








1 Department of Physiology and Pharmacology, University of Western Ontario, London, ON N6A 5C1, Canada
2 Institute of Cardiovascular Sciences, Albrechtsen Research Centre, St. Boniface Hospital, and Department of Physiology and Pathophysiology, Rady Faculty of Health Sciences, University of Manitoba, Winnipeg, MB R2H 2A6, Canada
3 Department of Biochemistry, University Alberta, Edmonton, AB T6G 2H7, Canada
§Retired.
Academic Editor: Fabian Sanchis-Gomar
Abstract
The mammalian Na
Keywords
- NHE1 regulation
- NHE1 inhibitors
- cardiac hypertrophy and remodelling
- ischemia/reperfusion injury
- pyrazinoyl guanidine
The ubiquitously expressed mammalian Na
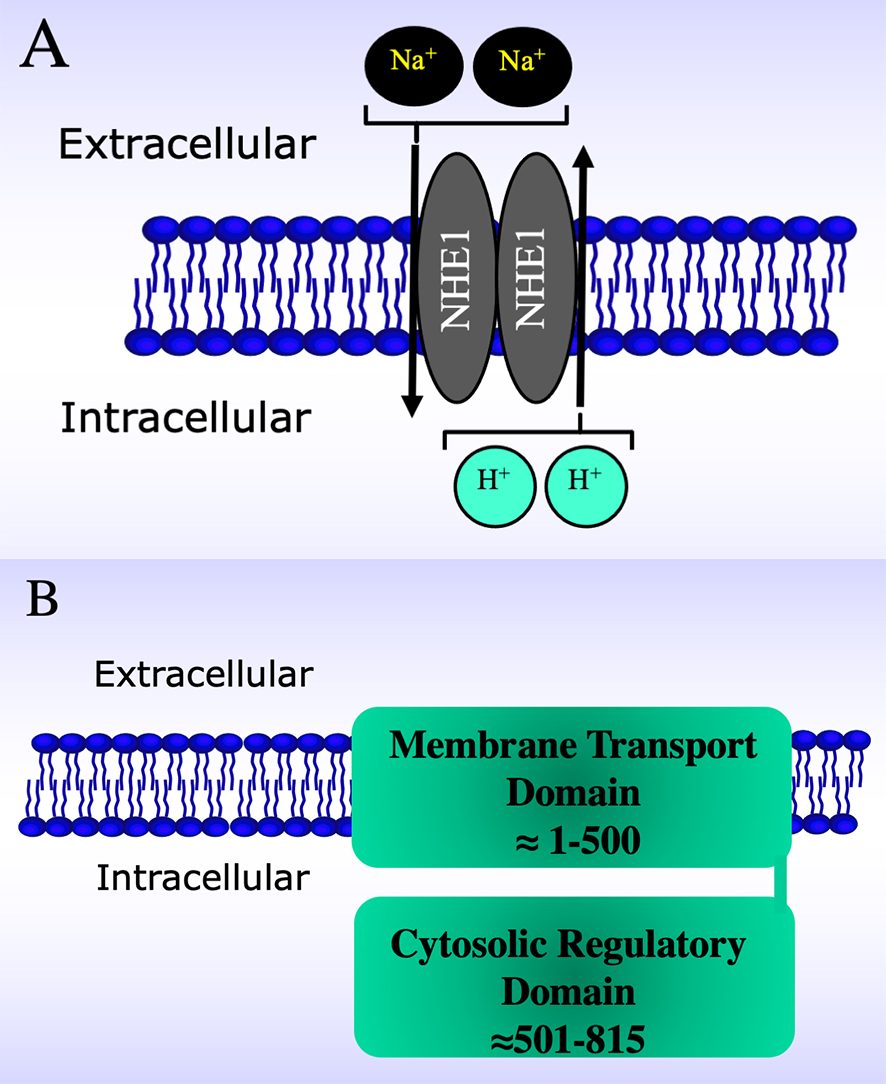 Fig. 1.
Fig. 1.Schematic diagrams of the Na
The physiological and pathological roles of NHE1 are many. Outside of the myocardium NHE1 plays a role in cell growth, proliferation and differentiation [2, 13, 14, 15, 16]. NHE1 is also an important trigger of growth and metastasis in cancer, notably as a trigger of metastasis in breast cancer [17, 18, 19, 20, 21]. Genetic mutations in NHE1 and its absence, have been shown to be responsible for the disease Lichtenstein-Knorr syndrome which manifests itself through many developmental defects and ataxia and hearing loss [22, 23].
In the myocardium, NHE1 is the only plasma membrane isoform of Na
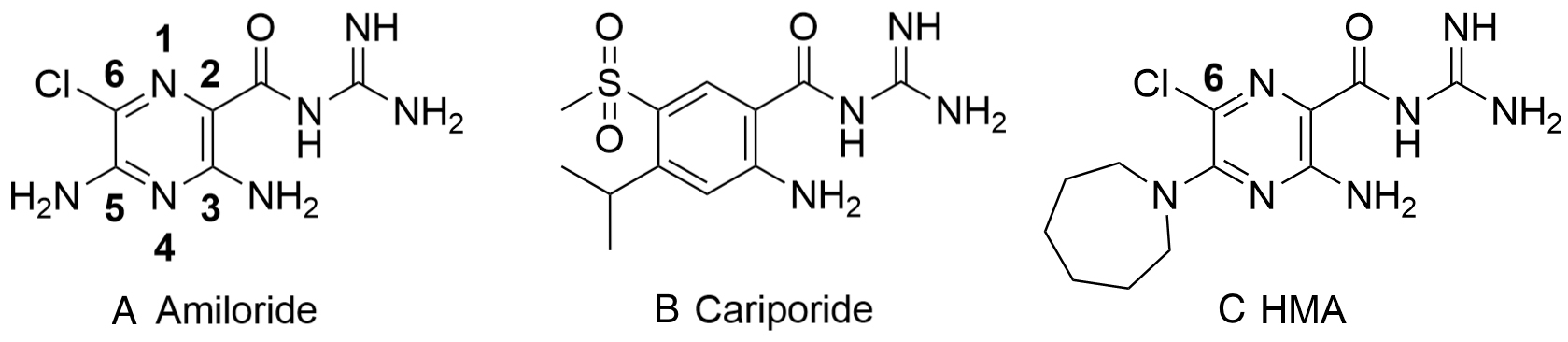 Fig. 2.
Fig. 2.Structure of amiloride, cariporide and hexamethylene amiloride (HMA) (A-C, respectively). Amiloride and HMA are pyrazinoyl guanidines that differ in the aromatic core from cariporide a benzoyl guanidine.
This review presents a discussion of the structure, chemistry and regulation of NHE1 in the myocardium as well as location of inhibitor binding sites and the potential development of novel NHE1 inhibitors. This is followed by assessment of the role of NHE1 in cardiac pathologies including ischemic and reperfusion injury, myocardial hypertrophy and remodeling resulting in heart failure as well as its role in diabetes related cardiac pathology. Finally, we discuss the clinical potential of NHE1 inhibitors to treat heart disease reflecting on completed, ongoing and future clinical trials. Our goal is to both update the field and to stimulate potential development of new inhibitors that can be useful in treating heart disease and indeed other common human afflictions.
NHE1 plays an important physiological role in regulating intracellular pH
(pH
NHE1 is normally quiescent in the myocardium at neutral pH, however the protein
is activated when intracellular pH decreases and is also activated by stimuli
such as growth factors, hormones and osmotic stress. These tend to shift the
activity curve such that the protein is active at more alkaline
pH
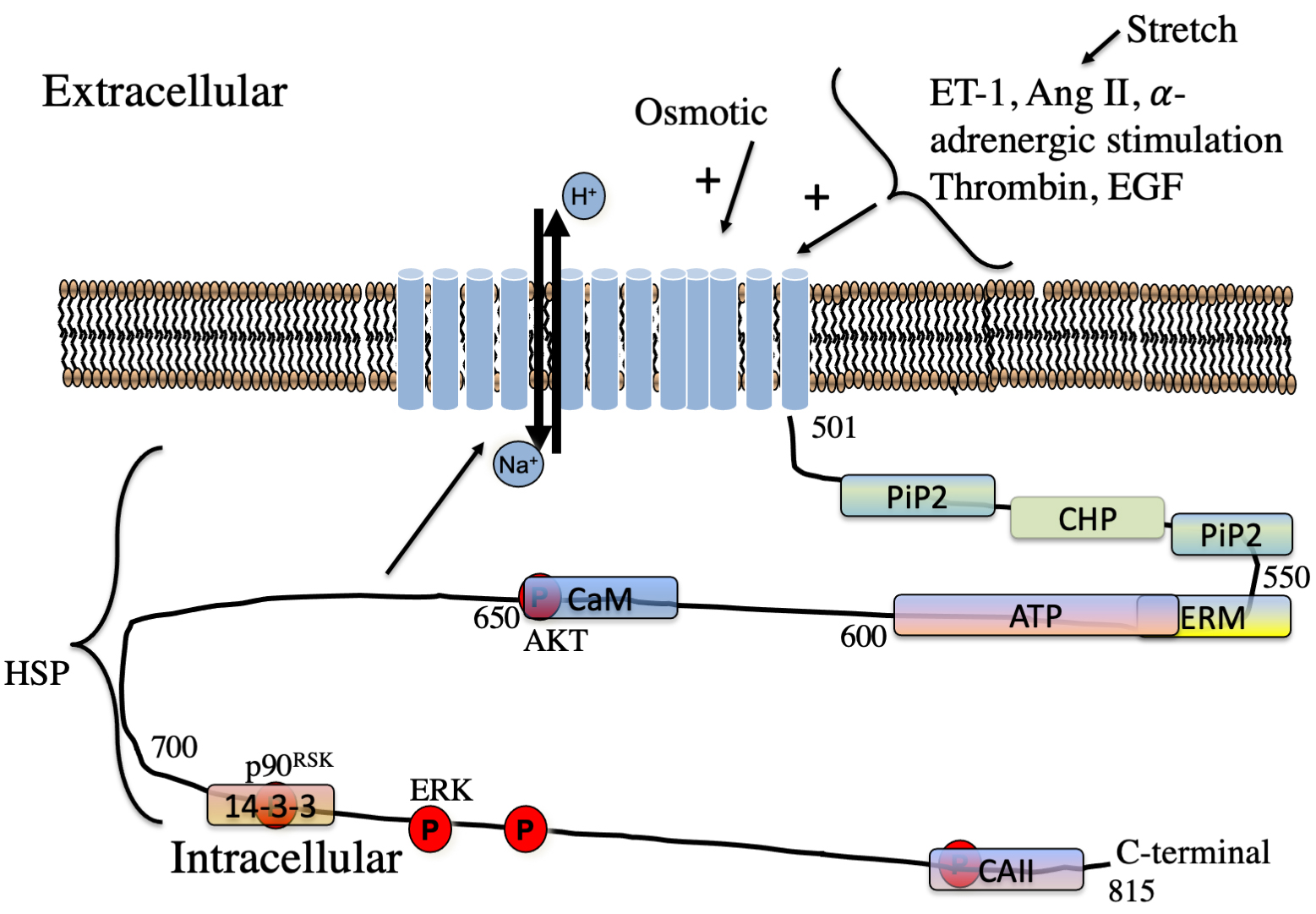 Fig. 3.
Fig. 3.Schematic illustration of regulators of NHE1 in the myocardium. Hormones regulating NHE1 are indicated. The approximate location of lipid,
protein and phosphorylation sites on the cytosolic regulatory tail is indicated.
P, phosphorylation sites. The kinases phosphorylation sites of AKT, ERK and
p90
Hormonal activation of NHE1 occurs at least partially through protein kinase-mediated phosphorylation or through interaction with other regulatory proteins (or lipids). While the exact percentage of activation in the myocardium that occurs through phosphorylation is not known, it has generally been estimated to be 50% of hormonal regulation though this surely varies with cell type (see reviews [12, 44]). A number of different protein kinases phosphorylate the regulatory tail which is thought to occur mainly in the C-terminal 180 amino acids [12, 60]. In brief, phosphorylation-mediated regulation of NHE1 in several tissues was described earlier [4, 10, 12, 44, 78]. Amino acids phosphorylated include Ser648 by Protein kinase B (PKB or Akt) and [79, 80] amino acids Thr718, Ser723, Ser726, Ser729 by p38 MAPK (equivalent human numbering) [81], and see also [4] for review).
The MAPK phosphorylation pathway was identified as being important in NHE1
regulation in the myocardium. This pathway is regulated by many hormones
including endothelin, angiotensin II, catecholamines and some cytokines [82].
This pathway has also been shown to play a role in ischemia reperfusion
activation of NHE1 leading to cardiac injury [83] and this p90
Some other protein kinases also phosphorylate NHE1 though these are less well
studied. Heart
The requisite dephosphorylation of NHE1 protein must occur sometime after
activation of the protein. It is not as well studied in the myocardium. Both
protein phosphatases 1 and protein phosphatase 2A (PP1 and PP2A) directly
associate with NHE1 [98, 99]. Colocalization of NHE1 and PP2A was shown in
ventricular cardiomyocytes [98]. The calcineurin A subunit also binds to NHE1
[100], but its role in dephosphorylation of NHE1 is not yet known. Its binding
may facilitate NHE1-induced translocation of NFAT and myocardial hypertrophy
progression [100]. An interaction between NHE1 and the Src homology 2
domain-containing protein tyrosine phosphatase (SHP-2) has also been confirmed.
Functionally, SHP-2 overexpression caused a higher steady state
pH
It is known that Na
Nitric oxide (NO) has been shown to regulate NHE1 activity in adult ventricular cardiomyocytes in a study where which NO levels were manipulated through various approaches [106]. The response was biphasic such that NHE1 flux was activated by low NO levels but inhibited by high NO amounts. These responses were dependent on two pathways, namely a cGMP-dependent NHE1 activation and a cAMP-dependent inhibition. The protein kinases PKG and PKA were tested for their ability to phosphorylate the NHE1 C-terminus and multiple residues were phosphorylated including Ser648 and Ser703. PKA was more selective for Ser648, possibly accounting for the inhibitory effect of cAMP. The biphasic effect of nitic oxide was specific to adult cardiomyocytes and was not observed in neonatal myocytes or in MDA-MB-468 breast cancer cells [106].
Protein mediated regulation of NHE1 also occurs through the cytosolic regulatory tail. Binding occurs by regulatory proteins, and also by other proteins that may be using NHE1 as a scaffold for other cellular functions aside from regulation of NHE1 activity. This regulatory mechanism has been suggested to account for 50% of the regulation of NHE1 (see reviews [12, 44]) though this is clearly difficult to quantitate and will surely vary with cell type. Most of this type of regulation has been studied in non-myocardial tissue and is briefly reviewed. The “scaffolding” of proteins may vary in response to cellular stimuli [107]. As noted above, protein phosphatases bind to the NHE1 tail and facilitate de-phosphorylation of the tail and have other physiological consequences [100]. In “opposition” to the phosphatases are kinases which also bind to the NHE1 tail. NHE1 acts as a scaffold for ERK and Raf [108]. ERK binds to the cytosolic domain at specific D-domain and F-sites binding sites. These binding sites, and the interaction of NHE1 with ERK affect not only the phosphorylation and activation of NHE1 but also the regulation and activation of ERK itself [96]. Direct binding of ERK to NHE in the myocardium has not, to our knowledge, been demonstrated.
Other interaction partners of NHE1 were reviewed [4] for all general tissues and will be briefly summarized. Several studies have also examined the “interactome” of the regulatory NHE1 cytosolic tail, describing protein that bind to the NHE1 C-terminus from the kidney [109] and from breast cancer cells [110]. Results from some of these are outlined below.
The calcium-binding second messenger protein known as calmodulin mediates
Ca
There are several isoforms of Calcineurin B homologous proteins (CHPs, CHP1,
CHP2 and CHP3) [112, 113, 114, 115]. These are Ca
Ezrin, radixin and moesin, the ERM family form links between NHE1 and actin filaments of the cytoskeleton [120, 121]. This linkage helps facilitate cell migration [122]. NHE1 has ERM binding motifs in amino acids 552-560 of its cytoplasmic tail [121]. While not a great deal has been studied with regards to ERM proteins and the myocardium, one study [123] examined the intracellular location of ERM proteins in left ventricular cardiomyocytes and found that they were localized predominantly at the intercalated disc regions. With intracellular acidification this localization changed, with more localization of activated phospho-ERM in the transverse tubules, which is where NHE1 was localized. This effect was blocked with the NHE1 inhibitor cariporide. These results suggested that ERM proteins may mediate at least some of NHE1 activation in the myocardium.
Heat shock proteins, both Hsp70 and Hsp90, have been shown to be associated with NHE1 in several studies [101, 109, 110, 124] and inhibition of heat shock proteins can affect NHE1 activity [109, 125]. Hsp90 may affect NHE1 function through alteration of phosphorylation of the protein via AKT kinase [109]. A role of the association in inflammatory response has been suggested [126] and though the association has been shown in different tissues such as kidney and breast cancer cells [109, 110], it has not been extensively studied in the myocardium. One study has examined cardiofibroblasts [127] and showed the NHE1 interacted with Hsp70 by immunoprecipitation.
Having an association with a membrane protein which moves ions such as those
which CAII or other proteins produce, is thought to facilitate transport of the
ions and is called a membrane transport metabolon [128]. CAII is a protein that
catalyzes the hydration of carbon dioxide which leads to production of
bicarbonate ions and protons. It occurs in a metabolon with NHE1 and other
membrane ion transport proteins [128, 129]. A CAII-NHE1 interaction of this type
has been studied in some detail in the myocardium. Initial studies characterized
the fundamentals of the interaction and demonstrated that CAII binds to NHE1
in vivo, at the penultimate 13 amino acids of the regulatory
cytosolic tail (Fig. 3). Ser796 and Asp797 form part of the CAII binding site on
NHE1. The association of NHE1 and CAII was dependent on NHE1 being phosphorylated
upstream of the CAII binding site [130, 131]. The association of these two
proteins was then studied in the myocardium when varying myocardial stretch.
Stretch is known to activate NHE1 (see Section 3.2.11) so the association of NHE1
with CAII was examined following stretch of rat papillary muscle by
co-immunoprecipitation of the two proteins. Stretch increased association of NHE1
and CAII and inhibition of p90
Phosphatidylinositol 4, 5-bisphosphate is a different binding cofactor of NHE1,
being a lipid and not a protein. It binds in two cationic juxtamembrane binding
regions of NHE1, at amino acids 513-520 and 556–564 of the rat protein which are
equal to amino acids 509-516 and 552-560 of human NHE1 [134]. Mutation of these
binding sites decreases NHE1 transport efficiency [134]. The second region
overlaps with a region between amino acids 542-598 which is called a lipid
interacting domain, with a hydrophobic sequence
NHE1 is an ATP binding protein. Early studies showed that depletion of
intracellular ATP levels inhibits NHE1 activity [139, 140, 141, 142, 143]. More recently, direct
binding of ATP to the NHE1 cytosolic domain was demonstrated by photoaffinity
labeling and equilibrium dialysis. The location of ATP binding was localized to
amino acids Gly542-Pro598 of human NHE1 (Fig. 3). ATP binding affected the pH
dependence of NHE1 activity, ATP depletion caused an acidic shift in the
pH
Stretch enhances myocardial contractility by two mechanisms. One rapid mechanism
is the classic Frank-Starling mechanism that is attributed to enhanced
myofilament calcium responsiveness. The second mechanism is the “slow force
response” which occurs more slowly, as its name suggests. It is due to an
increase in calcium transient size as a consequence of stretch activating
autocrine and paracrine mechanisms [148, 149]. It is in the slow force response
that NHE makes a significant contribution. Knockdown or inhibition of NHE1 blunts
the slow force response [150, 151]. The mechanism by which this response works
has been studied in several authors and was reviewed earlier [67, 148]. Briefly,
stretch caused release of Angiotensin II, and activation of the AT1 receptor.
This results in formation and release of endothelin which causes NHE1
hyperactivity. The increased NHE1 activity causes an increase in intracellular
sodium and results in an increase in intracellular calcium though the reversal
activity of the Na
Relatively little research has been carried out to identify the impact diet has on NHE1 function and expression. Generally, high fat diets have been shown to increase oxidative stress and heart dysfunction. This may be associated with an activation of NHE1 because long term ablation of NHE1 activity via the use of NHE1-null mice mitigated the deleterious cardiac effects of the high fat diet [157]. However, perhaps the most powerful dietary intervention that demonstrated NHE-1 inhibition was provided by ginseng, a widely used medicinal herb particularly in Asian societies. Ginseng provided a direct anti-hypertrophic action in cultured cardiomyocytes, and importantly, inhibited heart failure through an attenuation of the upregulation of NHE1 activity typically exhibited in hypertrophic responses [158].
The mechanism whereby diet alters NHE1 activity is unclear. However, one may
speculate that the membrane lipid composition which influences related membrane
transporters like the Na
The NHE1 gene has been cloned from several species including the human, porcine and rabbit forms of the promoter [165, 166, 167]. The human gene has 12 exons and 11 introns and the mouse gene has a similar design [168]. There is a very large (41.5 kb) intron between the first and second exon while the other introns are much smaller [169]. Studies in vitro and in transgenic mice with the 5’ flanking region of the mouse promoter showed that NHE1 expression in the myocardium is high during early embryonic development and showed that the NHE1 protein is at relatively high levels in the neonate and declines with age [58, 59, 170, 171]. This effect may be comparable to the myosin heavy chain where a switch to fetal type of gene expression during hypertrophy increases expression.
A number of studies characterized transcription factors and regions important in
expression in tissues outside the myocardium AP-1. The C/EBP family have been
shown to be important in some cell types [172, 173]. The porcine, rabbit and
mouse and human NHE1 promoter, are homologous particularly in the proximal 500 bp
of the 5’ flanking region [165, 166, 167, 169]. Consensus sequences for the
transcription factors AP-1, C/EBP and Sp1 are conserved between pig and human
[167]. A proximal AP-2 binding site in the mouse NHE1 promoter is important in
expression in fibroblasts and in P19 embryonal carcinoma cells [57, 165]. The
NHE1 promoter is activated in some models of cell differentiation including in
P19 cells or in L6 muscle cells [174, 175]. Some other regions of the promoter
important in expression are a poly (dA:dT) region of the NHE1 promoter is located
at bp -155 to -169 of the mouse gene which was tested in L6 and NIH 3T3 cells
[176]. Several other regions of the NHE1 promoter were studied in a variety of
cells. COUP-TF (Chicken ovalbumin upstream promoter transcription factor) type I
and II is more distal (-841 to -800 bp) and is also important in expression
[177]. Thyroid hormone receptor TR
Studies directly examining NHE1 promoter regulation in the myocardium are rare. A 1.1 kb region of the mouse promoter drove expression in cardiomyocytes that was stimulated by serum. Deletion of a proximal AP-2 site decreased promoter activity 4-fold [179]. Mutation of that site, combined with deletion of distal regions of the promoter, virtually eliminated promoter activity in cardiomyocytes. DNase I footprinting analysis showed that the poly(dA:dT) rich region (-155 to -169), is protected by heart nuclear extracts as is the COUP-TF element [176, 180]. Thyroid hormone may regulate the NHE1 gene in the heart. Treatment of heart cells with thyroid hormone increased protein binding of nuclear extracts in the COUP-TF region and treatment of cardiomyocytes with thyroid hormone increases NHE1 protein expression [178].
NHE1 levels have been shown to be elevated in the myocardium following ischemic
heart disease and some of this could be mediated through reactive oxygen species
(ROS). Increasing serum from 0.5 to 10% induces NHE1 promoter activity in NIH3T3
fibroblasts [179]. The increase correlates with increased O
Aside from these earlier studies, there has been a surprising dearth of recent work on the promoter in the myocardium. It would be interesting to examine NHE1 transcription in detail during cardiac hypertrophy and in response to ischemia and reperfusion.
The structure of NHE1 and the inhibitor binding site is of paramount interest as clearly the binding site will affect the efficacy and potency of inhibitors and the site itself will affect the design of novel inhibitors. Traditionally, human NHE1 has been thought of as a 12 transmembrane protein with a large cytoplasmic tail (Fig. 4A, Ref. [182, 183, 184]). This type of low-resolution model was initially based on hydrophobicity plots and later more experimental evidence was added, mostly using cysteine accessibility studies along with hydrophobicity plot [182, 183]. These studies initially suggested 12 transmembrane segments were present with two intracellular re-entrant loops, one between TM4 and TM5 and one between TM8 and TM9. There was a larger extracellular loop predicted between TM9 and T10.
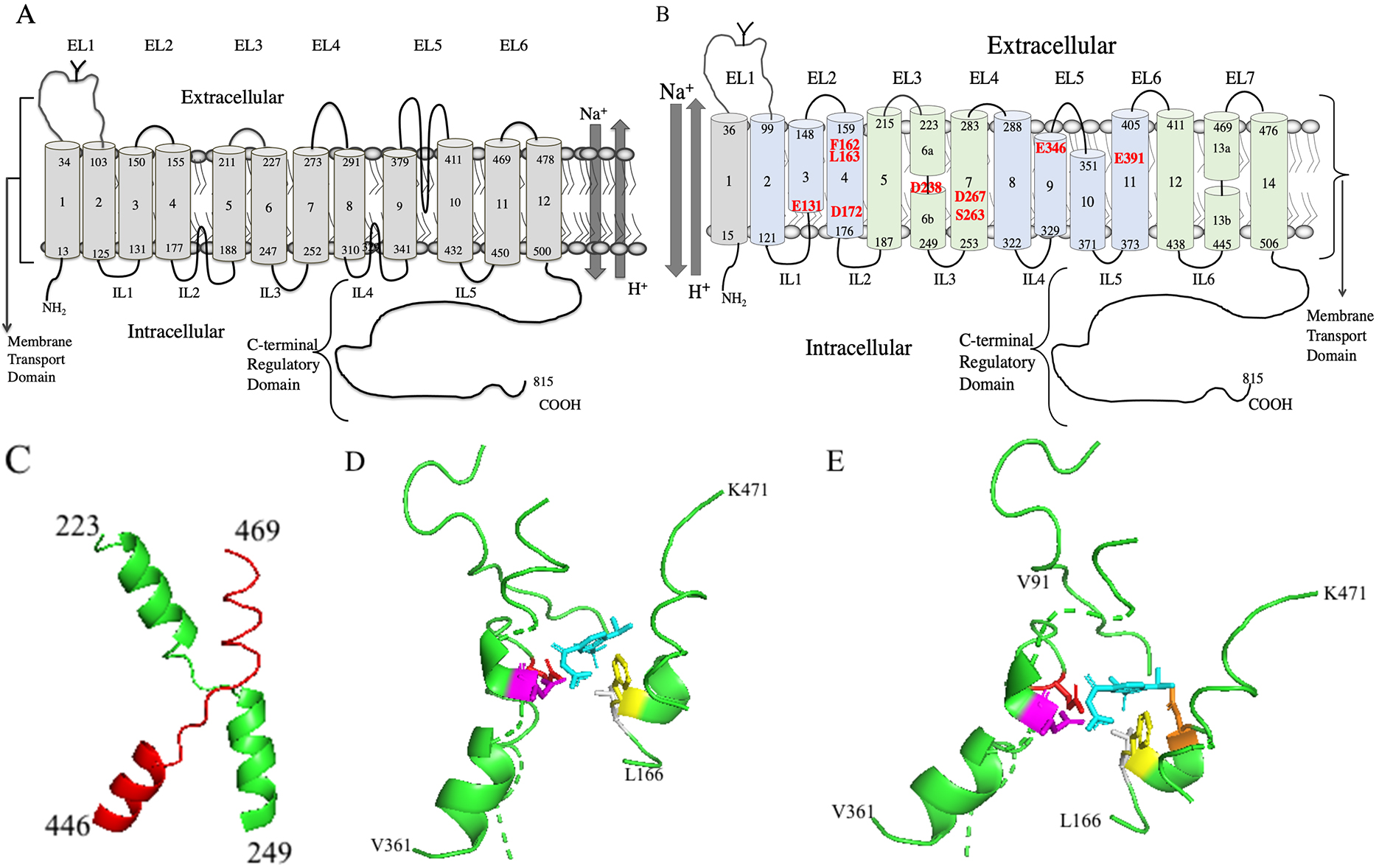 Fig. 4.
Fig. 4.Molecular models of human NHE1. (A) Two-dimensional topology model of human NHE1 based on hydrophobicity analysis and cysteine accessibility studies [182, 183]. (B) Novel two-dimensional topology model of human NHE1. TM’s 2-14 are based on the Cryo-EM structure [11]. TM1 is added as per [182, 183, 184]. The core domain TMs are colored light green and the dimerization domains TMs are light blue. Amino acid numbering indicates the end of the transmembrane segments. EL, extracellular loop; IL, intracellular loop. The transmembrane segment number is indicated beginning from the N-terminus and including the first TM. The unwound region of TMs 6 and 13 is indicated by a discontinuity. Amino acids important in cation coordination, proton sensing or inhibitor binding are indicated in red. (C) Structure of human NHE1 fold from [11]. TM 6 (green) and 13 (red) of human NHE1 are shown (amino acids 223-249 and 445-469 respectively). (D) Lateral view of cariporide binding site in the Cryo-EM structure [11] of human NHE1. Most of the protein is not shown. Cariporide cyan, E346 red, D267 magenta, F162 yellow, L163 grey. (E) Lateral/extracellular view, color scheme retained as in D, plus D159 orange.
The structure of the E. coli NhaA (Na
Structures of other Na
Very recently, the structure of human NHE1 in complex with the regulatory protein CHP1, was determined by cryogenic electron microscopy (Cryo EM) [11]. The structure was of amino acids 87-590, which formed 13 transmembrane helices and 3 cytosolic helices. The topology was generally consistent with previous models except a previous re-entrant loop between a previously labeled TM9 and TM10, is actually two TM helices. A simplified model of the topology is shown in Fig. 4B. The model is based on the Cryo EM structure of human NHE1. TM 1 was added based on an earlier molecular model of NHE1 and experiments involving cysteine labeling of TM 1 which showed that TM 1 was present in the intact protein [182, 183, 184] (Numbering of NHE1 TMs will from here on include TM1 as shown in Fig. 4B.)
NHE1 was a homodimer in the Cryo EM structure. Each monomer was made of a dimerization domain of TM segments (TMs 2-4, and 8-11, amino acids 99-176, plus 288-405) plus a core domain (TMs 5-7 and 12-14, amino acids 187-283 and 411-505) [11] (see Fig. 4B). TMs 6 and 13 (amino acids 223-253 and 445-469) have the NHE-fold with an unwound region in the middle and they cross each other (Fig. 4C). The helix breaks are thought to participate in forming the ion permeation pathway. In their structure there was a funnel between the dimerization domain and the core domain of each protomer. The funnel is formed by TMs 2,3,4,6,7 and 11. Proton-titratable residues in the funnel are E131, D172, D238, D267 and E391 (indicated in Fig. 4B). This results in a negatively charged cavity which was thought to participate in cation binding and proton sensing [11]. D267 was thought to participate in cation binding and is critical to activity [204, 205]. The S263 sidechain was also thought to participate in ion transport and D238 was thought to indirectly participate, coordinating a water molecule [11]. E391 is essential [204] but because of its location away from the cation binding site it may affect folding or stability of the protein and was not thought to directly binding cations. E131 was thought to be a pH sensor functioning when protonated to accelerate cation release [11]. Mutation of F162 has been shown to have a large effect, reducing efficacy of cariporide and mutation of I169 and I170 accentuate this effect [206]. Mutation of E346 has also been shown to have large effects on inhibitor efficacy [207, 208].
Considering the potential benefits of NHE1 inhibition in cardiac disease, there has always been great interest in the determination of the exact site of inhibitor binding. Classically, a number of studies carried out site specific mutagenesis on amino acids believed to be involved in inhibitor binding. Then any changes in inhibitor efficacy were examined. Some of these studies have suggested a number of amino acids with large effects on inhibitor potency as being important in human NHE1 inhibition that includes human F167 [209], L167 (rat) [208], Gly174, Leu163 [210] while other studies have showed smaller changes in drug potency with mutation of human L265 and L255 [211]. Other studies [208, 212, 213] used large scale replacement of pieces of NHE1 to try to determine which regions are critical in drug interaction, but these have the disadvantage of increased likelihood of disruption of the structure of NHE1. Molecular modeling of NHE1 and inhibitor docking was also attempted [184] and a number of amino acids were suggested to interact with inhibitors at different potential binding sites.
While these studies provided interesting information, the recent Cryo-EM structure directly showed the inhibitor binding site of the benzoylguanidine cariporide [11]. Cariporide bound from the extracellular side of NHE1 in a pocket located between the dimerization and core domain. It was surrounded by TMs, 4, 7, 9, 10 and 13. The extracellular loop #1 from the opposing subunit, and resides of TM 9 and 10 may be essential for the binding pocket. Amino acid D267 of TM7 points up into the inhibitor binding pocket and interacts with the positively charged group of the guanidine (Fig. 4D,E) [11]. The side chain of F162 (TM4) also interacts with the guanidine group and the phenyl ring of cariporide which agrees with mutational studies [206]. The guanidine group is also coordinated by the side chain of E346 (TM9) which agrees with another mutational studies [207]. Cariporide also has a methylsulfonyl group at the meta- and para- position of the phenyl ring (Fig. 2B). These are buried in a sub pocket formed by D159, L163, D95, H98 and V99. Mutational studies, including of L163, support the importance of these amino acids in inhibitor binding [209].
It is also important to note that it has long been suggested that inhibitors
such as amiloride, bind to the same or are overlapping with, the cation binding
site (reviewed in [214]). The Cryo-EM structure of NHE1 could not visualize the
precise location of the Na
Because of the extensive experimental evidence that NHE1 participates in cardiac pathologies including ischemia and reperfusion injury as well as myocardial remodeling and heart failure (discussed in Sections 6 and 7) there has been substantial interest in inhibition of NHE1 and in the development of clinically useful compounds for treatment of the diseased myocardium. Early pharmacological studies probing the function of NHE1 in biological systems including heart disease employed the use of amiloride (Fig. 2A), a potassium-sparing diuretic or its derivatives such as ethylisopropylamiloride (EIPA), methylisobutyl amiloride (MIA), hexamethyl amiloride (HMA), dimethylamiloride (DMA) (Fig. 2C) and others. While these agents are effective in inhibiting NHE1, they lack specificity against the NHE1 isoform, have non-specific effects on many aspects of cardiac performance [216] and are also effective against other ion regulatory systems. This concern regarding non-specificity was rectified to a large degree by the development of novel second generation NHE-1 specific inhibitors by the pharmaceutical industry. The first among these was the benzoylguanidine derivative HOE694 ((3-methylsulphonyl-4-piperidino-benzoyl) guanidine methanesulphonate developed by Hoechst AG (now part of Sanofi) and which was demonstrated to exert cardioprotective and antiarrhythmic effects in ischemic and reperfused hearts [32]. This was followed by the development of a new NHE1-specific inhibitor initially designated as HOE642 (N-(Aminoiminomethyl)-4-(1-methylethyl)-3-(methylsulfonyl)-benzamide) [31] and subsequently renamed as cariporide (Fig. 2B) in preparation for clinical development. As will be evident from the discussion below, cariporide is the most extensively studied of the NHE1-specific inhibitors both experimentally as well as in clinical studies particularly with respect to mitigating ischemic and reperfusion injury.
The development of these benzoyl guanidine derivatives has led to the rapid formulation of numerous newer and more potent NHE1-specific inhibitors. For an in-depth description of the development and chemistry of NHE1-specific inhibitors please refer to [217]. A partial list of these agents is provided in Table 1 (Ref. [32, 218, 219, 220, 221, 222, 223, 224, 225]). Virtually all of the agents listed in this table possess the monocyclic acylguanidine structure found in the amiloride-based inhibitors (with the exception of SL 59.1227) but demonstrate markedly enhanced specificity towards NHE1 as well as greater potency. Preclinical and clinical studies with a number of these agents are discussed in greater detail below.
| Drug | Chemical name | Developer | Reference |
| HOE694 | (3-methylsulphonyl-4- piperidino -benzoyl) guanidine methanesulphonate | Hoechst AG |
[32] |
| HOE642 |
(N-(Aminoiminomethyl)-4-(1-methylethyl)-3-(methylsulfonyl) -benzamide) | Hoechst AG | [218] |
| MS31-038 | 2-phenyl-8-(2-methoxyethoxy)-4-quinolyl carbonylguanidine bismethanesulfonate | Mitsui | [219] |
| EMD87580 |
N-carbamimidoyl-2-methyl-4,5-bis(methylsulfonyl) benzamide | Merck KGaA |
[220] |
| EMD85131 |
(2-Methyl-5-(methylsulfonyl)benzoyl)guanidine | Merck KGaA | [221] |
| T162559 | ((5E,7S)-[7-(5-fluoro-2-methylphenyl)-4-methyl-7,8-dihydro-5(6H)-quinolinylideneamino] guanidine dimethanesulfonate) | Takeda | [222] |
| BIX | N-[4-(1-acetyl-piperidin-4-yl)-3-trifluoromethyl-benzoyl]-guanidine | BI |
[223] |
| SL59.1227 | 3-[(cyclopropylcarbonyl)amino]-N-[2-(dimethylamino)ethyl]-4-[4-(5-methyl-1H-imidazol-4-yl)piperidin-1-yl]benzamide | Sanofi | [224] |
| Zoniporide | [1-(quinolin-5-yl)-5-cyclopropyl-1H-pyrazole-4-carbonyl] guanidine | Pfizer | [225] |
Recently one group has developed a novel series of compounds of 6-substituted
amiloride and hexamethylene amiloride (Fig. 2C) derivatives as inhibitors of
NHE1. Depending on the precise compound made, they may also inhibit human
urokinase plasminogen activator [226, 227, 228]. The reasoning behind modification of
amiloride (and its derivative) for use as an inhibitor is that amiloride is a
pyrazinoyl guanidine, a clinically safe compound used as a potassium-sparing
diuretic. Given the problems the benzoyl guanidine derivative cariporide for
treatment of heart failure (see below) it was reasoned that using a pyrazinoyl
guanidine core that differs in the aromatic core from cariporide (a benzoyl
guanidine, Fig. 2) and related NHE1 inhibitors might be a prudent approach to
avoid side effects and regulatory issues for drug development. 6-substituted
amiloride/HMA compounds have increased potency towards NHE1. Amiloride is an
ideal candidate for a scaffold to build a dual-targeting compound with
cardiovascular beneficial properties. HMA is both significantly more potent and
more specific towards NHE1; amiloride inhibits sodium channels but HMA much less
so [229]. Inhibitors have been developed with nM potency towards NHE1 and show no
effects on diuresis or urinary Na
It is of note that some of these compounds have a dual action, inhibiting urokinase plasminogen activator (uPA) [226, 227, 230]. In the myocardium, uPA induces cardiac fibrosis and human hearts with end stage failure and fibrosis have elevated plasminogen activator activity [231]. Excess uPA promotes cardiac fibrosis in association with M2 macrophages [232]. Blocking the uPA pathway reduces cardiac macrophage accumulation, excess collagen formation and heart fibrosis [233]. Thus, it may also be of interest to test the effect of dual inhibition of NHE1 and uPA, in heart failure models. To our knowledge this has not been done. Caution is advised though, as urokinase inhibition could increase thrombolysis and thrombosis. Fortunately, a large number of NHE1 inhibitory compounds have been made with varying degrees of uPA inhibition from none, to little, to potent inhibitors of both uPA and NHE1 [226, 227, 230].
Although the entry of excessive extracellular Ca
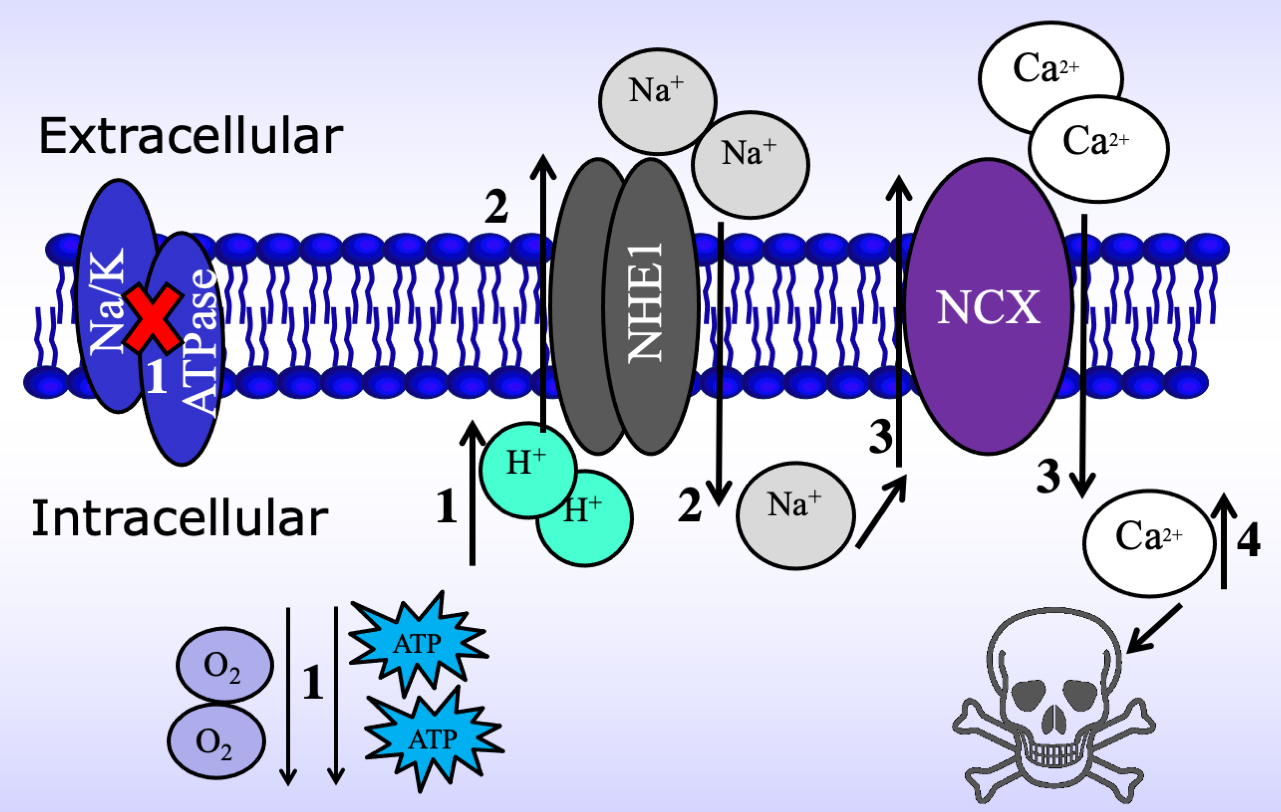 Fig. 5.
Fig. 5.Schematic of the mechanism by which the Na
Karmazyn’s laboratory was the first to demonstrate the likely validity of this
concept by demonstrating a cardioprotective effect of amiloride, in isolated
perfused rat hearts subjected to ischemia and reperfusion [240]. This seminal
observation using a well-known inhibitor of Na
A biochemical approach further indirectly implicated the NHE pathway in the
cardiac damage by modifying the extracellular acidity and Na
All of these studies were consistent with a critical involvement of NHE in the
cardiac damage [240, 241, 253, 254], cardiac contractile dysfunction [240, 241, 243], and arrhythmias [255, 256] that are evident during the ischemia and
reperfusion insult. NHE inhibitors were cardioprotective not only during ischemia
and reperfusion challenge but protected the myocardium during
hypoxia/reoxygenation insult [257], hypothermic conditions [258, 259], oxidative
stress [260, 261], and pacing induced-heart failure [262]. Conversely, NHE
inhibitors did not provide cardioprotection during ischemic pre-conditioning
[259] or the Ca
Earlier studies using amiloride or its analogues as cardioprotective agents presented some difficulty in interpretation as they lacked specificity in terms of targeting NHE1. Moreover, virtually all studies with these agents were carried out using in vitro cardiac preparations with no studies on more clinically-relevant in vivo animal models. This situation was rectified to large degree by the development of highly specific NHE1 inhibitors as discussed in section 5. The first of these agents to be developed was the benzoyl guanidine derivative HOE 694 which was shown to produce extensive protection in both isolated rat hearts as well as in rats subjected to coronary artery ligation [32]. Although HOE 694 was not destined for clinical development this was not the case for HOE 642 (cariporide) (Fig. 2B), a more potent NHE1 inhibitor, developed shortly thereafter [218] and which, in an initial study was shown to exert potent cardioprotective effects in both isolated hearts as well as in vivo coronary artery ligation followed by reperfusion [31]. This protection was manifested in terms of reduction in the incidence of arrhythmias, reduction in tissue injury as well as preservation of energy metabolites [31]. Cariporide subsequently emerged as the most widely studied of the newer generation of NHE1 inhibitors as a cardioprotective strategy. Indeed, these studies using cariporide have shown excellent cardioprotection across different animal species and experimental models [253, 254, 263, 264, 265, 266, 267].
The consistent protection seen with cariporide was clearly evident in subsequent studies utilizing newer NHE1 specific inhibitors including AVE 4890 [268], MS-31-038 [219], BIX [223], BIIB 513 [269], EMD 85131 [221] and others. Indeed, it can be safely stated that virtually all animal studies using these agents have consistently demonstrated excellent cardioprotection irrespective of experimental model or animal species. This consistency in demonstrating cardioprotective efficacy of NHE1 inhibitors is likely unmatched by any other strategy. Indeed, Garrett Gross’ laboratory at the Medical College of Wisconsin has reported that NHE1 inhibition with BIIB 513 was superior to ischemic preconditioning particularly when ischemic preconditioning was no longer effective as a result of prolonged ischemic duration [221].
A number of studies were undertaken to determine whether genetic modulation of NHE1 alters the cardiac response to ischemia and reperfusion. Surprisingly, overexpression of NHE1 in transgenic mouse hearts resulted in enhanced recovery after reperfusion of ischemic isolated perfused hearts which the authors attributed to improved metabolic parameters [270, 271]. Ironically, in one study the protection seen with NHE1 overexpression was similarly observed with cariporide [271]. In another study, cardiac overexpression of NHE1 resulted in modest protection against ischemia and reperfusion in vivo although this was not affected by zoniporide, a highly specific NHE1 inhibitor suggesting that the protection was not NHE1 dependent [272]. With aging, NHE1 overexpressing mice exhibited increased apoptosis, left ventricular contractile dysfunction, myocardial remodeling and premature death which the authors attributed to sustained endoplasmic reticulum stress [272]. While further work is required in this area, when taken together the results suggest that NHE1 overexpression per se may not be necessarily deleterious to the ischemic myocardium although inhibiting the exchanger to a critical level confers protection. This concept is reinforced by the finding that genetic ablation of NHE1 in mice confers protection against ischemic and reperfusion injury in isolated perfused hearts [273].
Successful cardiac resuscitation, particularly after out-of-hospital sudden cardiac arrest, represents a major medical challenge. This reflects the very poor outcome in terms of successful resuscitation which is seen in only about 10% or less of cardiac arrest victims, although these rates vary depending on various factors [274]. Cardiac resuscitation is in essence an ischemia (cardiac arrest) and reperfusion (resuscitation) scenario thus suggesting a potential benefit of cardioprotective agents in terms of improving resuscitation efforts. Indeed, NHE1 inhibitors have been proven to be of immense benefit for improving post-cardiac arrest resuscitation in experimental animal models. Much of the original and pioneering work in this area comes from Gazmuri’s laboratory in Chicago who first proposed this concept (see below) and who recently published a comprehensive review of the role of NHE1 in cardiac resuscitation [275]. As such only a brief discussion is presented here.
As just noted, the first report demonstrating a beneficial effect of NHE1 inhibition was presented by Gazmuri and colleagues who showed that cariporide improved post ventricular fibrillation recovery in isolated rat hearts as well as rats in vivo subjected to cardiac arrest [276]. It is interesting that with respect to the latter, cariporide reduced the degree of chest compression required to attain sinus rhythm and precluded the necessity for electrical defibrillation in six of eight animals studied whereas electrical defibrillation was required in all 8 control rats [276]. Moreover, and importantly, over 90% of cariporide treated animals survived the cardiac arrest-resuscitation protocol compared to just over 60% seen in control animals [276]. A similar beneficial effect of cariporide was also convincingly demonstrated by these and other investigators using a pig model of cardiac arrest followed by attempted resuscitation [277, 278]. Thus, cariporide improved hemodynamics following resuscitation compared to control animals, completely prevented mortality while reducing neurological deficit [278].
From a mechanistic perspective, the beneficial effects of NHE1 inhibition in improving cardiac resuscitation appear to be similar to mechanisms seen in classic cardioprotection seen in ischemic and reperfused hearts with NHE1 inhibitors. Thus, it has been reported that the NHE1 specific inhibitor AVE4454B, although less effective than cariporide in terms of improving cardiac resuscitation [279], improved cardiac resuscitation in the rat which was associated with diminished cardiac mitochondrial calcium overload [280]. The beneficial effects of cariporide were also associated with diminished arrhythmogenesis including ectopic activity and reduced the shortening of the action potential duration associated with resuscitation [277, 281]. Further mechanistic insights into the improved resuscitation produced by NHE1 inhibitors were provided in a study using the NHE1 specific inhibitor zoniporide. In this regard zoniporide, as expected, improved ventricular recovery following ventricular fibrillation-induced cardiac arrest in pigs, effects associated with improved myocardial metabolic status including preserved myocardial creatine phosphate to creatine ratios thus indicating improved oxidative phosphorylation [282]. Mitochondrial as well as neuronal protection with cariporide was also demonstrated in an asphyxia model of cardiac arrest in the rat although the protection was evidently dependent on cariporide dose [283]. Thus, while a protective response was seen using either 1 or 3 mg/kg cariporide no protection was evident when the cariporide dose was increased to 5 mg/kg [283]. The protective effect of the two lower cariporide doses was enhanced under hypothermic resuscitation conditions whereas no additional benefit was observed with 5 mg/kg cariporide [283]. Taken together, the latter results suggest that the optimal beneficial effect of NHE1 inhibitors in cardiac resuscitation may be dependent on various factors including drug dose and resuscitation conditions.
Finally, it should be added that a contribution to the beneficial effect of cariporide in cardiac resuscitation may reflect improved hemodynamics, particularly in conjunction with the initial chest compression. Thus, Gazmuri’s group has shown, using a rat closed chest ventricular fibrillation model, that cariporide enhances hemodynamic efficacy of resuscitation by producing comparable or higher systemic as well as regional blood flows while at the same time reducing depth of compression, a finding of substantial clinical relevance in terms of improving efficacy of closed chest cardiopulmonary resuscitation [284].
The role of sodium in the development of cardiac hypertrophy is well
established. High dietary sodium is associated with an increased incidence of
cardiac hypertrophy and heart failure. Indeed, heart failure patients exhibit
defective sodium cellular regulation which could compound the deleterious effects
of sodium on cardiac pathology [285]. Although one of the mechanisms associated
with sodium-induced cardiac hypertrophy involves the secondary response to
chronic hypertension, substantial evidence supports a direct effect of sodium on
myocardial hypertrophy thus directly contributing to myocardial remodeling and
the development of heart failure. Thus, elevations in sodium concentrations
directly produce hypertrophy in cultured myocardial myoblasts [286] and
administering a high sodium diet to rats produces cardiac hypertrophy
independently of blood pressure elevation [287]. Moreover, the sodium channel
blocker tetrodotoxin prevents isoproterenol-induced hypertrophy in cultured H9c2
cardiomyoblasts [288]. One of the multiple sodium-dependent direct mechanisms
proposed to induce cardiac hypertrophy involves the elevation of intracellular
calcium concentrations due to reduced calcium efflux via the NCX or
reverse mode NCX activity [289]. This mechanism is supported by studies showing
that selective chronic inhibition of NCX inhibits cardiac hypertrophy in
nephrectomized hypertensive rats, a model of heart failure with preserved
ejection fraction, independently of blood pressure reduction [290]. However,
elevations in intracellular sodium concentrations can directly produce
hypertrophy by stimulation of intracellular signaling molecules linked to the
hypertrophic program, including protein kinase C [291] as well as reactive oxygen
species [292]. Moreover, high intracellular sodium concentrations may also
contribute to cardiac hypertrophy by altering mitochondrial dynamics resulting in
metabolic remodeling although the precise mechanisms underlying these events have
not been fully elucidated [293]. As discussed below, studies using NHE1-specific
inhibitors have demonstrated a multitude of intracellular mechanisms potentially
contributing to a sodium dependent hypertrophic program. It is interesting to
point out that NHE-dependent cardiac hypertrophy manifests primarily as
pathological hypertrophy and does not seem to be involved in physiological
adaptive hypertrophic responses. In this regard, it has been proposed that AKT
(protein kinase B)-dependent NHE1 phosphorylation prevents NHE1 overactivation in
physiological hypertrophy [294] and thus it is likely that NHE1 activation does
not partake in the physiological hypertrophic program. High intake of dietary
sodium in experimental animals has also been linked to activation of the
adrenergic system, particularly that involving
A number of studies have documented an important role of NHE1 in mediating the enhanced intracellular sodium elevations, particularly under ischemic insult. For example, Bak and Ingwall showed that amiloride, a nonspecific NHE inhibitor, blunted the rise in intracellular sodium in ischemic isolated rat hearts [298]. However, as mentioned, amiloride is a nonspecific agent which affects other cellular processes in addition to NHE inhibition and thus, studies using this drug alone should be interpreted cautiously. However, Baartscheer and colleagues showed that cariporide markedly inhibited intracellular sodium overload and improved calcium regulation in rabbit hearts ex vivo in which the animals were subjected to 12 weeks of chronic combined pressure and volume overload [299].
While NHE1 activation likely represents a major mechanism for intracellular sodium elevation, other cellular processes may also contribute and should be mentioned. For example, inhibition of the late sodium current with the selective inhibitor ranolazine inhibited both hypertrophy and fibrosis as well as improving cardiac function in mice exposed to chronic pressure overload produced by aortic banding [238]. Importantly, this effect was associated with a suppression of sodium overload and improved intracellular calcium homeostasis [238].
A number of lines of evidence suggest that NHE1 is a major contributor to the
development of cardiac hypertrophy and heart failure. First, cardiomyocyte
hypertrophy is associated with upregulation of NHE1 expression and activity in
cardiomyocytes in a number of diverse experimental models [262, 299, 300, 301, 302, 303, 304, 305] as well
as in cardiomyocytes harvested from humans with end stage heart failure [306].
Secondly, many paracrine, autocrine and hormonal factors which are important
initiators of the hypertrophic program are also potent activators of NHE1
activity in the heart, among these being angiotensin II, endothelin 1 and
| Experimental model | NHE1 inhibitor | Reference | Main Result of inhibitor treatment |
| Rat 1 wk CAL | cariporide | [309] | Attenuate HY and HF |
| Rat 13–15 wk CAL | cariporide | [310] | Attenuate HY and HF |
| Rat PH/RVH | cariporide | [302] | Attenuate RV HY & fibrosis |
| SHR | cariporide | [33, 314, 315, 316] | Attenuate HY, apoptosis & antiarrhythmic |
| cariporide | [317] | Prevent HY, HF & fibrosis | |
| Rabbit P/V overload | cariporide | [299, 301, 311] | |
| Attenuate HY, regress remodeling | |||
| Isoproterenol treated rats | BIIB723 | [33] | Prevent HY & fibrosis |
| Aldosterone treated NRVM | EMD87580 | [318] | Prevent HY & Na |
| Rat 12 wk CAL | EMD87580 | [220] | |
| Mouse 5 wk TAB | cariporide | [319] | |
| LV paced rabbits | BIIB722 | [262] | |
| Rat 12 or 18 wk CAL | EMD87580 | [320, 321] | Protect mito function |
| Hamster HHC | EMD87580 | [322] | Prevent Na |
| [323] | Prevent early death | ||
| Rat 18 wk CAL | cariporide | [313] | |
| GC-A deficient mice | cariporide | [324] | Normalize pHi, Ca |
| PE-treated NRVM | EMD87580 | [325] | |
| ET1-treated NRVM | cariporide | [33, 326] | Prevent ET-1 HY & |
| NHE1 overexpressing TG mice | cariporide | [327] | block |
| Estrogen treated ARVM | AVE4890 | [328] | Block |
| ET1-treated NRVM | AVE4890 | [329] | |
| Glycoside-treated NRVM | AVE4890 or EMD87580 | [330] | |
| ISO-infused rats | BIIB723 | [331] | |
| Ang II-treated ACVM | cariporide | [332] | |
| Rat 6 wk CAL | BIX | [312] | |
| PE-treated NRVM | BIX | [312] | |
| Ang II-treated H9c2 cells | EMD87580 | [333] | |
The ability of NHE1 inhibition to attenuate cardiac hypertrophy is not
restricted to experimental models involving myocardial ischemia. For example,
Cingolani’s group showed that cariporide reduced the hypertrophic response as
well as myocardial fibrosis in the spontaneously hypertensive rat (SHR) which was
dissociated from blood pressure reduction [314, 334]. Moreover, cariporide
effectively prevented cardiac hypertrophy, fibrosis and left ventricular
dysfunction in a transgenic mouse model overexpressing the
NHE1 likely plays an important role in the development of heredity hypertrophic cardiomyopathy. Bkaily’s group has shown that dietary administration of the NHE1-specific inhibitor EMD 87580 (rimeporide) prevented the development of hypertrophy, necrosis, intracellular sodium and calcium overload, as well as preventing early mortality in a dystrophic hamster model [322, 323]. Moreover, rimeporide administration to dogs with muscular dystrophy resulted in a reduction in left ventricular function deterioration in these animals [335]. Such promising results in animal models has led to clinical testing of rimeporide in young boys with Duchenne muscular dystrophy (DMD, N = 20). A phase 1B clinical trial revealed that four-week treatment with rimeporide is well tolerated and produces no safety concerns [336]. Coupled with encouraging, although preliminary biomarker data suggesting some clinical efficacy, further larger scale placebo-controlled studies are planned to demonstrate the effectiveness of rimeporide in reducing cardiomyopathy associated with DMD. The mechanisms underlying the beneficial effects of NHE1 inhibition in dystrophic cardiomyopathy are not known with certainty but, as already alluded to, likely involve attenuation of calcium and sodium overload. Moreover, as DMD is associated with mitochondrial dysfunction [337, 338], protection by NHE1 inhibitors may involve mitochondrial protection as observed in other models of heart failure (see Section 7.5).
Direct effects of various hormonal as well as paracrine and autocrine factors,
some of which have already been referred to, can produce hypertrophy either
directly on cultured cardiomyocytes or via chronic in vivo
infusion through NHE1-dependent mechanisms. Among these are aldosterone [318, 339], estrogen [328], cardiac glycosides [330], isoproterenol [331] and
angiotensin II acting via the angiotensin AT
A non-pharmacological mode of NHE1 upregulation involves genetic modification of the antiporter resulting in enhanced NHE1 activity. In this regard, Fliegel’s group showed that transgenic mice expressing an overactive form of NHE1 exhibit cardiac hypertrophy in the absence of any pro-hypertrophic insult, when compared to mice expressing the wild type NHE1 [64, 65]. Additionally, infection of neonatal rat ventricular myocytes with an adenoviral vector expressing a constitutively active NHE1 resulted in a hypertrophic response in the absence of any other pro-hypertrophic intervention [63].
The mechanisms by which NHE1 activation induces cardiomyocyte hypertrophy are likely complex and associated with the stimulation of a number of intracellular pathways. As discussed above, NHE1 interacts with various intracellular cofactors and binding partners which in general, enhance the antiporter’s activity. With respect to the development of hypertrophy specifically, it has been reported that the ubiquitous multifunctional protein osteopontin originally identified in bone may be an important cofactor in mediating the hypertrophic influence of NHE1 activation. Thus, a close relationship between NHE1 and osteopontin expression was identified in cultured cardiomyocytes and silencing osteopontin in these cells supressed the hypertrophic effect of NHE1 overexpression [62]. Furthermore, osteopontin expression upregulation and the hypertrophic response in H9c2 cardiomyoblasts treated with angiotensin II was prevented by rimeporide [347].
Among the strongest candidates as a key factor in mediating the pro-hypertrophic effect of NHE1 activation is stimulation of calcineurin, a serine/threonine protein phosphatase which is an activator of transcriptional factors well known to be important in the pathological hypertrophic program as well as evolution to heart failure, among these being nuclear factor of activated T cells (NFAT) and myocyte enhancer factor 2 (Mef2) [348, 349]. As calcineurin can be activated by increased intracellular calcium concentrations or more specifically by formation of a calcium/calmodulin complex, it is not surprising that stimulation of NHE1 is a likely contributor to increased calcineurin activity leading to cardiomyocyte hypertrophy based on the concepts discussed in section 7.1 related to increased intracellular calcium concentrations. Indeed, overexpression of cardiac NHE1 per se has been shown to be sufficient to increase intracellular calcium levels, upregulate the calcineurin pathway and induce cardiomyocyte hypertrophy, thus demonstrating a strong link between NHE1 and the calcineurin pathway in promoting the hypertrophic program [327]. Inhibition of cardiac hypertrophy both in vivo as well as in cultured cardiomyocytes is associated with concomitant regression of calcineurin/NFAT expression [304]. Kilić et al. [312] showed that early and transient NHE1 inhibition was sufficient in preventing the hypertrophic response and calcineurin activation both in rats subjected to sustained coronary artery ligation as well as myocytes exposed to phenylephrine treatment. The relationship between NHE1 and calcineurin activity has also been reported with other antihypertrophic strategies including treatment with ginseng [158] or a chimeric natriuretic peptide [350]. Thus, NHE1-dependent calcineurin activation and subsequent cardiomyocyte hypertrophy likely follows the series of events summarized in Fig. 6.
 Fig. 6.
Fig. 6.Proposed pathway for calcineurin-mediated hypertrophy following NHE1 activation.
The evidence for calcineurin notwithstanding, other intracellular mechanisms may
also contribute to protection of the hypertrophied myocardium shown by NHE1
inhibitors particularly concerning mitochondrial preservation during the
remodeling process. Recent evidence, for example, suggests that the
antihypertrophic effect of NHE1 inhibition with rimeporide in cultured H9c2 cells
treated with angiotensin II is associated with suppression of the activation of
Cathepsin B, a cysteine protease involved in cardiovascular and other pathologies
[333]. Mitochondrial protection as demonstrated by reduced mitochondrial
dependent generation of reactive oxygen species may also be of importance in
understanding the anti-remodeling effects of NHE1 inhibition [351]. Karmazyn’s
laboratory has previously shown that rimeporide reduced MAPK activity, preserved
mitochondrial membrane potential, attenuated permeability transition pore opening
and reduced superoxide generation in phenylephrine-treated neonatal rat
cardiomyocytes while supressing the hypertrophic response [320, 325]. Further
evidence suggests that NHE1 inhibition reduces mitochondrial dysfunction in the
hypertrophied heart by attenuating phosphorylation of AMP-activated protein
kinase (AMPK)/glycogen synthase kinase 3
It is interesting to note that NHE1 inhibition may also contribute to the antihypertrophic and remodelling effects of drugs not initially developed for this purpose. An example of this are the sodium-glucose cotransport 2 inhibitors, the so-called gliflozins, developed for the treatment of type 2 diabetes mellitus but which exert a number of beneficial effects on the heart not related to glucose regulation but which likely involve NHE1 inhibition [353, 354, 355, 356, 357]. For example, dapagliflozin reduced fibrosis, inflammation and left ventricular dysfunction in db/db diabetic mice chronically (30 days) infused with angiotensin II [358]. These beneficial effects were associated with a number of cellular effects including decreased intracellular calcium transients, decreased inflammation, decreased ROS production as well as decreased expression of the voltage-dependent L-type calcium channel and decreased NCX levels while inhibiting NHE1 [328]. The relative contribution of each of these mechanisms awaits concrete elucidation. Gliflozins, are drugs that work primarily on the kidney to aid in glucose homeostasis in diabetic patients and may have effects through NHE1 (see section 8.1). In addition to the gliflozins, teneligliptin, a dipeptidyl peptidase-4 inhibitor used for the treatment of type 2 diabetes in some countries has been shown to reduce cardiac hypertrophy and the concomitant increase in NHE1 expression in spontaneously hypertensive rats that was attributed to normalization of the elevated plasma angiotensin II levels observed in these animals [359].
Genetic polymorphisms of NHE1 have been reported and this includes at least one that results in human disease (recently reviewed in [22]). Briefly, the first genetic defect in humans that was found to be attributable to NHE1 was reported by Guissart et al. [23]. Lichtenstein-Knorr syndrome is one of several autosomal recessive cerebellar ataxias with a variety of neurological symptoms, cardiomyopathies and ataxias [360]. It is of juvenile or adolescent onset with ataxia and sensorineural hearing loss [361]. Guissart et al. [23] showed that the SLC9A1 (NHE1) gene, is responsible for the defect in this disease. A mutation in the SLC9A1 gene changed Gly305 to Arg. The Gly305Arg mutation causes reduced expression and decreased protein glycosylation. It also caused almost a complete absence of targeting of the protein to the cell surface and virtually no protein activity at the cell surface [23]. After this report another study [362] reported a different human mutation resulting in similar symptoms including cerebellar ataxia. In this case the mutation at amino acid Ile288 caused a premature truncation of most of the protein.
NHE1 knockout mice have also been characterized. One spontaneous mutation of NHE1 in mice was a change causing Lys442 to become a stop codon and terminate NHE1 within the transmembrane domain. Homozygous defective mice had a slow-wave epilepsy (swe) mutation. They also had an ataxic gait including locomotor ataxia that was prominent in their hind limbs Mutant homozygous mice were of small size and less than half survived to weaning [363]. A second study in mice confirmed the above physiological effects with a targeted disruption of NHE1 [364]. The genetic knockout of NHE1 allowed an interesting insight into NHE1 physiology in the myocardium. When mice with the genetic knockout of NHE1 were subjected to cardiac ischemia reperfusion injury, they were resistant in comparison to the controls [273]. This confirmed the role of NHE1 in ischemia reperfusion damage.
Various specific genetic polymorphisms have been identified in the NHE1 gene though a thorough study of their total incidence and effect on the myocardium is lacking. One polymorphism was a change of Asn266 to His [365]. The mutation was not fully characterized clinically. Mutant N226H protein was expressed and targeted properly however, the N266H protein had no detectable activity. The NHE1 cytoplasmic tail is responsible for regulation of NHE1. Another study characterized the effect of stop codon polymorphisms in the regulatory tail [366]. Stop codons at amino acid 321, 449 and 735 were examined (mutations at 321 and 449 were actually within the membrane domain). Mutants stopping at amino acids 321 and 449 lost NHE1 activity and did not target properly to the plasma membrane. They were also more rapidly degraded than wild type protein. The mutant protein ending at amino acid 735 had reduced expression and activity.
Another study [367] examined the effect of change of two polymorphisms in the phosphorylatable amino acids in the regulatory tail. It examined the Ser703 and Ser771, to proline polymorphisms [367]. Ser703 is critical to 14-3-3 binding to NHE1 and to NHE1 activation by growth factors [368, 369] (Fig. 3). Ser771 is also important in Erk 1/2 dependent activation of NHE1 [370, 371] (see section 3.1.1). The Ser703Pro mutant had virtually the same activity, targeting and expression levels as the wild type NHE1 protein. However, the Ser771Pro mutant protein had reduced activity and expression levels, but normal cell surface targeting. The Ser771Pro mutant showed abnormal regulation. It was not strongly activated by sustained intracellular acidosis, but was activated partially even by very short periods of acidosis. It was hypothesized that insertion of a Pro in this location leads to an abnormal conformation that alters synthesis or degradation of the protein and causes an abnormal regulation of the protein by conformational changes in the tail, and by the inability to be phosphorylated [367]. It is not known how mutation of these regulatory amino acids could affect NHE1 function in humans. However, Ser703 is phosphorylated by p90 ribosomal S6 kinase. A mouse heart dominant negative p90 ribosomal S6 kinase mutant was resistant to myocardial injury induced by left coronary artery occlusion [90]. One might postulate that humans carrying the Ser703Pro polymorphism might also be resistant to coronary artery occlusion, but this remains to be demonstrated.
Because of the ubiquitous expression of the NHE throughout the tissues and cell
types of the body [1, 2] and its important function in each of these tissues, it
is perhaps not surprising that changes in NHE expression and function have been
implicated in a variety of diseases in a wide range of tissues and cell types.
This includes brain development and function [372], dental pulp [373], immune
function and inflammation [374], kidney function [375], epilepsy [363, 376],
gallstone formation [377], cataracts [378] and muscular dystrophies [379]. These
areas of NHE lesions have, in some cases, led to heart related problems as well.
For example, it has been suggested that the heart failure associated with Becker
and Duchenne muscular dystrophies may be in part due to NHE1 over-activation and
the subsequent Na
A significant change in how diabetic heart disease was viewed was first proposed with experimental evidence in the 1970’s and early 1980’s [381, 382, 383, 384, 385, 386, 387, 388, 389]. Instead of diabetic heart disease and failure being viewed as a primarily vascular lesion [390], the aforementioned works clearly identified a subcellular basis for a cardiomyopathy independent of vascular complications. The cardiac dysfunction was demonstrated in both insulin dependent and non-insulin dependent models of diabetes [382, 383, 391, 392].
Insulin dependent models of diabetes exhibited hearts resistant to
ischemic/reperfusion insult [393]. A reduced [pH]
However, whereas type 1 diabetic animals are more resistant to ischemia, insulin
resistant Type 2 diabetic animals were conversely more sensitive to ischemic
challenge. The decreased post-ischemic cardiac performance exhibited by hearts
from late-stage insulin-resistant models of diabetes, may be due to greater
endogenous stores of glycolytic substrates and the resultant excessive production
of lactate and H
As noted above in section 7.6, sodium/glucose cotransporter 2 (SGLT2)
inhibitors, gliflozins, are drugs that work primarily on the kidney to aid in
glucose homeostasis in diabetic patients. Empagliflozin has been reported to
attenuate both sodium and calcium dysregulation in mouse ventricular myocytes
treated with ouabain potentially via NHE1 inhibition [402]. It is also
important to note that empagliflozin has recently been shown to reduce oxidative
stress in cultured human umbilical vein endothelial cells and coronary artery
endothelial cells through a mechanism involving NHE1 inhibition [403] thus
providing further supporting evidence for the beneficial effects of gliflozins on
the heart through NHE1 inhibition. However, several recent studies have suggested
that these compounds also inhibit cardiac NHE1 activity [353, 354, 358, 404] and
expression [358]. This inhibition appears to be the mechanism for a lowering of
cytosolic Na
The glifozins are not the only non-specific drug interactions that may have their biological action via a primary effect on the NHE. NHE3 in the kidney is affected by incretin-based agents, antagonists of the renin-angiotensin system, insulin and insulin sensitizers, statins and spironolactone [409].
The robust experimental data demonstrating substantive cardioprotective properties of NHE1 inhibitors, unmatched by other cardioprotective strategies, rapidly progressed to clinical evaluation of NHE1 inhibitors in patients with coronary artery disease, mostly employing cariporide as the drug of choice. The first such study recruited a total of 100 patients who had experienced an acute myocardial infarction (AMI) and who were subjected either to percutaneous transluminal coronary angioplasty (PTCA) with cariporide administered at the time of reperfusion or with a placebo [410]. Patients receiving cariporide exhibited improved left ventricular function three weeks post-PTCA and reduced plasma enzyme levels within 72 hours after reperfusion, the latter indicating an inhibition of reperfusion injury by cariporide [410].
The results of the above study were somewhat surprising in view of the small
number of patients recruited but also because experimental studies have
demonstrated that optimal protection by NHE1 inhibitors occurs when the drug is
also present during the ischemic period before the onset of reperfusion.
Clinically, this can be achieved under controlled I/R conditions such as in
coronary artery bypass grafting (CABG, discussed below). Indeed, a Phase II
clinical study named “Evaluation of the Safety and Cardioprotective Effects of
Eniporide in AMI” (ESCAMI) study was conducted to investigate the hypothesis that
eniporide would reduce injury given as an adjunct to reperfusion performed either
by thrombolysis or PTCA [411]. This was a phase 2 international multicenter
randomized, double-blinded, placebo-controlled, dose-finding trial which was
carried out in two stages: in Stage 1 (433 patients), eniporide was administered
at 50 mg, 100 mg, 150 mg or 200 mg whereas in Stage 2 (978 patients), based on
the results of Stage 1 eniporide was further studied at doses of 100 mg or 150
mg. For both stages, eniporide or placebo was administered over a 10-minute
infusion period. Specifically, in patients subjected to thrombolytic therapy,
infusion was completed at least 15 minutes after starting thrombolytic treatment,
whereas in angioplasty, the patient’s infusion was completed at least 10 minutes
before the start of PTCA. The results of the Stage 1 study were encouraging in
that there were significant reductions in infarct size, the primary efficacy end
point of the study, of 25.7% and 41.7%, were found with 100 mg and 150 mg
eniporide, respectively. This effect was more evident in the PTCA treated
patients. However, no protection was observed in Stage 2 of this trial using
these two eniporide doses. It is interesting to add that in a subgroup of over
300 patients who were subjected to delayed reperfusion (
Further evaluation of the protective effects of NHE1 inhibitors was then carried out in two important clinical trials. The first of these was the Guard During Ischemia Against Necrosis (GUARDIAN) trial which was designed to determine whether cariporide could reduce mortality and MI in patients at risk of myocardial necrosis as well as to determine the drug’s safety [412]. GUARDIAN was a combined phase 2/phase 3 international multicenter, double-blind, randomized and dose-finding study in which the primary objective was to evaluate the efficacy of cariporide in reducing all-cause mortality and/or MI across the various entry populations 36 days after randomization. This study recruited 11,590 patients who were either hospitalized for an acute coronary syndrome (unstable angina or non–Q-wave myocardial infarction) or who were subjected to either PTCA or CABG. Patients were randomized to receive either one of three cariporide doses of 20 mg, 80 mg or 120 mg or placebo which were administered every eight hours for two to seven days as a 60-minute infusion. Starting time for cariporide administration differed based on the underlying condition and as decided by individual investigators. Generally, cariporide was initiated as soon as possible after admission in patients with acute coronary syndrome and between 15 minutes and 2 hours before PTCA or CABG. Doses of 20 and 80 mg were ineffective across all clinical settings. However, at day 36 CABG patients who were treated with 120 mg cariporide exhibited a significant 25% risk reduction in either death or myocardial infarction which primarily reflected a 32% risk reduction in nonfatal infarctions.
The GUARDIAN study, while showing no overall benefit of cariporide when assessed
across all clinical settings, did demonstrate substantial benefit, as noted
above, with fewer end-point events when administered at the 120 mg dose to
high-risk CABG patients. This encouraging result was subsequently used as the
major basis for the phase 3 Na
The results from the EXPEDITION trial were promising vis a vis the cardioprotective effects of cariporide in that the incidence of death or MI was reduced from 20.3% in the placebo group to 16.6% in patients treated with cariporide as was the incidence of MI alone (18.9% in the placebo group vs 14.4% in cariporide-treated patients), both highly significant reductions. These beneficial effects were maintained at 6 months follow up. Unfortunately, the beneficial cardiac effects of cariporide were associated with increased mortality from 1.5% in the placebo group to 2.2% with cariporide. These increases were statistically significant at 5 days and 3 months follow up but not at 6 months and were caused almost exclusively by a significantly higher incidence of thromboembolic strokes in patients receiving cariporide [413].
This paper has presented an overview of NHE1 in terms of its chemistry, regulation and its role in cardiac pathologies, the latter pertaining primarily to myocardial ischemic and reperfusion injury as well as myocardial remodeling resulting in heart failure. Much has been learned about the role of the antiporter as a critical regulator of intracellular pH but of greater relevance to the present discussion, its potential as a target for pharmacological intervention for cardiac therapeutics. The robust experimental evidence demonstrating salutary effects of NHE1 specific inhibitors has led to a rapid evaluation of these agents in the clinical setting particularly with respect to the assessment of cariporide as a cardioprotective agent in patients subjected to reperfusion protocols. Needless to say, the overall results seen in clinical trials with cariporide, as well as with eniporide, have been disappointing as evidenced by lack of efficacy and unexpected side effects as seen in the EXPEDITION study (even though a cardioprotective influence was demonstrated). A thorough evaluation of these results has previously been presented [414] and will, therefore, be discussed briefly here.
The results of the initial small study [410] notwithstanding, the failure to demonstrate efficacy in either the ESCAMI study or in either the thrombolysis or PTCA arms in the GUARDIAN trial, is surprising as animal data clearly demonstrated optimal protective efficacy of NHE1 inhibitors when the drug is present during the ischemic period, as noted in Sections 6.2 and 6.3. Indeed, when CABG patients were treated prior to surgery with the highest cariporide dose (120 mg group) in the GUARDIAN study, significant cardioprotection was observed [412]. Thus, expectations were high for favorable results in EXPEDITION as only CABG patients were recruited to this study with one standard cariporide dosing regimen. In fact, a significant cardioprotection was seen in EXPEDITION but the results were associated with a significantly increased incidence of ischemic strokes, thus resulting in an early cessation of the trial. The reasons for the increased incidence of strokes in cariporide-treated patients are not known with certainty. Importantly, we do not know for example whether this reflects a property of NHE1 inhibitors in general or cariporide specifically. The former is unlikely as NHE1 inhibitors have been extensively shown to exert cerebral protective effects and have been proposed as a potential treatment for strokes [415]. In addition, inhibitors of platelet NHE1 inhibit platelet aggregation [416].
There is a strong possibility that the increased incidence of strokes seen in EXPEDITION reflected the substantially different dosing regimen as well as total dose of cariporide administered when compared to that administered in the 120 mg CABG group in the GUARDIAN study. Thus, the total cariporide administered to patients in EXPEDITION was 1620 mg over a 48-hour period compared to 720 mg during the same time period in the GUARDIAN study. It is therefore possible, or even likely, that the increased incidence of strokes seen in cariporide-treated patients in EXPEDITION reflected an unnecessary overdosing with cariporide, particularly when compared with the highest dose CABG group in the GUARDIAN study which resulted in cardioprotection but no increase in the incidence of strokes [412]. As the treatment regimen in the highest cariporide dose CABG group in GUARDIAN resulted in cardioprotection, it is difficult to rationalize the more than two-fold increase in cariporide dosing in EXPEDITION during the pre-surgery 48-hour period. As outlined by the late Dr Gerald Buckberg, a renowned cardiac surgeon who was a participating investigator and a member of the EXPEDITION Steering Committee, the sponsoring company of the trial altered the steering committee’s recommendations regarding dosing by extending drug delivery duration and adding a high dose delivery. “They did this despite our emphasizing that these patients did not need elevated doses” [417].
The increased incidence of cerebrovascular events seen in EXPEDITION had a profoundly negative effect on the clinical development of NHE1 inhibitors as evidenced by a total cessation of all NHE1 inhibitor-related research and development by the pharmaceutical industry. Whether this action was justified is difficult to address as there is a complete paucity of data in the literature addressing the question of a possible pro-thrombotic effect of cariporide specifically or NHE1-specific inhibitors in general. For example, was the thrombotic effect of cariporide due to high dosing and can this be confirmed in the laboratory setting? Are there any insights into potential mechanisms for the pro-thrombotic effect of cariporide and does this involve NHE1 inhibition or non-specific effects of cariporide? This question would be particularly important to address since NHE1 activity likely contributes to platelet aggregation with an attenuation of the latter by NHE1 inhibitors, as already noted above. Do other NHE1-specific inhibitors share this pro-thrombotic effect particularly at high doses? Does the method of administration influence the deleterious effect of cariporide? In this regard, cariporide and other NHE1 inhibitors would obviously be most effective as cardioprotective agents in CABG patients, based on existing clinical data with cariporide. Would an oral preparation be effective in producing cardioprotection and would this minimize any possible prothrombotic risk compared to drug infusion? In view of the immense potential of NHE1 inhibitors for the treatment of heart disease as outlined in this review, the results of the EXPEDITION study should likely not have precluded the development of newer NHE1-specific inhibitors without addressing the issues just raised. The potential for NHE1 inhibitors to benefit patients with cardiovascular disorders warrants further research and possible clinical development of these agents. As stated by the EXPEDITION study authors “the use of NHE inhibitors could lead to significant improvement in medium- and long-term survival among patients undergoing heart surgery as well as those at risk of MI at any time” [413]. This potential benefit should not go unexplored.
With the recent elucidation of the 3D structure of NHE1 [11] and the development of novel inhibitors towards NHE1, there are also new opportunities. Understanding the precise location of the inhibitor binding pocket and its coordination may allow medicinal chemists to design novel inhibitors with even higher specificity and potency towards NHE1. Additionally, some such novel NHE1 inhibitors have recently been developed [226, 227, 228, 230] though they have not been tested in a cardiovascular disease setting. The advantage of novel inhibitors that are structurally different from cariporide is that they may avoid the stigma of the previous problems and it may be easier to obtain regulatory approval for them. There has also been little attention to altering NHE1 regulation in the disease state, which is another approach that has shown some promise [66, 83, 90], but has not been further developed in the clinic.
In summary, we propose that NHE-specific inhibition remains a worthwhile endeavour in the development of effective therapeutics for the treatment of heart disease. The scientific evidence for an effective approach to mitigating damage to the myocardium remains very strong as is the concept of NHE1 inhibition for the treatment of heart failure. Despite concerns with earlier clinical trials, the rationale and approach remain sound and new compounds for proposed treatment along with a more careful application based on experimental data could lead to useful clinical treatments for this major health problem.
LF, GP and MK designed the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
We thank B. Buckley, University of Wollongong, for his assistance drawing the structures of amiloride, cariporide and HMA. We thank the peer reviewers for their opinions and suggestions.
Work cited from the authors’ laboratory was funded by past Canadian Institutes of Health Research Operating Grants (MK, LF #MOP 97816) or a Foundation Grant (GNP) from the Canadian Institutes of Health Research.
The authors declare no conflict of interest. Morris Karmazyn and Grant N. Pierce are serving as the Guest editors of this journal. Grant N. Pierce is serving as one of the Editorial Board members of this journal. We declare that Morris Karmazyn and Grant N. Pierce had no involvement in the peer review of this article and have no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Fabian Sanchis-Gomar.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.