






1 Department of Cardiac, Thoracic and Vascular Sciences, University of Padua, 35128 Padova, Italy
Academic Editor: Stefan Peters
Abstract
Arrhythmogenic cardiomyopathy (ACM) is a rare heart muscle disease characterized by a progressive fibro-fatty myocardial replacement, ventricular arrhythmias, and increased risk of sudden cardiac death. The first diagnostic criteria were proposed by an International Task Force of experts in 1994 and revised in 2010. At that time, ACM was mainly considered a right ventricle disease, with left ventricle involvement only in the late stages. Since 2010, several pathological and clinical studies using cardiac magnetic resonance (CMR) imaging have allowed to understand the phenotypic expression of the disease and to reach the current idea that ACM may affect both ventricles. Indeed, left ventricular involvement may parallel or exceed right ventricular involvement. The main limitations of the 2010 criteria included the poor sensitivity for left ventricular involvement and the lack of inclusion of tissue characterization by CMR. The 2020 International criteria (the Padua criteria) were developed to overcome these shortcomings. The most important innovations are the introduction of a set of criteria for identifying left ventricular variants and the use of CMR for tissue characterization. Moreover, criteria for right ventricular involvement were also updated taking into account new evidence. According to the number of criteria for right and/or left ventricular involvement, the 2020 Padua criteria allows diagnosing three ACM phenotypic variants: right-dominant, biventricular and left-dominant. This review discusses the evolving approach to diagnosis of ACM, from the 1994 International Criteria to the 2020 Padua criteria.
Keywords
- arrhythmogenic cardiomyopathy
- cardiomyopathy
- ventricular arrhythmias
- cardiac magnetic resonance
- diagnosis
Arrhythmogenic cardiomyopathy (ACM) is a rare heart muscle disease pathologically characterized by a progressive replacement of the ventricular myocardium with fibro-fatty tissue, and clinically by life-threatening ventricular arrhythmias and sudden cardiac death (SCD) [1, 2].
At first, the disease was considered as only affecting the right ventricle (RV). The first report of ACM as heredo- familial disease was published in 1736 by Giovanni Maria Lancisi [3]. He described a family with disease recurrence in four generations, which occurred with palpitations, dilatation of the RV, heart failure and SCD. In 1982, Marcus et al. [4] introduced the term “arrhythmogenic right ventricular dysplasia”, by reporting 24 cases of adult patients with left bundle branch block morphology ventricular arrhythmias, in keeping with origin from an affected RV. It was considered a development defect of RV myocardium. Some years later, Thiene and colleagues [5] for the first time recognized this disease as a main cause of SCD in young people and athletes. Moreover, the post-mortem examination of the hearts brought the idea of a heart muscle disease developing after birth, because of the histopathological evidence of inflammation, degeneration, and necrosis foci, with progressive loss of myocardium. Therefore, the authors defined the disease “arrhythmogenic right ventricular cardiomyopathy” (ARVC) rather than “dysplasia”. The discovery of defects in genes encoding desmosomal proteins resulted in the definitively introduction of the ARVC in the WHO nomenclature and classification of cardiomyopathy [6]. Moreover, the absence of a single gold standard for reaching the diagnosis of ARVC led to the necessity of formal criteria aimed at facilitating and standardizing the clinical diagnosis.
In 1994 a group of experienced clinicians in cardiomyopathies published the first Task Force (TF) criteria [7]. The diagnosis was based on multiple parameters from six different categories, including global or regional dysfunction and structural alterations of the RV demonstrated on imaging, tissue characterization by endomyocardial biopsy (EMB), ECG repolarization abnormalities, ECG depolarization abnormalities, arrhythmias and family history. Each criterion was classified as “major” or “minor” according to its specificity for differentiating ARVC from conditions with an overlapping phenotype such as idiopathic right ventricular outflow tract (RVOT) ventricular tachycardia (VT) or biventricular dilated cardiomyopathy (DCM). It was proposed that the diagnosis of ARVC would be fulfilled by the presence from different groups of either two major criteria, or one major plus two minor criteria, or four minor criteria. At that time, ACM was mainly considered as a RV disease, with left ventricle (LV) involvement only in the late stages. Indeed, the morpho-functional RV abnormalities diagnostic criteria were met in presence of no or only mild LV impairment.
The 1994 TF criteria had some limitations. First, they resulted to be highly specific, but they lacked sensitivity when evaluating asymptomatic patients and relatives with early/minor ARVC. This is because the clinical experience was primary based on SCD victims and symptomatic index cases, with clear and severe manifestations of ACM [8]. Moreover, the criteria revealed faults in clinical application because of the qualitative and subjective assessment of the clinical parameters, rather than quantitative.
In 2010, the Revised International Task Force (ITF) criteria were published [9]. The organization in 6 different categories and the classification in major and minor criteria were maintained. In order to improve diagnostic accuracy, the 2010 ITF criteria provided quantitative imaging reference values for defining normal RV and various degree of structural and functional abnormalities, and also a quantitative histomorphometric definition of fibrofatty replacement of myocardium on EMB. In addition, ECG and ventricular arrhythmias criteria were updated, and the “family history” category was enriched with molecular genetic information [9, 10, 11, 12]. Another important element of novelty of the 2010 criteria is the introduction of “borderline” (1 major plus one minor or three minor criteria) and “possible” (1 major or two minor criteria) diagnostic categories.
Since 2010, several pathological and clinical studies have allowed to better understand the phenotypic expression of the disease and to reach the current idea of a cardiomyopathy that can be biventricular or exceed either in RV involvement (ARVC) or in LV involvement (left-dominant or arrhythmogenic left ventricular cardiomyopathy (ALVC)) [13, 14]. Subsequently, the original term “ARVC” was replaced with the broader definition of “arrhythmogenic cardiomyopathy” (ACM) [15].
In 2019, a group of International Experts in cardiomyopathies published an extensive critical review [16] of the clinical performance of the 2010 criteria, emphasizing three major points:
(1) They lacked specific criteria for diagnosing left-sided variants;
(2) Cardiac magnetic resonance (CMR) has become crucial not only for assessing volumes, systolic function and wall motion abnormalities, but especially for tissue characterization using late gadolinium enhancement (LGE) technique, which is essential for diagnosing LV involvement that can be characterized by subepicardial fibrosis or fibro-fatty scars with or without ventricular wall motion abnormalities [17, 18];
(3) Genetic testing was considered a major diagnostic criterion also in probands, potentially allowing to reach the diagnosis also in absence of morpho-functional ventricular abnormalities or tissue changes. Instead, in all the other cardiovascular diseases, genetic testing is recommended in probands that already fulfill clinical diagnostic criteria.
In addition to these three points, the experts underlined that some RV criteria needed revision.
On the basis of this critical appraisal, in 2020 an international expert consensus document upgraded diagnostic criteria for ACM [19].
The 2020 International criteria (“Padua Criteria”) maintained the same organization in 6 sets of criteria, the differentiation of the criteria in major and minor depending on their specificity for diagnosing ACM and the three diagnostic categories (“definite”, “borderline” or “possible”). However, there were several innovations: (i) because ACM is a structural disease, at least one morpho-functional or structural criterion needed to be satisfied; (ii) tissue characterization through CMR was introduced; (iii) two different sets of criteria for identification of RV (updated criteria) and LV involvement (new criteria) were provided (Table 1, Ref. [19]).
| Criteria for RV involvement | Criteria for LV involvement | |
| I. Morpho-functional ventricular abnormalities | By 2D echocardiogram, CMR or angiography: | By 2D echocardiogram, CMR or angiography: |
| Major | Minor | |
| Regional RV akinesia, dyskinesia, or bulging plus one of the following: | Global LV systolic dysfunction (depression of LV EF or reduction of echocardiographic global longitudinal strain), with or without LV dilatation (increase of LV EDV according to the imaging test specific nomograms for age, sex, and BSA) | |
| -global RV dilatation (increase of RV EDV according to the imaging test specific nomograms for age, sex and BSA) | ||
| or | ||
| -global RV systolic dysfunction (reduction of RV EF according to the imaging test specific nomograms for age and sex) | ||
| Minor | Minor | |
| Regional RV akinesia, dyskinesia or aneurysm of RV free wall | Regional LV hypokinesia or akinesia of LV free wall, septum, or both | |
| II. Structural myocardial abnormalities | By CE-CMR: | By CE-CMR: |
| Major | Major | |
| Transmural LGE (stria pattern) of |
LV LGE (stria pattern) of | |
| By EMB (limited indications): | ||
| Major | ||
| Fibrous replacement of the myocardium in |
||
| III. ECG repolarization abnormalities | Major | Minor |
| Inverted T waves in right precordial leads (V |
Inverted T waves in left precordial leads (V | |
| Minor | ||
| -Inverted T waves in leads V1 and V2 in individuals with completed pubertal development (in the absence of complete RBBB) | ||
| -Inverted T waves in V1, V2, V3 and V4 in individuals with completed pubertal development in the presence of complete RBBB | ||
| IV. ECG depolarization abnormalities | Minor | Minor |
| -Epsilon wave (reproducible low amplitude signals between end of QRS complex to onset of the T wave) in the right precordial leads (V1 to V3) | Low QRS voltages ( | |
| -Terminal activation duration of QRS |
||
| V. Ventricular arrhythmias | Major | Minor |
| -Frequent ventricular extrasystoles ( |
Frequent ventricular extrasystoles ( | |
| Minor | ||
| -Frequent ventricular extrasystoles ( |
||
| VI. Family history/genetics | Major | |
| -ACM confirmed in a first-degree relative who meets diagnostic criteria | ||
| -ACM confirmed pathologically at autopsy or surgery in a first-degree relative | ||
| -Identification of a pathogenic or likely pathogenetic ACM mutation in the patient under evaluation | ||
| Minor | ||
| -History of ACM in a first-degree relative in whom it is not possible or practical to determine whether the family member meets diagnostic criteria | ||
| -Premature sudden death ( | ||
| -ACM confirmed pathologically or by diagnostic criteria in second-degree relative | ||
| ACM, arrhythmogenic cardiomyopathy; BSA, body surface area; CE-CMR, cardiac enhanced-cardiac magnetic resonance; CMR, cardiac magnetic resonance; EDV, end diastolic volume; EF, ejection fraction; EMB, endomyocardial biopsy; LBBB, left bundle branch block; LGE, late gadolinium enhancement; LV, left ventricle; RBBB, right bundle branch block; RV, right ventricle; RVOT, right ventricular outflow tract. Adapted from Corrado et al. [19]. | ||
Finally, according to the number of LV and RV criteria that are fulfilled, the 2020 criteria provide a classification of ACM in three different phenotypic variants: “dominant right” variant, which is the classical form with RV involvement; “biventricular disease” variant, with involvement of both ventricles; “dominant-left” variant, with involvement of only the LV (Fig. 1, Ref. [20]).
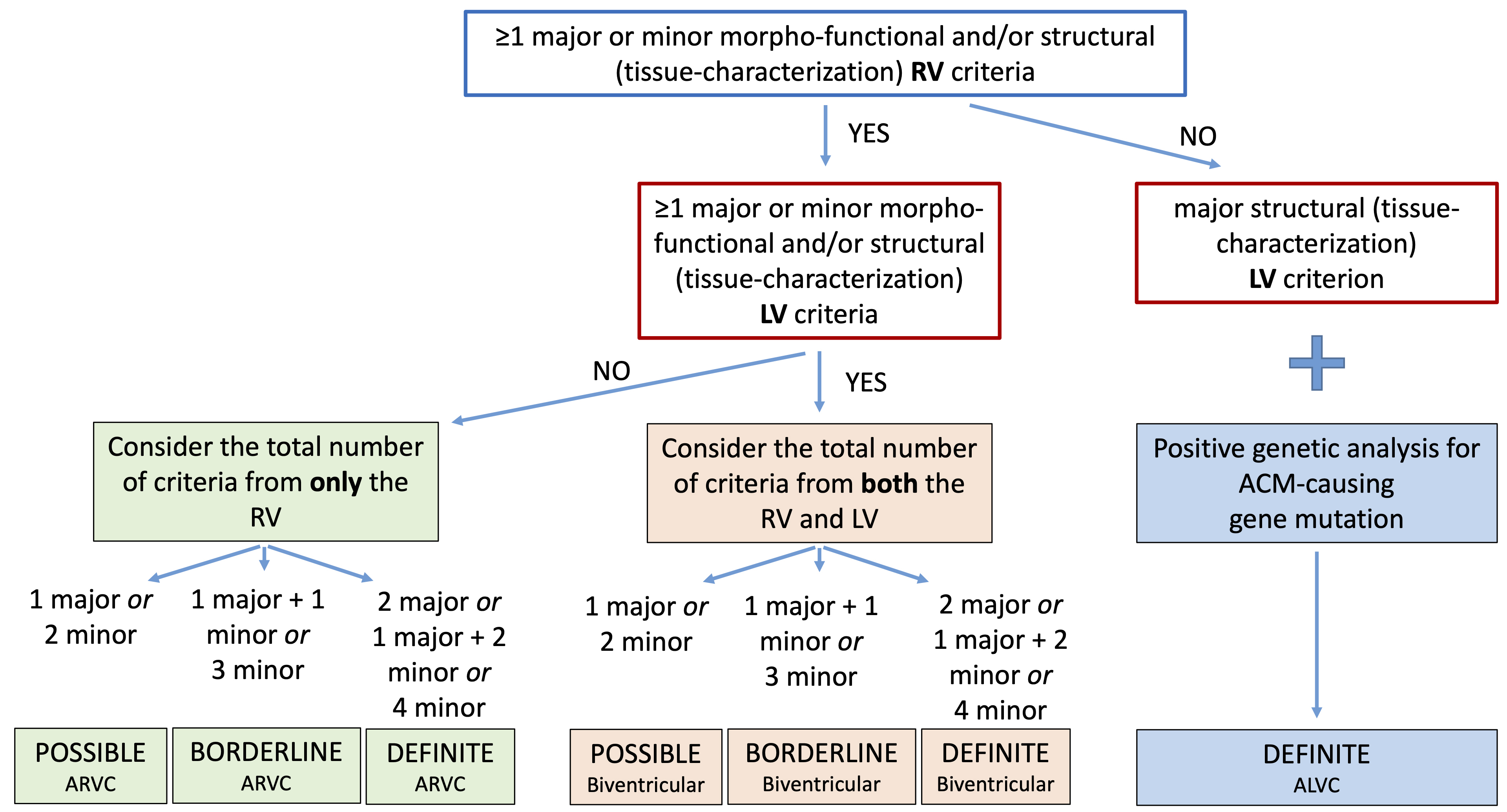 Fig. 1.
Fig. 1.Flowchart for phenotypic characterization of ACM. The diagnosis of ACM requires at least 1 morpho-functional or structural abnormalities criterion, either major or minor. The diagnosis of the specific phenotypic variant depends on the ventricle interested on alterations (see text for details). Moreover, the likelihood of disease is defined by the combination of the major and minor criteria fulfilled. ACM, arrhythmogenic cardiomyopathy; ALVC, arrhythmogenic left ventricular cardiomyopathy; ARVC, arrhythmogenic right ventricular cardiomyopathy; LV, left ventricle; RV, right ventricle. Adapted from Corrado et al. [20].
Once the diagnosis is reached, genetic testing and cascade family screening allow to classify the etiology of the disease into four categories: due to desmosomal gene mutation, due to non-desmomal gene mutation, familial but gene-elusive and non-genetic/non-familial. In this last case differential diagnosis with disease phenocopies must be considered, such as cardiac sarcoid (Fig. 2, Ref. [19]).
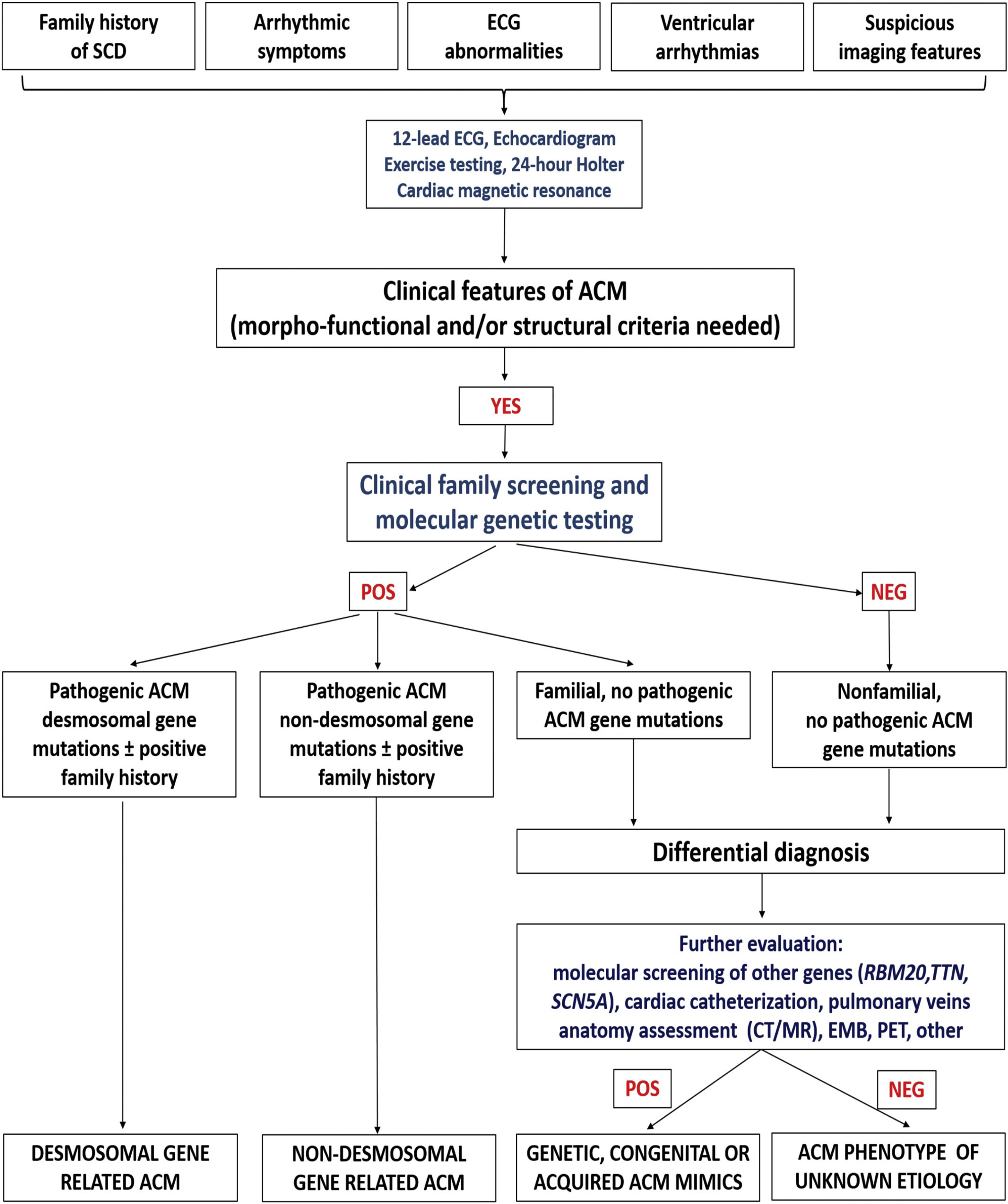 Fig. 2.
Fig. 2.Flow-chart for etiology assessment of ACM. After the diagnosis is reached in a proband, cascade family screening and molecular genetic testing may allow to identify patients with identified gene mutation in a desmosomal or non-desmosomal gene. In patients with negative genetic testing, cascade family screening may allow to identify other affected family members: in this case, the diagnosis is a familial disease with still unknown genetic basis (so-called “gene elusive”). In case both genetic testing and family screening are negative, further testing may be performed to exclude phenocopies such as congenital heart disease or myocarditis. Adapted from Corrado et al. [19].
Therefore, the use of the 2020 International criteria can be considered as a three-step process. The first step is represented by a systematically research of signs of ACM through the multiparametric approach involving functional and structural ventricular abnormalities, tissue characterization findings, depolarization and repolarization ECG alterations, ventricular arrhythmias, family history and genetic findings. The second step is the identification of the specific phenotype and the likelihood of the disease according to the combination of the criteria fulfilled. The third step is to investigate the disease aetiology and differentiate ACM from phenocopies.
I. Morpho-functional ventricular abnormalities
Such as in 2010 ITF criteria, echocardiography, CMR and angiography were indicated as possible tools for assessing morpho-functional ventricular abnormalities. However, in attempt to identify the disease earlier, any degree of RV dilation or dysfunction in association with regional wall motion abnormalities was considered a major RV criterion while the presence of regional wall motion abnormalities without RV dilatation or dysfunction was introduced as minor criterion. This finding is due to the regional nature of ACM that can affect the segmental contractility because of local fibro-fatty replacement, without compromising the global hemodynamics of the RV [14, 21]. At the same time, it has been classified as a minor criterion because of the misinterpretation of some normal variants of the RV wall motion [18].
The two morpho-functional criteria introduced for the LV are both minor because of the low specificity for diagnosing left-sided ACM variants. They include the detection of global LV systolic dysfunction with or without dilatation, and regional LV hypokinesia or akinesia. The use of strain echocardiography is recommended, because of the ability to detect subtle changes, especially in the early stages of the disease [22, 23].
Instead of using fixed cut-off values, the Padua criteria recommend the use of current reference values for cardiac chamber size and function, normalized for sex, age and body surface area, recommended by international societies of cardiovascular imaging [24, 25], and proper reference values for athletes, especially if engaged in sports associated with the greatest RV remodeling, such as canoeing and rowing [26].
II. Structural myocardial abnormalities
Fibrous or fibro-fatty replacement of myocardium is the pathological manifestation of ACM, and its detection through EMB has been indicated since 1994 [7]. However, because of its invasive nature with potential serious complications, the 2020 criteria have limited EMB indication to selected cases of non-familial ACM with negative genotyping to exclude phenocopies, such as myocarditis, sarcoidosis or DCM [15]. The demonstration of fibrous replacement of the RV myocardium in at least 1 sample, with or without fatty tissue, represents a major criterion. EMB is particularly helpful in ARVC, because of the peculiar topographic and histological features of the disease with fibro-fatty replacement reaching the subendocardium. A negative EMB do not exclude the diagnosis of ACM because of the possibility of sampling error. Moreover, the LV EMB is not indicated at present because the risk/benefit ratio is not yet known [27].
The introduction of non-invasive tissue characterization with CMR is one of the most important innovations of the 2020 criteria. The major CMR criterion is the presence of transmural LGE in at least 1 RV segment, confirmed in 2 orthogonal views. Currently, the diagnostic specificity of RV LGE is considered high, instead the sensitivity is low. This is due to the CMR technology characterized by a poor spectral resolution and suboptimal contrast/noise ratio in quantifying the thin RV wall [17, 28, 29, 30]. The highest specificity is reached when wall motion abnormalities and pre/post contrast signal alterations are considered together [29] (Fig. 3, Ref. [31]).
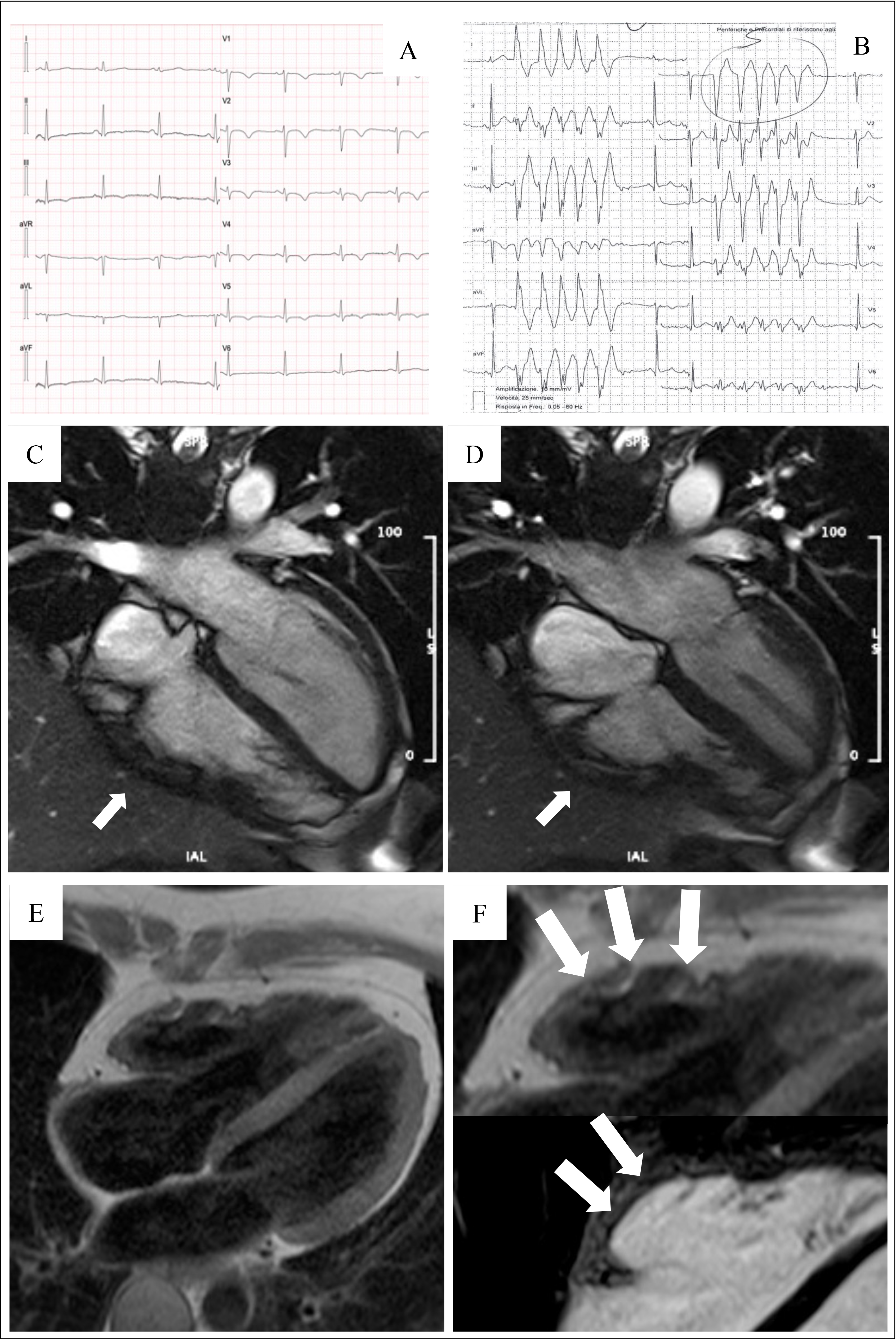 Fig. 3.
Fig. 3.Clinical features of ARVC. Basal ECG, exercise testing ECG and CMR findings in a 38-year-old woman hospitalized for sustained VT. Basal ECG showed TWI in V1–V5 and flattened T wave in inferior leads (A). Exercise testing revealed frequent PVBs and a non-sustained VT with LBBB/superior axis morphology, originating from RV free wall (B). CMR revealed mild RV dilatation, moderate RV systolic disfunction, a wide peritricuspid aneurysm, with an extreme thinning of the wall (four-chamber cine view in diastolic phase, C, and systolic phase, D). The PD-TSE sequences revealed fatty infiltration of the RV wall, especially in the subtricuspid region (E, and magnified on the top of F). No RV LGE was identified, not even in the same regions of RV fatty infiltration, maybe because an extreme thinning of the RV wall (F on the bottom). The diagnosis was “definite ARVC”. ARVC, arrhythmogenic right ventricular cardiomyopathy; CMR, cardiac magnetic resonance; LBBB, left bundle branch block; LGE, late gadolinium enhancement; PD-TSE, positron density-turbo spin echo; PVBs, premature ventricular beats; RV, right ventricle; TWI, T wave inversion; VT, ventricular tachycardia. Adapted from Graziano et al. [31].
In the LV, the presence of a stria of LGE with a non-ischemic distribution (subepicardial and/or midmyocardial, most affecting the inferolateral region) in at least 1 LV Bull’s Eye segment, confirmed in 2 orthogonal views (excluding junctional LGE, that is considered non pathologic) is a major criterion. Moreover, the circumferential involvement of septum and LV free wall in short axis view is called “ring-like” pattern, and it is considered as highly specific for ALVC [32]. Nonetheless, at present there is no gold standard for differentiating non-ischemic LGE secondary to ACM or to other diseases such as myocarditis: for this reason, in the absence of concomitant RV involvement, the diagnosis of left-dominant ACM in a proband requires positive genetic testing (Fig. 1). Fatty tissue infiltration is often observed on dedicated CMR sequences, but it is not considered a diagnostic criterion in isolation because of its low specificity.
In the early stages of LV involvement, the typical non-ischemic distribution of fibro-fatty replacement sparing the subendocardial layer can explain the absence of wall motion abnormalities, dilatation, or dysfunction of the LV. Thereby, the absence of LV functional abnormalities on echo, cine-CMR or angiography cannot rule out LV involvement, and CE-CMR characterization plays a key role in detection of left-sided ACM [14, 16, 33, 34, 35, 36] (Fig. 4, Ref. [14]).
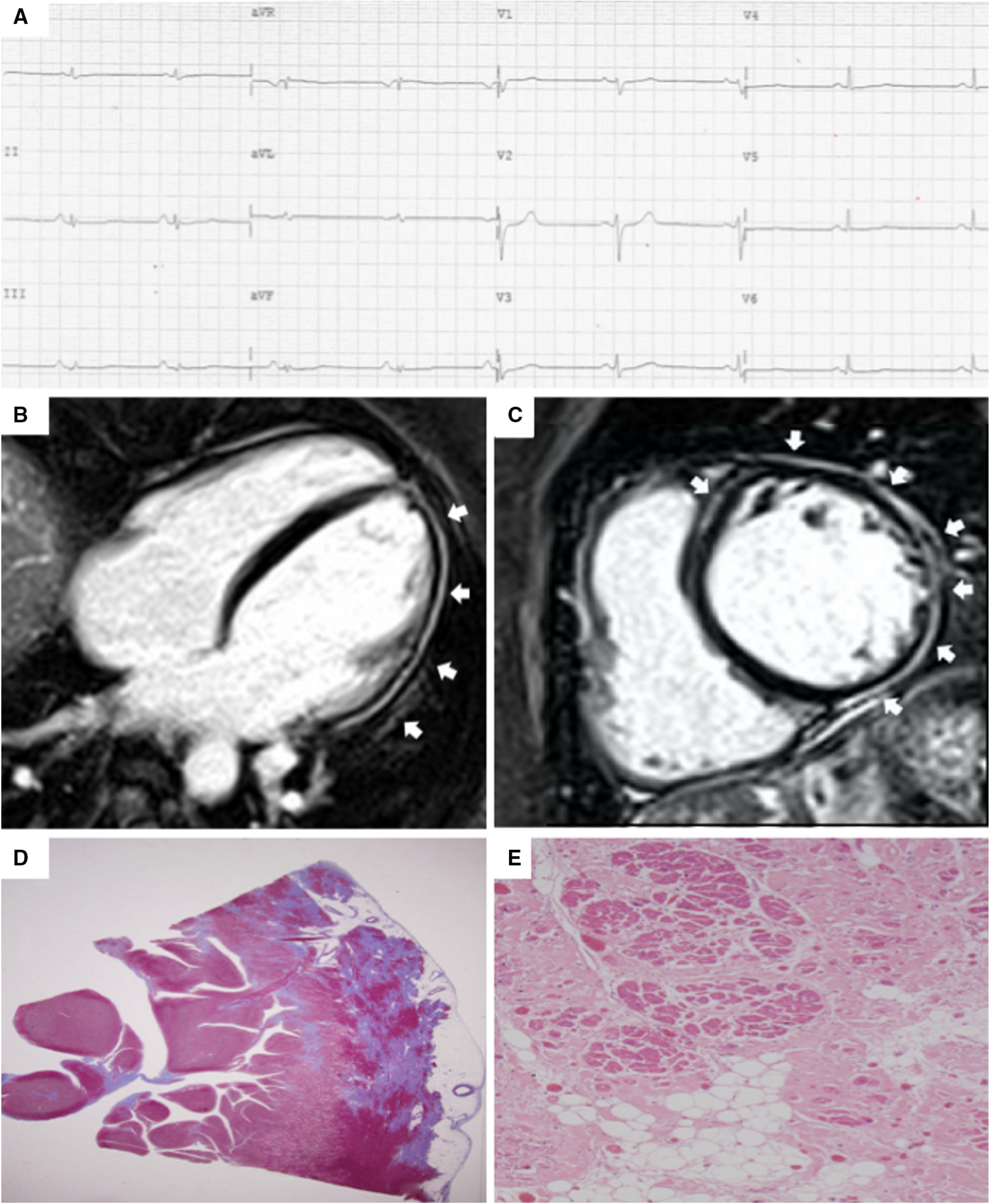 Fig. 4.
Fig. 4.Clinical and histopathological features of ALVC. Basal ECG and CMR findings in a patient who underwent cardiac transplantation because of ALVC related to a DSP gene mutation. Basal ECG revealed low QRS voltages in limb leads and flattened T-waves in infero-lateral leads (A). Post-contrast sequences on CMR (four-chamber view, B, and short-axis view, C) revealed subepicardial LGE involving the anterior septum and the whole LV free wall (“ring like” pattern) from basal to apical regions. Histology in LV inferolateral region demonstrated fibrofatty myocardial replacement in the subepicardial layer (D); a magnification of residual myocytes embedded within fibrous and fatty tissue (hematoxylin and eosin stain) (E). The diagnosis was “definite ALVC”. ALVC, arrhythmogenic left ventricular cardiomyopathy; CMR, cardiac magnetic resonance; DSP, desmoplakin gene; LGE, late gadolinium enhancement; LV, left ventricle. Adapted from Cipriani et al. [14].
III. Repolarization abnormalities
As regards to repolarization abnormalities, the detection of T wave inversion (TWI) in right precordial leads (V1–V3) or beyond, or in V1–V2 only, remain major and minor criteria respectively. These findings require the absence of a complete right bundle branch block (RBBB). Otherwise, in the presence of RBBB, TWI in V1–V4 is a minor criterion. These criteria are valid in patients with complete pubertal development. A remarkable consideration is that TWI extension from right precordial leads (V1–V3) to left ones (V4–V6) is the expression of a more severe RV dilatation, with its displacement toward the axilla, rather than of LV involvement [34]. LV involvement can be only predicted with TWI in left precordial leads (V4–V6) in absence of complete LBBB, but it is a minor criterion because of its low specificity [15, 33, 34].
IV. Depolarization abnormalities
Signal averaged ECG is no more included among Padua criteria based on the
experts’ opinion that they lacked specificity and showed low diagnostic accuracy
[16]. Moreover, the epsilon wave in right precordial leads has been downgraded to
minor criterion, because its identification and interpretation are highly
influenced by ECG filtering and sampling rate, with a consequently large
interobserver variability [37]. The ECG pattern in right precordial leads of a
terminal activation duration (TAD) of the QRS
The presence of low QRS voltages in limb leads (peak-to-peak QRS amplitude
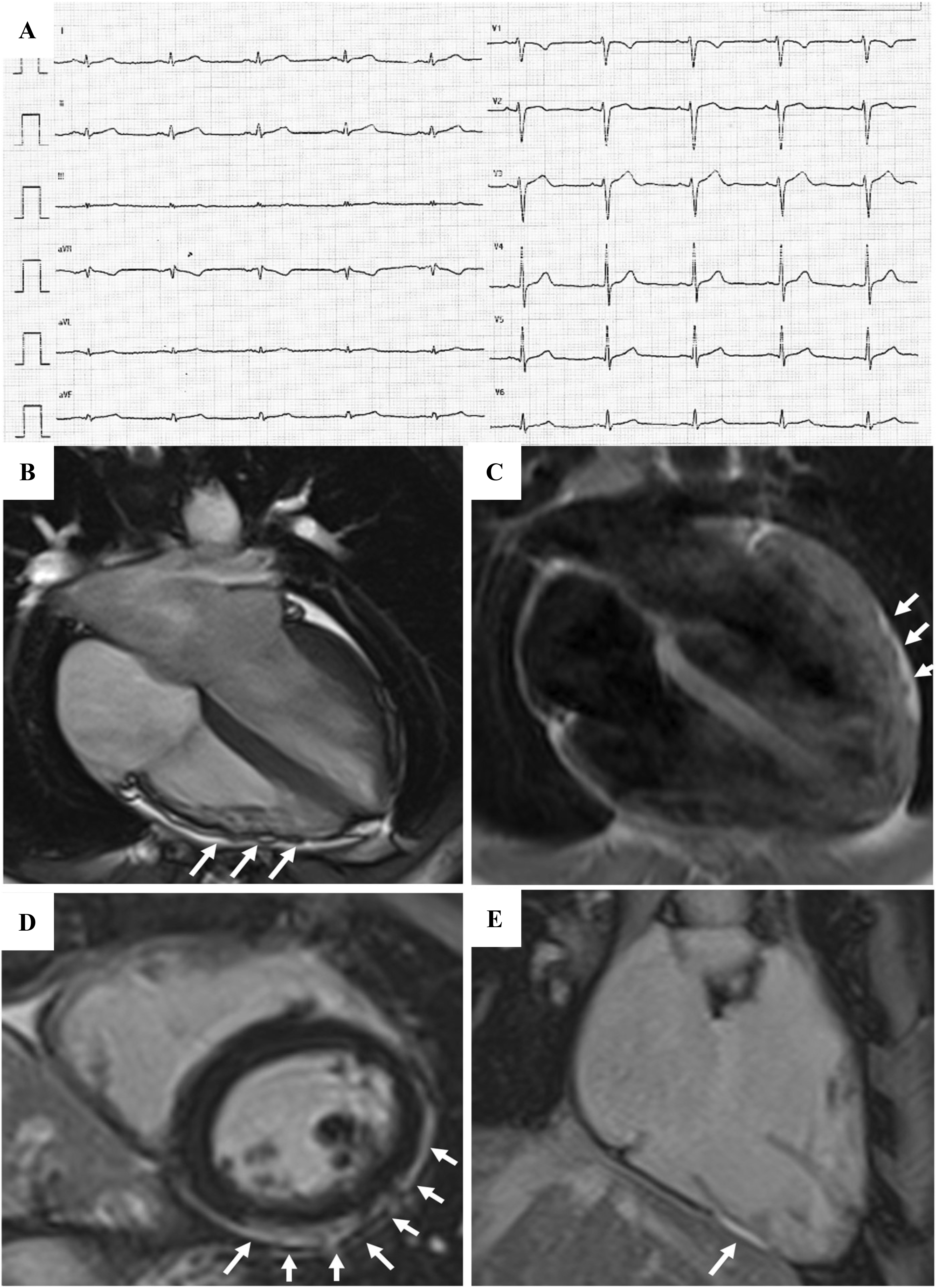 Fig. 5.
Fig. 5.Clinical features of biventricular ACM. Basal ECG and CMR findings in a 28-year-old elite athlete. ECG revealed low QRS voltages in limb leads (A). Exercise testing demonstrated PVBs with a RBBB/superior axis morphology, isolated and in couples. CMR cine-sequences showed hypokinesis of the mid-apical lateral wall and multiple small bulging of the RV free wall (B, four-chamber view). The PD-TSE sequences revealed epicardial fatty infiltration of the lateral and inferior LV walls (C, four-chamber view). Post-contrast sequences on CMR demonstrated a subepicardial stria of LGE involving the antero-lateral, infero-lateral and inferior LV walls (D, short- axis view) and also the RV inferior wall (E, RV inflow-outflow view). The diagnosis was “definite biventricular ACM”. ACM, arrhythmogenic cardiomyopathy; CMR, cardiac magnetic resonance; LV, left ventricle; PVBs, premature ventricular beats; PD-TSE, proton density-turbo spin echo; RBBB, right bundle branch block; RV, right ventricle. Adapted from Zorzi et al. [38].
V. Ventricular arrhythmias
ACM is characterized by premature ventricular beats (PVBs) with origin from or
around the fibro-fatty tissue. PVBs are considered in terms of absolute number
(
Electrophysiology study (EPS) is not considered a diagnostic criterion; however, it can be useful in selected patients for differential diagnosis. In particular, response to programmed ventricular stimulation and isoproterenol may allow to differentiate between idiopathic right-ventricular outflow tract (RVOT) VT or Brugada syndrome and the scar re-entrant VT of ACM. Addition of RV endocardial voltage mapping may be of incremental diagnostic value for differential diagnosis with idiopathic RVOT VT, as demonstration of low-voltage areas suggest the presence of fibro-fatty scars [16].
VI. Family history and molecular genetics
Compared to the 2010 ITF criteria, in the Padua criteria the category family history and molecular genetics has not changed. Moreover, the category is shared between RV and LV criteria. This is because the manifestation of ACM and the predominant ventricular involvement may vary among members of the same family and with the same gene mutation. However, more restricted indications for genotyping have been proposed to the new criteria, trying to avoid misinterpretation of molecular genetic results and misdiagnosis. The genetic test is recommended in probands with a definite biventricular or ARVC diagnosis, in order to screen family members [40]; it may be considered in borderline forms to reach the definite diagnosis, provided that the results of genetic test are interpreted by experts in ACM; it is mandatory to reach the diagnosis of non-familial ALVC to exclude phenocopies [16].
Major criteria are the detection of a pathogenic or likely pathogenic ACM gene mutation in the patient under evaluation, the history of a first-degree relative with ACM confirmed pathologically at autopsy or surgery or who reached the criteria necessary for ACM diagnosis. The minor criteria are the suspicion of ACM without confirmation in a first-degree relative, the suspicion of ACM in a first-degree relative who suddenly died before the age of 35, the confirmed diagnosis of ACM in a second-degree relative.
The second step is the identification of the specific ACM phenotype according to the number of criteria for the RV and LV involvement that are fulfilled. According to the 2020 ITF criteria, any diagnosis of ACM requires that at least 1 criterion from category I (morpho-functional abnormalities) or II (structural abnormalities) must be reached, either major or minor, and only these two categories are taken into consideration to classify the phenotypic variant.
If these criteria are only fulfilled for the RV, the diagnosis is the classical right-dominant variant (ARVC). Whereas, if the criteria are fulfilled for both RV and LV, the diagnosis is “biventricular” form. Moreover, it is possible to define the likelihood of disease according to the number of major and minor criteria reached from all categories. So, the diagnosis can be “definite” if either 2 major criteria, or 1 major and 2 minor criteria or 4 minor criteria are fulfilled, “borderline” if either 1 major and 1 minor criterion or 3 minor criteria are reached, and “possible” if either 1 major criterion or 2 minor criteria are satisfied.
The diagnosis of ALVC is reached in patients with structural LV abnormalities (major criterion) and no RV involvement, when a pathogenic or likely pathogenic ACM-causing gene mutation is identified. In this case, the diagnosis of ALVC is always definite. The need for positive genotyping testing is due to the possible overlap of these morpho-functional and structural findings with phenocopies, such as DCM, cardiac sarcoidosis or myocarditis.
After the clinical diagnosis of ACM and the definition of the specific phenotype, the third step is to define the aetiology of ACM and to exclude phenocopies. This purpose can be reached thanks to molecular genetic testing and cascade family screening. Indeed, ACM is generally transmitted as an autosomal dominant trait, with variable expressivity and incomplete penetrance. So, the molecular genetic test can identify either desmosomal or non-desmosomal gene defects causing ACM.
Desmosomes are proteins forming the area composita of the intercalated disc that
are structure crucial for electromechanical connection of cardiomyocytes and
intracellular signaling cascades. These structures are also composed by adherens
junctions, gap junctions and ion channels. Pathogenic mutations of gene encoding
desmosomal proteins such as plakophilin (PKP2), desmoplakin (DSP), desmoglein
(DSG2) and desmocollin (DSC2) are identified in
ACM left-sided variants are also associated with mutations in non-desmosomal
genes encoding for ion channels and cytoskeletal components, such as lamin A/C
(LMNA), filamin C (FLNC), transmembrane protein 43 (TMEM 43), desmin (DES), titin
(TTN), phospholamban (PLN), the cardiac ryanodine receptor-2 (RYR2), sodium
voltage-gated channel alpha subunit 5 (SCN5A) and transforming growth factor
beta-3 (TGF
In case of a negative molecular genetic testing but positive clinical family screening, ACM is defined familial but “gene elusive”. Also in this case, the presence of affected relatives allows to rule out non-hereditary conditions mimicking ACM.
If both the genetic testing and the cascade clinical family screening for ACM are negative, it is essential to perform further evaluations in order to exclude mimics, both acquired (sarcoidosis, DCM, pulmonary artery hypertension, myocarditis, Chagas disease) and congenital (left-to right shunt or Ebstein anomaly) phenocopies.
In a cohort of 87 patients from the University of Padua who fulfilled the 2010 International Criteria for definite ACM, the application of the Padua criteria allowed to re-classify 51 of them as biventricular ACM because they also fulfilled either the morpho-functional or the structural criteria for LV involvement. Moreover, 5 of 15 patients with borderline diagnosis according to the 2010 ITF criteria were re-classified as definite ACM according to the Padua criteria. Finally, 9 patients with desmosomal-gene mutations but no signs of RV involvement met the major LV structural criterion and were thus re-classified as ALVC [14].
The additional value of the Padua criteria compared to the 2010 ITF was particularly evident among carriers of gene mutations characterized by predominant LV involvement such as desmoplakin, phospholamban and filamin-C genes. In a pooled analysis of patients with FLNC cardiomyopathy, 60 were diagnosed with ACM. Based on the 2010 ITF criteria, only a minority of patients fulfilled the criteria for definite ACM but according to the Padua criteria more than half of cases were diagnosed with definite left-dominant ACM [43]. Of 72 probands with DSP-gene mutations, Bariani et al. [44] showed that 26 had pure LV involvement and 7 biventricular involvement, but only 20 a classical ARVC. Overall, the number of patients reaching a definite diagnosis raised from 32 to 49 patients by using the 2020 Padua criteria compared with the 2010 ITF criteria. Moreover, Cicenia et al. [45] demonstrated that the application of the Padua criteria increased the sensitivity for ACM compared to the 2010 ITF criteria also in a small pediatric cohort, by demonstrating LV in half of the study sample.
These preliminary studies suggest the accuracy of the Padua criteria, but future studies on large populations are necessary to confirm their validity in diagnosing, and to assess their possible use for risk stratification and management of patients, especially in variants involving LV.
However, potential drawbacks of the Padua Criteria should be recognized. The first and most important is that they were proposed by a group of authors from the University of Padova and endorsed by several external experts, but they do not represent the result of an international consensus conference such as the 2010 ITF criteria. For this reason, they are still not universally accepted. There are then specific criteria that were based on experts’ opinion and thus require that their diagnostic accuracy is evaluated in the clinical practice. For example, evaluation of isolated wall motion abnormalities, particularly LV hypokinesia, is subject to high inter-observer variability; the acceptance of fibrotic changes in only one biopsy without any further quantification may potentially give rise to overestimation; and the exclusion of SAECG from Padua criteria was based on the experts’ opinion and was not supported by scientific data.
The development of the 2020 International criteria was a necessary step to improve the capability of diagnosing ACM. The most important innovation is the recognition and characterization of left-sided variants, which were underdiagnosed with the previous criteria. Because the typical ACM lesion is the subepicardial scar that may not cause wall motion abnormalities (particularly in the LV), the tissue characterization ability of CMR has become crucial. Preliminary data suggest that the diagnostic accuracy of ACM has improved thanks to the clinical use of the Padua criteria [14].
FG, AZ, AC, MDL, BB, IR, GB, KP, CB, MPM and DC contributed to design the research study. FG, AZ and DC wrote the manuscript. FG, AZ, AC, MDL, BB, IR, GB, KP, CB, MPM and DC contributed to editorial changes in the manuscript. FG, AZ, AC, MDL, BB, IR, GB, KP, CB, MPM and DC read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest. Alessandro Zorzi is serving as one of the Editorial Board members of this journal. We declare that Alessandro Zorzi had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Stefan Peters.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.