
Academic Editors: Kazuhiro P. Izawa, Peter H. Brubaker and Giuseppe Santarpino
Heart failure with reduced ejection fraction is associated with increased exercise intolerance, morbidity, and mortality. Importantly, exercise intolerance in heart failure with reduced ejection fraction is a key factor limiting patient quality of life and survival. Exercise intolerance in heart failure with reduced ejection fraction stems from a multi-organ failure to maintain homeostasis at rest and during exercise, including the heart, skeletal muscle, and autonomic nervous system, lending itself to a system constantly trying to “catch-up”. Hemodynamic control during exercise is regulated primarily by the autonomic nervous system, whose operation, in turn, is partly regulated via reflexive information from exercise-stimulated receptors throughout the body (e.g., arterial baroreflex, central and peripheral chemoreceptors, and the muscle metabo- and mechanoreflexes). Persons with heart failure with reduced ejection fraction exhibit malfunctioning autonomic reflexes, which lead to exaggerated sympathoexcitation and attenuated parasympathetic tone. Chronic elevation of sympathetic activity is associated with increased morbidity and mortality. In this review, we provide an overview of how each main exercise-related autonomic reflex is changed in heart failure with reduced ejection fraction, and the role of exercise training in attenuating or reversing the counterproductive changes.
Chronic heart failure (HF) is a complex clinical syndrome [1] that 26 million
people live with worldwide [2]. HF is characterized by severe exercise
intolerance (i.e., low peak oxygen uptake (VO
The autonomic nervous system regulates cardiovascular and hemodynamic adjustments to changes in energy demand (e.g., exercise) and in times of stress. Here we will discuss the pathways and mechanisms by which the autonomic nervous system controls our hemodynamic responses to exercise, including the exercise pressor reflex (muscle metaboreflex and muscle mechanoreflex), the arterial baroreflex, and the chemoreflexes (central and peripheral) in the situation of HFrEF, i.e., autonomic dysfunction.
The autonomic nervous system has two primary branches: (1) the parasympathetic; and (2) the sympathetic nervous systems [21, 22]. The balance between parasympathetic and sympathetic nerve activity modulates cardiovascular homeostasis and the response to exercise [21, 23]. For example, at dynamic exercise onset, parasympathetic withdrawal causes a nearly instantaneous increase in heart rate. Conversely, increased sympathetic activation at exercise onset causes a gradual rise in heart rate. The dominant controller of heart rate thus shifts from parasympathetic actvity at rest (i.e., high activity) and during initial exercise onset (i.e., withdrawal) to sympathetic activity (“sympathoexcitation”) during sustained exercise[23, 24]
Exaggerated sympathoexcitation in HFrEF may play a large role in disease progression [25] and is a marker of prognosis and survival [20, 26]. At the onset of left ventricular systolic dysfunction, the balance between parasympathetic and sympathetic nerve activity shifts in response to: (a) an increase in left ventricular end-diastolic pressure; or (b) a decrease in stroke volume [20, 26, 27, 28]. This shift in balance favours sympathoexcitation to temporarily re-establish hemodynamic equilibrium [21]. As HFrEF advances, parasympathetic outflow progressively wanes while sympathetic outflow increases [29], independent of HFrEF etiology [30, 31]. Concurrently, renin-angiotensin-aldosterone system activity increases [30, 31]. These autonomic and neurohormonal responses may initially be “adaptive” to maintain cardiac function [21, 30], but may subsequently and progressively become maladaptive and worsen HFrEF severity.
Autonomic dysfunction in HFrEF combines cardiac, ventilatory, neurohormonal, and vascular factors [6, 28, 30, 32] that are also related to poor prognosis [33] and increased mortality [20, 34]. Cardiac factors may include decreased tonic and reflex vagal heart rate modulation, increased resting heart rate [34, 35], chronotropic incompetence [36], and decreased heart rate variability [9, 36, 37, 38]. Elevated resting minute ventilation [12] and ventilation-to-perfusion mismatch are also consistently found in HFrEF [6, 33, 39, 40]. Neurohormonal changes in HFrEF include increased venous plasma norepinephrine, cardiac and renal norepinephrine spillover, cardiac norepinephrine stores, and plasma brain natriuretic peptides [6, 41]. Increased renal sympathetic nerve activity reduces renal blood flow, which may in turn exacerbate the renin-angiotensin-aldosterone system [42]. Finally, increased resting muscle sympathetic nerve activity (MSNA) burst frequency in HFrEF [9, 10, 11, 12, 13, 14] elicits tonic vasoconstriction and increased peripheral resistance at rest. Exaggerated sympathoexcitation in HFrEF is most common with severe exercise intolerance [15, 19] or ischemic heart disease [9].
Contrary to initial conceptual models that emphasized a generalized, non-selective increase in sympathetic nerve activity to all vasculature in response to left ventricular systolic dysfunction [27], evidence suggests a selective alteration in autonomic cardiovascular changes in the early stages of HFrEF progression (e.g., increased cardiac norepinephrine spillover versus no change in total or renal norepinephrine spillover) [28]. Autonomic dysfunction in HFrEF may be examined as the exaggeration or blunting of cardiovascular regulating autonomic reflexes in response to exercise: the exercise pressor reflex, the arterial baroreflex, and the central and peripheral chemoreflexes [21, 22] (Fig. 1, Ref. [21]). The current review is focused on extra-cardiac reflexes, therefore, the cardiopulmonary baroreflex and cardiac chemoreflex are not discussed herein. We refer the reader to this review for further detail [43].
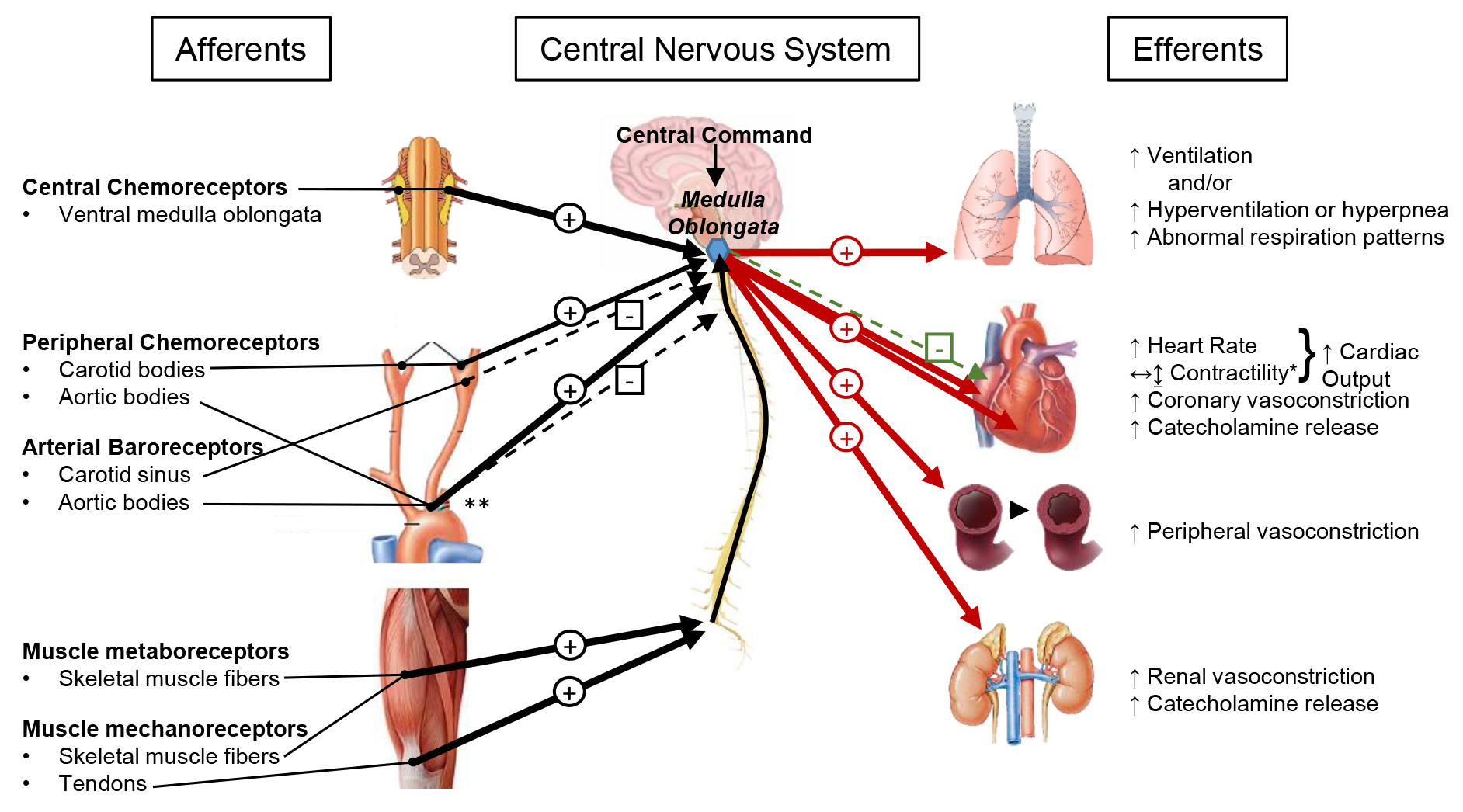 Fig. 1.
Fig. 1.Integration of exercise-related, cardiovascular autonomic reflexes in HFrEF. Afferent feedback is collected and integrated at the medulla oblongata of the brain stem (blue hexagon), where from efferent signals are sent to various organs in response to the integration of the afferent and central information. Excitatory (+) and inhibitory (-) stimuli are shown for afferent feedback (black arrows), efferent sympathetic nerve activity (red arrows) and efferent parasympathetic nerve activity (green arrows). Exaggerated stimuli due to HFrEF are indicated as thick arrows, while blunted stimuli due to HFrEF are indicated as dashed thin arrows. *Note: the effects of sympathoexcitation on cardiac contractility in HFrEF are generally to increase contractility, but some research suggests stroke volume remains the same or decreases during the activation of the metaboreflex in HFrEF. **Note: the aortic bodies contain both baroreceptors and chemoreceptors, which emit contrasting afferent signals. The baroreflex stimulus is inhibitory and is blunted in HFrEF; the chemoreflex stimulus is excitatory and is exaggerated in HFrEF. Figure concept and representation of afferent and efferent pathways was inspired by figure 4 in reference [21].
Curiously, muscle reflex physiology may play a large role in HFrEF exercise
pathophysiology. First described by Alam and Smirk in 1937, the exercise pressor
reflex is stimulated by skeletal muscle contraction to generate an increase in
mean arterial pressure and ventilation during exercise in healthy humans [44, 45].
The exercise pressor reflex is stimulated via group III and IV muscle afferents
and encompasses two separate reflexes: (1) the muscle mechanoreflex (mainly group
III afferents); and (2) the muscle metaboreflex (mainly group IV afferents).
Their purpose is to increase sympathoexcitation during exercise to maintain the
balance between cardiac output and peripheral O
The exercise pressor reflex is thought to be the “neural link” between
peripheral muscle abnormalities and exaggerated hemodynamic responses to exercise
in HFrEF [51]. When group III and IV muscle afferents were blocked via lumbar
intrathecal fentanyl administration during knee extension exercise in patients
with HFrEF, there was attenuated sympathetic outflow (i.e., lower total
norepinephrine spillover) and lower exercise cardiac output, but there was also
increased femoral blood flow and vascular conductance and less exercise fatigue
[52]. Thus, the exercise pressor reflex may modulate central hemodynamic control
and elicit exaggerated sympathoexcitation that impairs femoral blood flow during
exercise in HFrEF but not in controls [52]. The exaggerated exercise pressor
response may be related to impaired muscle quality and function (e.g., muscle
atrophy, altered enzyme concentrations, lower mitochondrial density, rapid
exercise high energy phosphate decline [53, 54, 55]), impaired muscle O
The muscle mechanoreflex may independently contribute to HFrEF pathophysiology.
Deformation of the skeletal muscle mechanoreceptors increases the muscle afferent
signalling rate to the central nervous system [46]. Mechanoreceptors rapidly
adjust their firing rate to changes in muscle tension consistency and respond
earlier and faster than metaboreceptors [47]. The mechanoreflex elicits an
increase in sympathoexcitation that increases arterial and renal vasoconstriction
[58], and that debatably/negligibly increases ventilation [3, 4, 47]. Thus, muscle
contraction indirectly increases perfusion pressure and O
Renal vasoconstriction during exercise is exaggerated during isolated mechanoreflex but not metaboreflex activation in HFrEF [58]. In contrast, the mechanoreflex has little effect on the exercise ventilatory response in both HFrEF and health, while the metaboreflex has a large regulatory role in ventilation modulation during exercise [3, 4]. In rodents with dilated cardiomyopathy (coronary ligation), the exercise pressor response and isolated mechanoreflex activity (i.e., mean arterial pressure and heart rate responses) progressively increased as left ventricular function decreased over a 10-week period [59]. Thus, an exaggerated mechanoreflex response in HFrEF may be systolic function dependent [59]. MSNA increased similarly during normal exercise in HFrEF compared to controls, both with dichloroacetate (which reduces lactic acid production) or saline (placebo) transfusion, but increased earlier during exercise in HFrEF compared to controls during placebo [50]. Further, mechanoreflex isolation via passive arm stretch caused an exaggerated early and sustained elevation of MSNA in HFrEF compared to no effect in controls [50]. Thus, there may be a prominent role for increased mechanosensitivity in the exaggerated sympathoexcitation during exercise in patients with HFrEF [50]. Additionally, the findings suggest muscle acidity does not affect mechanoreceptor sensitization in patients with HFrEF [50], which contrasts other reports [46]. In summary, the mechanoreflex may be exaggerated in HFrEF, such that renal vasoconstriction, mean arterial pressure, heart rate, and MSNA responses to exercise are elevated.
The muscle metaboreflex independently contributes to exaggerated sympathoexcitation in HFrEF [3, 4, 19, 47, 60]. The metaboreflex occurs when increased metabolite concentration (e.g., ATP [61], lactic acid, adenosine [10], phosphate, kinins, and cations [47, 62]) stimulates skeletal muscle receptors, whose afferent firing rate increases to yield hemodynamic, ventilatory, and sympathetic changes [21]; these changes include increases in MSNA, peripheral vasoconstriction [47], mean arterial pressure, pulmonary vascular resistance [63], and ventilation or hyperpnea [47]. During exercise, the metaboreflex increases cardiac output via stroke volume in health [64] and potentially heart rate [65] depending on the state of exercise, recovery, or ischemia [47]. However, the presence of HFrEF alters these effects.
The metaboreflex is consistently elevated in patients with HFrEF [21, 66] such that MSNA during ischemic and nonischemic handgrip exercise is greater and occurs earlier [19]; exercise ventilation is amplified due to an exaggerated metaboreflex response [3, 66, 67] in symptomatic but not in asymptomatic patients with HFrEF [68]; post-exercise heart rate recovery is slower [60]; and arterial pressure is elevated during post-exercise circulatory occlusion [60, 64, 67, 69, 70, 71]. While increased arterial pressure is primarily due to increased cardiac output in health, metaboreflex activation in HFrEF elicits negligible cardiac output changes and thus elevates mean arterial pressure via peripheral vasoconstriction and elevated systemic vascular resistance [63, 64, 69, 70, 71]. A dose-response relationship for this phenomenon was recently reported across three metaboreflex exercise intensities [72]. Cardiac output was limited during metaboreflex activation because of increased coronary vasoconstriction in dogs with HFrEF, but this has yet to be confirmed in humans with HFrEF [73]. Thus, the metaboreflex mechanisms in HFrEF may be related to the impaired ability to increase stroke volume during exercise [64, 70, 71].
In HFrEF, metaboreflex sensitivity and activity are negatively related to peak
VO
Some research suggests that HFrEF, perhaps only in its more severe form, attenuates [46] or has no effect on the metaboreflex [50]. Patients with mild HFrEF and controls have elevated MSNA and blood pressure during metaboreflex activation but not in those with severe HFrEF [75]. Further, one HFrEF group observed a decrease in MSNA from exercise to post-exercise ischemia while controls observed an increase [11]. The same group later published directly contrasting results in addition to earlier fatigue and greater accumulation of muscle metabolites in HFrEF, thus supporting the overall metaboreflex exaggeration in HFrEF [76]. Conflicting results on metaboreflex activity in HFrEF may result from different measured physiological indices (e.g., MSNA vs. ventilation vs. blood pressure) [77], exercise modes (e.g., rhythmic or isometric), muscle group (e.g., forearm or knee extensor), and/or HFrEF severity (e.g., muscle quality, muscle afferent sensitization or desensitization). For more on this, we refer the reader to the Wang et al. [77] and the give and take discussion between the blunted and exaggerated hypotheses. Taken together, we know muscle metaboreflex dysfunction in HFrEF alters exercise and post-exercise MSNA (mixed views on blunted vs. exaggerated responses [47]), and increases exercise and post-exercise blood pressure, vasoconstriction, and ventilation. Thus, dysfunctional metaboreflex may play a critical role in manifesting HFrEF exaggerated sympathoexcitation, early fatigue, and exercise intolerance.
The attenuated arterial baroreflex control of heart rate in patients with HFrEF [28, 32, 78, 79, 80] is associated with poor survival [81]. The arterial baroreceptors, located in the aortic arch and carotid sinuses, facilitate a negative feedback reflex that responds to stretch from a sudden increase in blood pressure by signalling afferents to the medulla oblongata. This will: (a) decrease sympathetic activity to the heart and vessels (i.e., lower heart rate, contractility, and vasoconstriction); and (b) increase parasympathetic activity to lower heart rate [22, 32]. Baroreflex activation thus lowers blood pressure via cardiac output, peripheral resistance, and venous return; the opposite effects would occur with a drop in baroreceptor activity [32].
How arterial baroreflex function is altered in HFrEF is debated. Some propose that the baroreflex responds appropriately to detected decreases in stroke volume or diastolic pressure by increasing sympathoexcitation in HFrEF [21]. Others describe normal baroreflex circuits in HFrEF with, however, exaggeration of other sympatho-excitatory reflexes that may override baroreflex function, such as the exercise pressor and chemoreflexes [82]. Conversely, somewhat conflicting evidence suggests arterial baroreflex dysfunction: heightened baroreflex sensitivity (i.e., the response magnitude when the baroreflex effectively drives the sinus node) in HFrEF [83]; reduced baroreflex sensitivity in optimally-treated patients with HFrEF [84]; reduced baroreflex sensitivity being related to pulse wave velocity and not EF, suggesting that structural alterations of the arterial wall may modulate baroreflex function [84]; and depressed baroreflex effectiveness index (i.e., the number of times the baroreflex effectively drives the sinus node) in a sample with no abnormal baroreflex sensitivity in HFrEF [85]. The baroreflex effectiveness index may be a better measure in HFrEF as it accounts for non-baroreflex control of heart rate, such as central command, ventilation, and neurohormonal factors, each of which may be abnormal in HFrEF [85]. Notwithstanding the preferred measure, a recent publication reported not only reduced baroreceptor sensitivity in patients with HFrEF, but a relationship between ejection fraction and baroreceptor sensitivity such that as ejection fraction declines, baroreceptor sensitivity declines [86]. Indeed, patients with HFpEF exhibited no such relationship nor any decline in baroreceptor sensitivity. Other functional relationships with baroreceptor sensitivity including MSNA, heart rate, and catecholamine levels [86]. Therefore, it may be surmised from the bulk of the literature that baroreflex dysfunction exists in those with HFrEF, however, the degree of dysfunction may be dependent upon the severity of the HFrEF state and level of neuroadrenergic activation.
Baroreflex modulation of heart rate, cardiac output, and blood pressure is depressed in HFrEF dogs at rest [87, 88, 89, 90, 91], during mild and moderate exercise [92, 93], and during metaboreflex activation [94], but without effect on the baroreflex modulation of vascular resistance at rest [91]. Thus, the ability of the baroreflex to buffer the metaboreflex is diminished in HFrEF dogs [93, 94]. Further, baroreflex unloading via bilateral carotid occlusion during concurrent muscle metaboreflex activation in dogs with HFrEF causes vasoconstriction in all vascular beds, non-ischemic and ischemic active tissue alike, resulting in a surge in mean arterial pressure as a result of a great pressor response [95]. This heightened sympathetic outflow as a result of baroreflex unloading in HFrEF initiates a “vicious cycle” of vasoconstriction in active tissue, which enhances the muscle metaboreflex, which causes more vasoconstriction. The authors speculate that baroreflex dysfunction coupled with muscle metaboreflex exaggeration may be a feedback loop exacerbating exercise intolerance in patients with HFrEF [95]. The interaction between these dysfunctional reflex systems in HFrEF appears to play a leading role in HFrEF pathophysiology and exercise intolerance. However, this stream of evidence comes with validity limitations. Exercise heart rate dominantly controls cardiac output in healthy dogs, whereas exercise stroke volume modulates cardiac output in HFrEF dogs [93]. This directly opposes previous work in HFrEF dogs by the same group [71], and therefore should be further investigated in patients with HFrEF of various severity and etiology.
Baroreflex sensitivity is improved after exercise training in HFrEF [30] and after simulating exercise training with electrical stimulation of the peroneal and tibial nerves [96]. Baroreflex activation therapy (carotid sinus stimulation) safely improved exercise capacity, functional class, and quality of life, and reduced NT-proBNP levels in patients with HFrEF already optimally treated with guideline therapy [97]. Vagal nerve stimulation therapy in NYHA class II—III HFrEF patients yielded similar but inconsistent findings to baroreflex activation therapy, with no apparent benefit to survival or heart function [98, 99, 100]. Recently, a series published in the Lancet reviewed baroreceptor stimulation as a non-pharmacological treatment of autonomic dysfunction in heart failure, however, outcomes were minimal, showing improved exercise capacity but no improvement in cardiac function, and trial control patient groups were not included [101], lending themselves to low-impact findings and implications. To summarize, baroreflex control of heart rate is impaired in HFrEF, however, the mechanisms may be outside the baroreceptors themselves [21, 82] or related to arterial stiffness [32, 84]. Improved arterial baroreflex sensitivity impacts exercise tolerance and quality of life [82, 97] but not necessarily prognosis.
The chemoreflex may be exaggerated in patients with HFrEF, observed as abnormal
ventilation at rest [102] and blunted vasodilation during exercise due to
exaggerated MSNA [12]. The chemoreflex modulates homeostasis under conditions of
hypercapnia, acidosis, or hypoxemia [32]. The chemoreflex has two levels: (1) the
central chemoreflex; and (2) the peripheral chemoreflex. The chemoreceptors
respond to hypoxemia and/or hypercapnia by stimulating increases in ventilation
and sympathetic outflow, with the goal of increasing tissue perfusion, O
The central chemoreceptors are located adjacent to the respiratory centres in
the medulla oblongata and detect blood CO
Exaggerated peripheral chemoreflex activity is common in HFrEF and may
contribute to exaggerated sympathoexcitation, hyperventilation, breathing
instability (e.g., Cheyne-Stokes) [102, 108], and autonomic dysfunction in HFrEF
[30]. The peripheral chemoreceptors (“glomus cells”) in the carotid bodies and
aortic arch detect blood O
Hypoxemia increases ventilation at rest and during exercise, and decreases the
peak work rate but not peak VO
The carotid chemoreflex interacts with the muscle metaboreflex to regulation minute ventilation in a breathing frequency dependent manner in patients with HFrEF, potentially contributing the exercise hyperventilation and exercise intolerance in HFrEF [111]. In other words, the activation of the muscle metaboreflex worsens the effects of the already hyperactive carotid chemoreflex in patients with HFrEF; as exercise stimulates both reflexes, this interaction may be a key player in functionally limiting exercise tolerance, which in turn is related to quality of life and mortality in HFrEF. The interaction between these situationally distant reflexes poses a therapeutic problem pharmacologically, however, exercise therapy (see section 1.6) may be used to address both reflexes individually and thus improve the nature of their interaction in patients with HFrEF.
Carotid body ablation, resection, or denervation have demonstrated improved
autonomic function (reduced renal sympathetic nerve activity or MSNA, increased
renal blood flow, increased heart rate variability, reduced peripheral
chemosensitivity, increased baroreflex sensitivity) in animal HFrEF models
[112, 113, 114]. A 2017 first-in-man study demonstrated that carotid body resection (4
patients unilateral resection, 6 patients bilateral resection) in HFrEF decreased
both MSNA and peripheral chemosensitivity and improved exercise tolerance 1-month
post-resection [115]. However, safety concerns arose in those with bilateral
resection who trended lower of oxygen saturation at night [115]; carotid bodies
stimulate protective reactions to hypoxia, therefore, further investigation
balancing patient safety and clinical benefit is required prior to wider usage of
carotid body resection or alteration. Although still debated, hemodynamic,
neurohumoral, and local metabolic mechanisms underlying HFrEF pathophysiology may
collectively progress peripheral chemoreceptor sensitization [102]. Despite the
high O
Exercise training is a highly effective therapy that increases not only
functional capacity, but enhances quality of life, resting heart rate,
ventilatory efficiency, and ejection fraction, as well as reduces the incidence
of major cardiovascular events, hospitalizations, and cardiac mortality
[120, 121, 122] and thus shows promise as an effective therapy for autonomic
dysfunction [42]. Further, exercise training reduced the rate of new-onset atrial
arrhythmias, but not ventricular arrhythmias, in patients with HF. Indeed, lower
peak VO
A recent meta-analysis (study n = 16) by Pearson and Smart [126]
reported exercise training improved short-term heart rate variability (study
n = 11; 2 studies with HF with preserved ejection fraction), decreased
MSNA (study n = 6), and increased heart rate recovery at 1-min
post-exercise (study n = 5) in exercise-trained patients with HFrEF
compared to non-exercise trained HFrEF controls. Several animal HFrEF models have
reported decreased sympathetic activation or improved hemodynamic control
following exercise training [113, 127, 128, 129, 130, 131, 132]. Exercise training in rat models
of HFrEF increased parasympathetic tone and intrinsic heart rate compared to
untrained rats with HFrEF, such that post-training values achieved no difference
with healthy sham rats [133]. Further, normalized parasympathetic activity was
related to training-induced preservation of preganglionc vagal neurons [133].
Early evidence in patients with HFrEF exhibited a shift from sympathetic to
parasympathetic dominance after 5–8 weeks of exercise training [134, 135, 136].
Similarly, 9 months of exercise training shifted the sympathovagal balance toward
a greater parasympathetic tone in patients with HFrEF, but this effect required a
long time course as it was not seen at 3 months [137]. Improved sympathovagal
balance, as measured by a decrease in the ratio of low frequency to high
frequency with power spectral heart rate analysis, was inversely correlatd with
increased peak VO
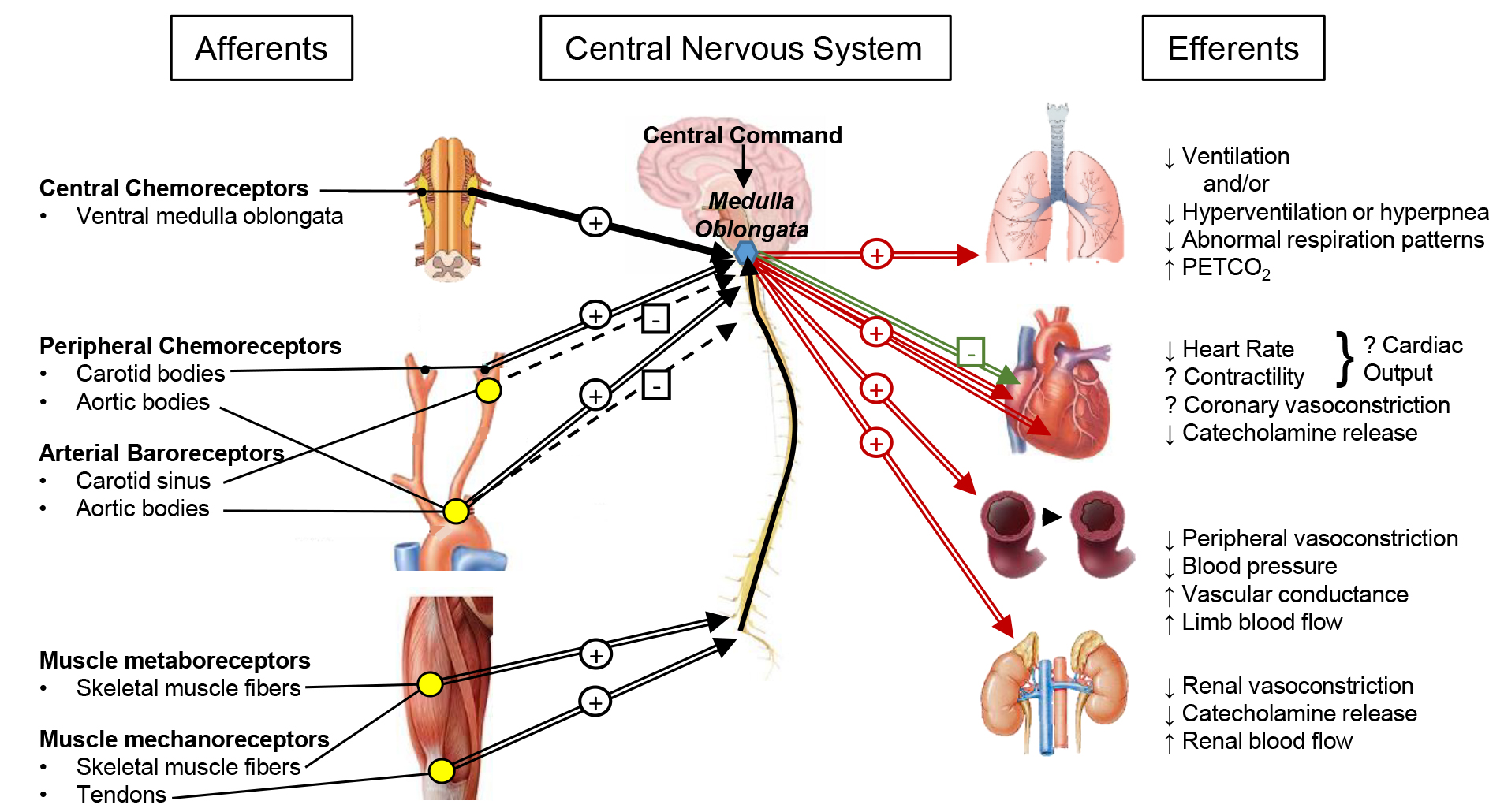 Fig. 2.
Fig. 2.Integration of cardiovascular autonomic reflexes in HFrEF after chronic exercise training. Afferent feedback is collected and integrated at the medulla oblongata of the brain stem (blue hexagon), where from efferent signals are outputted to various organs in response to the integration of the afferent and central information. Excitatory (+) and inhibitory (–) stimuli are shown for afferent feedback (black arrows), efferent sympathetic nerve activity (red arrows) and efferent parasympathetic nerve activity (green arrows). Exaggerated stimuli due to HFrEF are indicated as thick arrows, while blunted stimuli due to HFrEF are indicated as dashed thin arrows. Based on limited evidence, Fig. 2 represents the proposed changes following exercise training. The normalized signaling found following chronic exercise training is shown by parallel lines. Improved sensitivity of reflex receptors are indicated by a yellow dot.
It is imperative to consider that improved exercise capacity, cardiac function,
and/or skeletal muscle function does not equal improved autonomic function.
Similar to how exercise regime (moderate intensity continuous training, high
intensity interval training, resistance training, etc.) yields different health
and clinical benefits improve autonomic function, although the majority include
some form of aerobic training. Low intensity cardiac rehabilitation in patients
with HFrEF [143], whether or not the exercise regime improves autonomic function
in HFrEF may depend greatly on the mode, duration, and intensity of exercise
[30]. Nonetheless, it appears that a wide variety of regimes restored autonomic
tone and reactivity to sympathetic and parasympathetic stimuli in patients with
HFrEF [137]. An aerobic, resistance-band training study targeting each muscle
group one at a time lowered circulating catecholamine levels at rest and during
exercise in HFrEF patients [144]. Combined interval and resistance training had
statistically similar effects on respiratory drive compared to interval training
alone in patients with HFrEF, but data indicated that combined training may have
improved resting and exercise breathing pattern indices and P
Exercise training alters both the muscle metaboreflex and the muscle
mechanoreflex in patients with HFrEF [147, 148]. Six weeks of forearm training
reduced the systolic and diastolic pressure, minute ventilation, respiration
rate, and leg vascular resistance responses and increased the leg blood flow
response to post-handgrip exercise circulatory occlusion in patients with HFrEF
more so compared to healthy controls [148]. Further, these changes were
accompanied by a decrease in sympathetic activation but without change in
parasympathetic activation as measured by power spectral analysis of blood
pressure low frequency components [148]. Antunes-Correa et al. [147]
reported lower resting MSNA following exercise training (4-months combined
aerobic, resistance, and flexibility training) compared to an untrained control
HFrEF group; interestingly, however, the nature of the metaboreflex improvement
further complicates the known literature surrounding muscle metaboreflex
dysfunction in patients with HFrEF. During muscle metaboreflex activation via
post-exercise circulatory occlusion, exercise trained patients with HFrEF
reported heightening MSNA as well as increased transient receptor potential
vanilloid type-1 (TRPV1) receptors and cannabinoid receptor type-1 (CB1) in the
vastus lateralis (trained muscle) whereas the untrained group saw no change in
MSNA or the expression of either receptor compared to baseline testing [147].
This latter finding suggests that the blunted muscle metaboreflex response, found
in some HFrEF studies [11, 75, 149] but not all [21, 47, 51, 66, 148], and confirmed at
baseline testing in this study, is augmented with exercise training, thereby
returning muscle metaboreflex dysfunction closer to normal function. It may be
that HFrEF severity dictates the nature of muscle metaboreflex dysfunction [75],
such that mild cases (e.g., NYHA class I—II) exhibit exaggerated responses that
deteriorate as disease severity worsens, resulting in a blunted muscle
metaboreflex response in more severe HFrEF conditions (e.g., NYHA class III–IV).
In addition to muscle metaboreflex isolation, authors isolated the mechanoreflex
with passive exercise and found decreased MSNA and expression of cyclooxygenase-2
(COX-2), prostaglandin-E2 receptor-4, and thromboxane-A2 receptors [147], each of
which is involved in the COX pathway, a hypothesized mechanism behind
mechanoreflex dysfunction in HFrEF. Recently, 6-months of exercise training
cardiac rehabilitation in patients with HFrEF not only improved peak VO
Two studies by Wang et al. [151, 152] demonstrated that exercise training targets both the muscle metaboreflex and mechanoreflex in rat HFrEF models. Exercise training normalized pre-exercise blunted muscle metaboreflex (isolated via capsaicin injection) and decreased the pre-exercise exaggeration of the muscle mechanoreflex (isolated via contraction and passive stretch) in rats with HFrEF, with no effects seen in sham rats post-training [152]. Mechanistically, exercise training improved the sensitization of both group III and IV afferents in the HFrEF rat models, corresponding to improved function of certain muscle metaboreflex (TRPV1) and mechanoreflex (P2X) receptors [151]. A review by the same group discussed the discrepancy between the observations of both blunted and exaggerated muscle metaboreflex responses in HFrEF, as well as potential mechanisms underlying the improved exercise pressor reflex response after exercise training, to a greater detail than may be covered herein [77].
The effect of exercise training on baroreflex function in patients with HFrEF
is, surprisingly, vastly understudied in humans. To the knowledge of the authors,
the only known study in human patients with HFrEF showing improved baroreflex
sensitivity post-exercise training lacked a control group [153]. Fortunately,
6-months of combined aerobic and circuit training significantly improved
baroreflex sensitivity with concomitant improvements in heart rate variability,
cardiac presynaptic sympathetic innervation, increases in myocardial blood flow
where it was reduced pre-training, however, without a statistically significant
increase in peak VO
Despite the surprising lack of controlled, exercise training trials in patients with HFrEF on baroreflex sensitivity, evidence strongly suggests that exercise therapy improves baroreflex function in patients on the pre-HFrEF spectrum (e.g., hypertension, coronary artery disease, post-myocardial infarction, etc. [155, 156]) as well as in animal HFrEF models [128, 129, 157, 158, 159], and thus strongly warrants clinical trials investigating the efficacy of improved baroreflex function and corresponding health benefits from exercise. However, given the lack of patient evidence, this section of the review will focus on animal based HFrEF studies.
Exercise training improved arterial baroreflex control of both renal sympathetic nerve activity and heart rate in HFrEF rabbits [158] and rats [160]. The latter finding was accompanied by improved aortic depressor nerve activity sensitivity following exercise training in rats with HFrEF [160], suggesting a role not only of carotid baroreceptors but of aortic baroreceptors in modulating heart rate and sympathetic outflow in HFrEF. Indeed, work by Liu et al. [159] demonstrated that atropine and metoprolol infusion modulated baroreflex control of heart rate through increased vagal tone, rather than reduced sympathetic outflow, following exercise training in HFrEF.
Exercise training normalized sympathetic outflow (resting renal sympathetic nerve activity) and arterial baroreflex function (attenuated maximum gain of heart rate and renal sympathetic nerve activity curves) in rabbits with HFrEF [128, 157]. Further, angiotensin II type 1 receptor blockade in non-exercise trained HFrEF rabbits raised baroreceptor sensitivity approximating that of those exercise-trained rabbits with HFrEF; exercise in HFrEF rabbits also lowered circulating angiotensin II levels [157]. Mousa et al. [158] attributed their observed reduction in renal sympathetic nerve activity and elevation in baroreflex sensitivity in rabbits with HFrEF to concurrent reduction in circulating angiotensin II and angiotensin receptors in the central nervous system that occurs following exercise training. Thus, angiotensin II and the RAAS pathway is mechanistically tied to the dysfunctional pathophysiology of the arterial baroreflex in HFrEF. In rats, only endurance training improved cardiac autonomic control (increased baroreflex sensitivity) compared to moderate interval training, despite improved cardiac function and exercise capacity in both programs [129].
Patients with HFrEF following carotid body resection exhibited significantly improved exercise time compared to pre-resection exercise time, reduced MSNA, and decreased peripheral chemosensitivity [115]. Although these are encouraging findings in human patients, this approach has been contested, for many reasons, in favour of exercise training [161]. Exercise training poses a less-invasive, more cost-effective, and in most cases, simpler alternative that may produce similar or better results, as well as more global physiological and health benefits (endothelial function, peripheral blood flow distribution, skeletal muscle factors, and metabolic factors associated with reduced mortality).
Animal models have confirmed the efficacy of exercise training on attenuating exaggerated chemoreceptor sensitivity in HFrEF. Exaggerated peripheral chemosensitivity was attenuated by exercise training in rabbits with HFrEF [162]. Renal blood flow is classically reduced in the HFrEF condition due to exaggerated renal sympathetic nerve activity and consequent renal vasoconstriction, mediated in part by heightened peripheral chemoreceptor sensitivity. Exercise training attenuated peripheral chemoreflex-mediated tonic reductions in renal blood flow in rabbits with HFrEF, as assessed by compared carotid-body chemoreceptor denervated animals with carotid-body intact animals; further, exercise training also attenuated hypoxia induced reductions in renal blood flow in the same HFrEF sample [163]. Along with improved exercise capacity, rabbits with pacing-induced HFrEF returned to pre-pacing levels of renal blood flow and vascular conductance during acute hypoxia after exercise training [127]. Thus, animal models have demonstrated that exercise training attenuates exaggerated peripheral chemoreceptor sensitivity found in HFrEF and improves physiological functions (increased peripheral blood flow) and autonomic signalling (reduced renal sympathetic nerve activity, increase baroreflex sensitivity). Unfortunately, in human patients with HFrEF, data supporting direct involvement and improvement of peripheral chemoreceptor function is lacking. Both interval training and combined interval and resistance training improved respiratory drive in patients with HFrEF [145]. Coats et al. [134] demonstrated reduced minute ventilation and the ventilation-carbon dioxide production slope post-exercise training, along with improved heart rate variability and reduced catecholamine levels, indicative of a shift in sympathovagal from sympathetic toward greater vagal tone, in patients with HFrEF.
Central command is critical to the autonomic response to exercise initation in healthy humans and is involved in arterial baroreflex resetting during exercise [164]. Despite the instrumental difficulty in differentiating between changes in central command from changes originating in other autonomic reflex segments (e.g., the mechanoreflex), evidence suggests central command is a modifiable and trainable portion of the autonomic control centre [165]. Using electrically evoked skeletal muscle contractions to isolate central command, routine exercise training was shown to decrease central command input during exercise without a concomitant change in exercise pressor reflex in healthy humans [165]. However, evidence of central command changes and the effects of exercise training in HFrEF is limited. Central command was shown to contribute to the exaggerated sympathetic nerve activity during exercise that is characteristic of HFrEF by simulating exercise with mesencephalic locomotor region stimulation in decerebate, ischemic HFrEF rats, and observing fictional-exericse-induced elevated renal and lumbar sympathetic nerve activity [166]. In humans with severe HFrEF, the methodology was unable to differentiate between central command and the mechanoreflex as the primary driver of elevated MSNA during exercise [75]. Interestingly, central command in patients with HF and preserved ejection fraction (HFpEF) was not different from healthy controls despite a lower exercise heart rate [167]; however, the such investigations to better understand the role of central command on exercise pathophysiology in patients with HFrEF is lacking and merits further attention.
Exaggerated sympathoexcitation and depressed vagal tone, as a result of exercise autonomic reflex dysfunction, are proven contributors to prognosis, exercise intolerance, and survival in patients with HFrEF [21, 32, 47]. It may be clinically prudent to treat the cause(s) of the sympathoexcitation in HFrEF rather than, or at least in addition to, its consequences (e.g., increased MSNA, vasoconstriction, blood pressure) [47, 48]. As discussed, potential mechanisms for each reflex may include skeletal muscle abnormalities, arterial stiffness, chronic or intermittent hypercapnia, respiratory acidosis or hypoxemia, and chronic oxidative stress. Exercise training may target some or all of these potential causes for afferent dysfunction, and may attenuate or reverse chronic sympathoexcitation and enhance vagal tone [21]. Other therapies targeting afferent, central, and efferent portions of the reflex loop (Fig. 1) are emerging [21].
Exercise training is an effective and low-cost non-pharmacologic therapeutic option for people living with HFrEF. Exercise training improves exercise capacity, which is related with better outcomes in patients with HF [168, 169]. This non-invasive, affordable, and readily accessible therapy has the potential to be more successful compared to devices and medications for a number of reasons, including but not limited to (a) the effects of exercise target multiple organs and signaling systems, resulting in a “global” improvement; (b) regular exercise improves perceived quality of life and mental health, and (c) exercise training impacts not only the HFrEF sequelae, but other comorbidities and thus lifts the overall burden of health.
The effect of exercise training on the function of each reflex [170], rather than the physiological consequence (e.g., MSNA, heart rate variability, heart rate recovery, etc. [126]) is relatively understudied and not completely known. For example, the recent meta-analysis reports only heart rate recovery, MSNA, and heart rate variability: all indirect measures of autonomic nervous system activity and not used to differentiate between sympathetic and parasympathetic activity, nor tied to any one reflex pathway [126]. This may be due to the technical difficulty and invasiveness in obtaining direct measures, coupled with the challenge of obtaining meaningful data across a wide variety of comorbidities (e.g., a recent mean of 5 comobidities [169]) that often accompany HFrEF. Medication usage presents another challenge with evaluating the effectiveness of exercise training trials as certain medications (e.g., beta-blockers) may negate or mask favourable sympathovagal shifts. The effect of exercise training on peripheral chemoreflex function and regulation of cardiovascular hemodynamics is understudied in humans with HFrEF. Further, Pearson and Smart [126] report that the majority of studies and randomized controlled trials on exercise therapy on autonomic function in HFrEF lack sufficient allocation concealment, intention-to-treat analysis, control group physical activity monitoring, and considerations regarding relative exercise intensity and accurate energy expenditure. Clearly, both study design and the primary outcome have room for improvement in future randomized controlled trials in exercise training effects on autonomic function in patients with HFrEF.
Mechanistic animal studies have yet to be confirmed in patients with HFrEF; cardiac catheterization during exercise reflex activation may determine the extent of reflex modulation of coronary vasoconstriction and altered ventricular function during exercise in HFrEF [73]. As well, despite the mounting evidence supporting oxidative stress involvement in chemoreflex exaggeration, there is limited data on the effects of exercise training on chemoreflex function and oxidative stress in patients with HFrEF [30]. Other therapies currently under or for future investigation that modulate reflex physiology may include carotid body resection, ablation or desensitization (early findings suggest unilateral vs. bilateral for patient safety [115]), arterial baroreceptor stimulation, vagal nerve stimulation, central acting parasympathetic-mimicking or sympatho-modulatory medications, efferent renal nerve ablation, acetylcholinesterase inhibition, nicotinic receptor agonists, and beta-adrenergic receptor blockers [21].
Randomized controlled trials are still needed to phenotype autonomic dysfunction to select the appropriate therapeutic approach in HFrEF. The issue of autonomic dysfunction in HFrEF becomes more complicated when comorbidities that also relate to autonomic dysfunction (e.g., hypertension, obesity, diabetes, etc.) [126, 171] are added to the equation. The combined effects of multiple morbidities are not well understood and thus requires larger spectrum of inclusion criteria in experimental designs to increase our body of evidence. Patient sex is often overlooked in heart failure research despite the well-acknowledged fact that cardiovascular health and autonomic function is different in females compared to males. As the majority of heart failure studies are primarily male participants, future trials with sex-based comparisons are needed to elucidate any potential sex differences to better treat female patients in the future. Finally, HFpEF and mid-range ejection fraction (HFmEF) are increasingly clinically recognized and studied. However, the evidence around autonomic dysfunction is vastly disproportionally in patients and animals with HFrEF compared to their phenotypic counterparts. Future research should address the gap in literature surrounding HFpEF and HFmEF autonomic function, as well as phenotype-based studies to liken and differentiate the diseases from one another so as to improve future therapy targets on a phenotype-specific basis.
Fundamentally, reflex dysfunction may be adaptive at the onset of cardiac failure, but inter-reflex coordination is progressively lost, and thus autonomic dysfunction occurs. The exercise pressor reflex is exaggerated and enhances mean arterial pressure, MSNA, peripheral and likely cardiac vasoconstriction during exercise in HFrEF. Conversely, the exercise pressor reflex may be attenuated in more severe HFrEF. Baroreflex control of heart rate is blunted in HFrEF, as is its ability to buffer the metaboreflex, resulting in elevated resting and exercise heart rate and mean arterial pressure. Chemoreflex hyperactivity induces respiratory disorders, elevated heart rate, and greater vasoconstriction at rest and during exercise. Cumulatively, cardiovascular autonomic reflex dysfunction in HFrEF exaggerates sympathoexcitation to cardiac, respiratory, renal, vascular, and skeletal muscular tissues. The cardiovascular autonomic reflex dysfunction thus critically contributes to exercise intolerance and prognosis in HFrEF patients.
Conceptualization—NGB, DDM, HH, and CRT; methodology—NGB, DDM, HH, and CRT; investigation—NGB; data curation—NGB; writing—NGB; review and editing—NGB, DDM, HH, and CRT; visualization—NGB and CRT; supervision—CRT; funding acquisition—NGB and CRT. All authors have read and agreed to the published version of the manuscript.
Not applicable.
Not applicable.
NGB is funded by a Natural Sciences and Engineering Research Council of Canada Doctoral Postgraduate Scholarship. CRT is funded by the Saskatchewan Health Research Foundation (Establishment Grant #3468) and the Heart and Stroke Foundation of Canada Grant-in-Aid (G-16-00014255).
The authors declare no conflict of interest.