping Pill, Shengmai Capsule, Qili Qiangxin Capsule, Qishen Yiqi Dripping Pill, Danhong Injection, Huangqi Injection, Shenmai Injection, Shenfu Injection, Shengmai Injection | Baishao, Baizhu, Bingpian, Chaihu, Chenpi, Chishao, Chuanxiong, Danggui, Danshen, Dazao, Fuling, Fupian, Fuzi, Gancao, Guizhi, Honghua, Hongshen, Huangqi, Jiangxiang, Jiegeng, Maidong, Maimendong, Niuxi, Renshen, Rougui, Sanqi, Shaoyao, Sharen, Shengdihuang, Shengjiang, Shudi, Tanxiang, Taoren, Tingli, Tinglizi, Wuweizi, Xiangjiapi, Yuzhu, Zexie, Zhiqiao |
HF is a complex, multifactorial clinical syndrome with heterogeneity. Despite the disappointing results of current targeted therapy studies and targeted therapy being influenced by many factors, targeted therapy remains a promising and attractive direction for the treatment of HF. The potential factors leading to negative outcomes should be carefully analyzed, and further research and optimization should be promoted to discover new molecular targets with more significant therapeutic potential to exert cardioprotective effects and become new approaches to HF treatment.
LM conceived the study, participated in the design and drafted the manuscript. YLL reviewed relevant literature, collected data for the manuscript, drew diagrams and created tables. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank all the staff in the laboratory.
The authors acknowledge the essential role of the funding of National Natural Science Foundation of Guangxi (2020GXNSFAA297003), National Natural Science Foundation of China (NSFC: 82060072 and 81760073), Project of Liuzhou Science and Technology (2020NBAB0818, 2021CBC0106 and 2021CBC0108), the project of Liuzhou people’s Hospital (LYRGCC202107 and LYRGCC202203), Guangxi Medical and health key discipline construction project and Guangxi Health Commission Key Laboratory of Clinical Biotechnology (Liuzhou People’s Hospital).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.