
† These authors contributed equally.
Academic Editor: Brian Tomlinson
The aim of this study was to investigate the correlation between v-set and transmembrane domain-containing 1 (VSTM1) expression and incidence of major adverse cardiac events (MACE) in patients with coronary heart disease (CHD). A total of 310 patients were divided into a non-acute coronary syndrome (non-ACS) group (containing the stable angina group, and the asymptomatic coronary artery diseaseand other patients group) and an ACS group (containing unstable angina and acute myocardial infarction patients). Monocytic VSTM1 expression levels (assessed via average fluorescence intensity derived from antibody binding to VSTM1) in each group were detected and analyzed. The cut-off value of monocytic VSTM1 expression to predict the onset of ACS and MACE was confirmed. VSTM1 expression in monocytes from the ACS group was lower than that of the non-ACS group. The incidence of MACEs in the high VSTM1-expression group was much less than that of those in the low VSTM1 expression group at the 1 year follow-up stage. VSTM1 expression had an independent-inversed association with increased incidence of MACE and ACS. VSTM1 expression in monocytes may help to predict the occurrence of ACS in patients with CHD, and moreover it may provide the means to evaluate MACE prognosis during CHD patient follow-up.
Coronary heart disease (CHD), also known as coronary artery disease (CAD), is one of a series of coronary artery diseases affecting hundreds of millions of people worldwide. It is the leading cause of death in the world and typically develops as the result of atherosclerosis (AS) build-up in the arteries [1, 2]. The unitability of atherosclerotic plaque primarily contributes to acute cardiovascular events, such as ST-elevation acute coronary infarction, non-ST-elevation myocardial infarction and unstable angina, these diseases are collectively known as acute coronary syndrome (ACS) [3, 4]. Monocytes/macrophages are a critical cell type in the pathological process of AS, which involves the initiation, progression and eventually rupturing of atherosclerotic atheroma [5, 6]. The formation of foam cells, which have a monocyte/macrophage origin, are the hallmark of atherogenesis [7]. Recent studies have shown the core status of monocytes/macrophages in the formation and progression of AS. High monocyte/macrophage biological function is one of the principal factors associated with plaque instability which induces ACS [8].
V-set and transmembrane domain-containing 1 (VSTM1), also known as signal
inhibitory receptor on leukeocytes-1 (SIRL1), is a recently discovered cell
membrane receptor. VSTM1 is located on human chromosome 19q. 13.4, adjacent to
the region of the leukocyte receptor complex, which is highly similar to many of
the receptor proteins regulating leukocyte function, such as LAIR-1 [9]. As a
membrane receptor, VSTM1 consists of extracellular, transmembrane, and
intracellular sequences. Its intracellular sequence is highly conserved between
different species and genera, suggesting the importance of its function. VSTM1 is
composed of two ITIM (immunoreceptor tyrosine-based inhibitory motif) regions,
which affect its downstream targets through tyrosine phosphorylation [10]. VSTM1
is mainly distributed in myeloid cells, especially on the surface of monocytes
[11, 12, 13]. It has been reported that VSTM1 expression is negatively correlated with
the severity of the inflammation [9, 14]. Simultaneously, similar phenotypic
changes of proinflammatory cytokine-tumor necrosis factor (TNF-
To test this hypothesis, monocytes were isolated from the peripheral blood of CHD patients to assess the relationship between monocytic VSTM1 expression and severity of CHD, in addition to investigating the value of VSTM1 as a tool for prognosis evaluation in ACS patients.
Patients were enrolled between October 2018 to March 2019. CHD diagnosis was
confirmed according to coronary artery branch diameter loss of
The patient inclusion criteria were: (1) Age
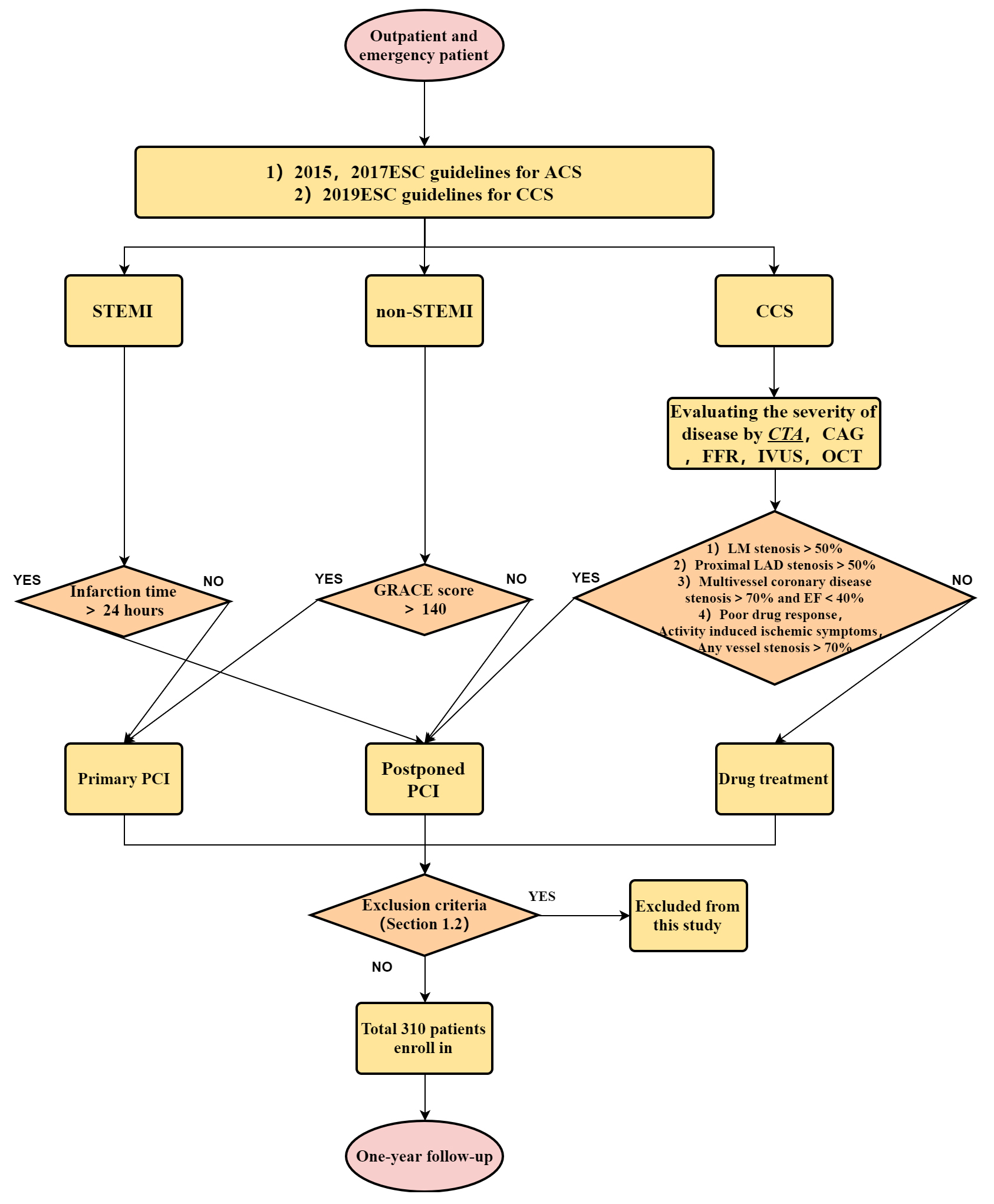 Fig. 1.
Fig. 1.Flow chart of the study.
A total of 310 patients that met the inclusion criteria but not the exclusion criteria were included in this study. These patients were divided into a non-ACS group and an ACS group based on clinical symptoms, laboratory examination and CAG results. The non-ACS group included the stable angina group, and the asymptomatic coronary artery disease and other patients, whereas the ACS group included unstable angina (UA) and acute myocardial infarction (AMI) patients. The AMI group contained non-ST-elevation myocardial infarction (NSTEMI) and ST-elevation-myocardial infarction (STEMI) patients.
Stable angina was defined as a rapid, temporary, reversible ischemia-hypoxia
syndrome due to increased myocardial load on the basis of coronary artery
stenosis. Asymptomatic coronary artery disease or ‘others’ were defined as
patients who did not have chest pain and myocardial ischemia when conducting
daily activities while the CAG showed coronary stenosis and an insufficient
coronary blood supply. Unstable angina was defined as angina at rest,
accelerating angina, or new-onset angina, without concomitant elevation of
cardiac markers. NSTEMI was defined as ischemic symptoms with elevated cardiac
markers in the absence of ST-elevation on ECG. STEMI was defined as continuous
chest pain lasting
The one year follow-up MACE events included cardiac death, non-fatal myocardial
infarction, target vessel revascularization (TVR) and target lesion
revascularization, (TLR). Cardiac death was defined as any death attributable to
a cardiovascular event (such as myocardial infarction, low output failure, fatal
arrhythmias). Non-fatal MI was defined as, after 8 hours of PPCI, a two-fold
elevation of the upper limit of normal creatine kinase myocardial band (CK-MB) or
troponin levels after 8 hours of PPCI. In addition, this was accompanied by one
or more of the following symptoms: new/recurrent sustained ischemic chest pain,
hemodynamic decompensation, or new/recurrent ST elevation/depression of
The basic data about each patient was retrieved from the medical records, this included gender, age, history of hypertension, diabetes, cerebral infarction, smoking, glycosylated hemoglobin, low-density lipoprotein (LDL), high-density lipoprotein (HDL), and other details pertinent to the study. CAG was performed using an FD20 cardiovascular angiography instrument (Philips, the Netherlands). The standard Judkins technique or right radial artery approach was used to determine the location and degree of stenosis.
The expression of VSTM1 in monocytes was detected using flow cytometry. After
admission, the relevant clinical data collection and biochemical index
examination were conducted for each patient. Briefly, 2 mL of venous blood was
collected from each patient on their admission day, this was mixed with EDTA-K
anticoagulants. Following centrifugation for 5 min, the upper aqueous phase
containing the leukocytes was collected. Thereafter VSTM1 within/on the monocytes
was labeled using an indirect immunofluorescence antibody. The leukocyte
suspension was mixed with human anti-CD14, followed by incubation at 4
The VSTM1 fluorescence intensity associated with the monocytes was determined via flow cytometry using a Becton Dickinson FACSCalibur. The excitation wavelength was set at 488 nm. Primarily, leukocytes with strained with anti-CD14 (ab183322, Abcam, Boston, MA, US) and isotype control (SA00006-2, proteintech, Chicago, IL, US), were sorted according to cell size, intracellular particle density, forward angle scattering (FSC), and lateral angle scattering (SSC), the leukocytes were divided into three subgroups as follows: neutrophils, lymphocytes, and monocytes. The monocytes gate underwent further study and the purity of monocytes was confirmed based on the fluorescence intensity of CD14. Subsequently leukocytes strained with anti-VSTM1 (ab189494, Abcam, Boston, MA, US) were used to detect the degree of VSTM1 protein expression on the cell membrane, and mean fluorescence intensity was applied to assess the expression of VSTM1 in the monocytes subgroup. Approximately 10,000 monocytes were analyzed using this predetermined gate. The average fluorescence intensity was visualized using a histogram.
Statistical analysis was performed using SPSS software
(version 22.0, IBM Corp., Chicago, IL, USA). Categorical variables were shown as
frequencies and compared using the chi-square or Fisher exact test as
appropriate. Normally distributed continuous variables are shown as mean and
standard deviation, and the groups were compared using a Student’s
t-test. Non-normally distributed continuous variables were shown as
median with interquartile ranges, and compared using a Mann-Whitney U test. A
2-sided P-value of
The baseline demographic characteristics and laboratory data for both the
non-ACS and ACS groups are displayed in Table 1. There were no significant
differences in any of the factors such as age, gender, history of hypertension,
history of diabetes, and triglyceride between the groups
(P
| Non-ACS | ACS | P | |||||||
| SA (N = 154) | N% | Asyp (N = 51) | N% | UA (N = 49) | N% | AMI (N = 56) | N% | ||
| Age | 64.6 |
/ | 68.18 |
/ | 70.06 |
/ | 64.80 |
/ | 0.229 |
| Male | 88 (57.14%) | 28.39 | 33 (64.71%) | 10.65 | 30 (61.22%) | 9.68 | 40 (71.43%) | 12.90 | 0.661 |
| Smoke | 31 (20.13%) | 10.00 | 19 (37.25%) | 6.13 | 18 (36.73%) | 5.81 | 27 (48.21%) | 8.71 | 0.024 |
| Hypertension | 97 (62.99%) | 31.29 | 27 (52.94%) | 8.71 | 30 (61.22%) | 9.68 | 30 (53.57%) | 10.00 | 0.631 |
| Diabetes | 31 (20.13%) | 10.00 | 16 (31.37%) | 5.16 | 15 (30.61%) | 4.84 | 19 (33.93%) | 6.13 | 0.422 |
| Total cholesterol (mg/dL) | 172.9 |
/ | 168.7 |
/ | 170.7 |
/ | 190.5 |
/ | 0.031 |
| Triglycerides (mg/dL) | 124.2 |
/ | 130.9 |
/ | 132.8 |
/ | 137.8 |
/ | 0.682 |
| LDL cholesterol (g/dL) | 95.5 |
/ | 91.2 |
/ | 109.4 |
/ | 119.5 |
/ | 0.004 |
| HDL holesterol (mg/dL) | 41.4 |
/ | 39.8 |
/ | 39.4 |
/ | 37.9 |
/ | 0.401 |
| ACS, acute coronary syndrome; SA, stable angina group; Asyp, asymptomatic coronary artery disease or others group; UA, unstable angina group; AMI, acute myocardial infarction group; LDL, low density lipoprotein; HDL, high density lipoprotein; the same abbreviations are used in the other tables. | |||||||||
As shown in Fig. 2, anti-CD14 and its isotype control were initially used to
identify monocytes with the aid of flow cytometry (FCM) (Fig. 2A). In subsequent
studies, anti-VSTM1 was applied to detect the differential expression of VSTM1 in
monocytes from the non-ACS and ACS groups. The FCM results revealed that VSTM1
expression was higher in the non-ACS than the ACS group (P
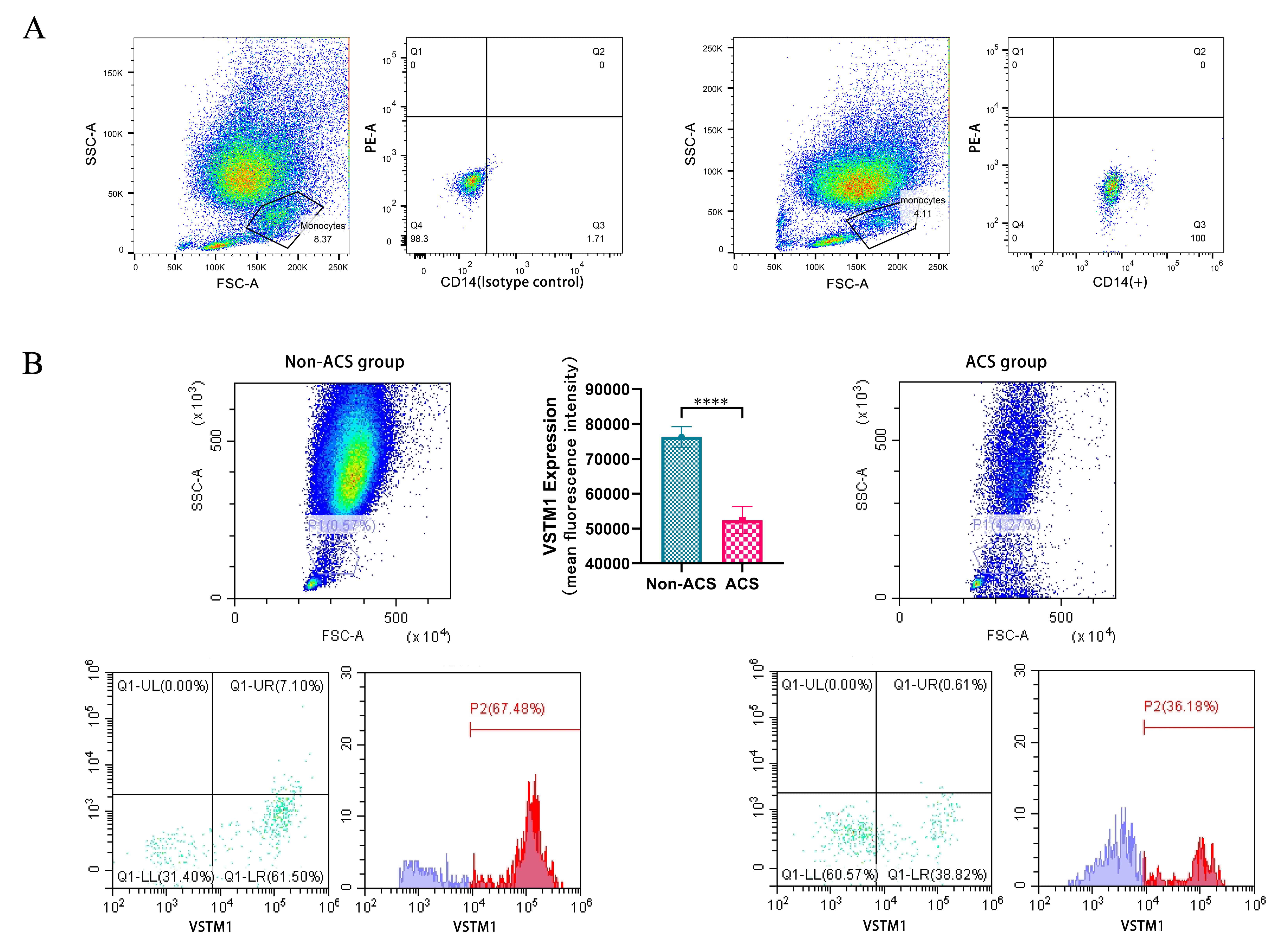 Fig. 2.
Fig. 2.Monocyte identification and VSTM1 expression detection in
monocytes using FCM. (A) Identification of monocytes using anti-CD14 and its
isotype control. (B) Differential expression of VSTM1 in monocytes from the
non-ACS and ACS groups was assessed by FCM (mean fluorescence intensity) using
anti-VSTM1. The results revealed that VSTM1 expression in the non-ACS group was
higher than the ACS group (P
| Non-ACS | ACS | P | |||
| SA (N = 154) | Asyp (N = 51) | UA (N = 49) | AMI (N = 56) | ||
| VSTM1 | 76284.52 |
77704.48 |
52396.69 |
53179.35 |
|
| 76637.78 |
52814 |
||||
In Fig. 3, it was found out that the cut-off value of VSTM1 expression to
differentiate patients with versus those without MACE was 66657 (area under the
curve =0.658 (0.619–0.747), P
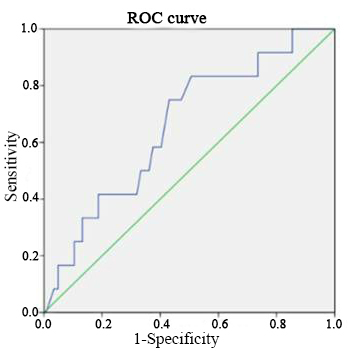 Fig. 3.
Fig. 3.The receiver operating characteristic (ROC) curve to
detect the best cut-off value of VSTM1 expression in predicting patients with and
without MACE.
Best cut-off: 66657; AUC: 0.658; Sensitivity: 83.3%; Specificity: 49.3%; PPV:
61.94%; NPV: 74.7%.
| P-value | |||
| Age | 67.21 |
66.54 |
0.627 |
| Male | 66 (69.5%) | 125 (58.1%) | 0.059 |
| Smoke | 46 (48.4%) | 49 (22.8%) | |
| Hypertension | 62 (65.3%) | 122 (56.7%) | 0.159 |
| Diabetes | 22 (23.2%) | 59 (27.4%) | 0.429 |
| LDL cholesterol, mg/dL | 116.8 |
91.9 |
|
| Triglycerides, mg/dL | 136.0 |
130.0 |
0.618 |
| SA | 30 (31.6%) | 124 (57.7%) | |
| Asyp | 8 (8.4%) | 43 (20.0%) | 0.011 |
| UA | 35 (33.3%) | 14 (6.5%) | |
| AMI | 48 (50.5%) | 8 (3.7%) |
After the traditional risk factors had been adjusted for, smoking and serum
LDL-c level were risk factors that increased the incidence of ACS (odds ratio
[or]: 2.325; 95% confidence interval [CI]: 1.409–3.837, P
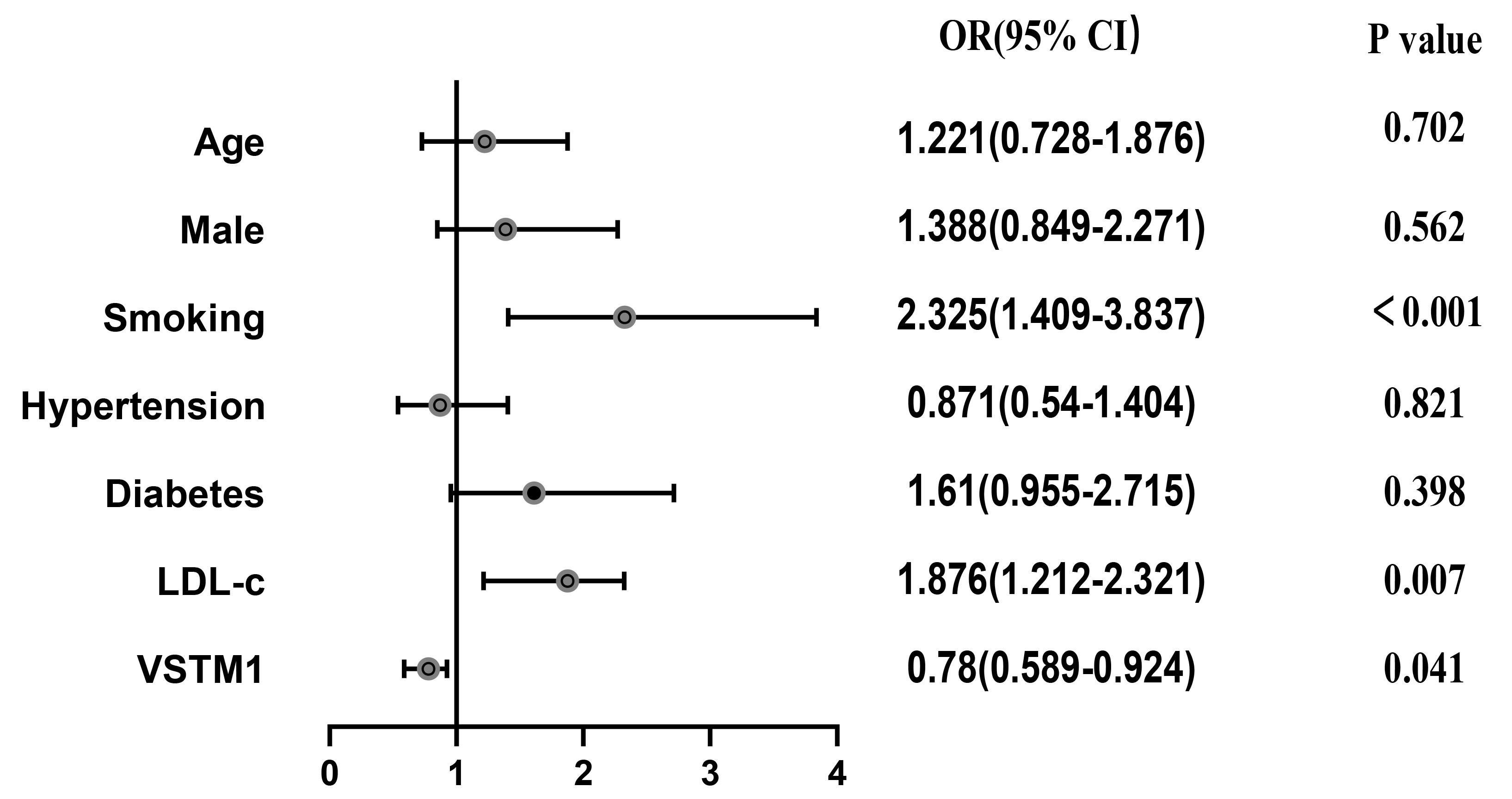 Fig. 4.
Fig. 4.Coronary heart disease risk factors. Smoking and serum LDL-c
levels were considered risk factors that increased the incidence of ACS (odds
ratio [or]: 2.325; 95% confidence interval [CI]: 1.409–3.837, P
A total of 11 patients experienced MACE during their 1 year of follow-up (Table 4). The Kaplan-Meier curve (Fig. 5) showed that the high-VSTM1 group had
a lower incidence of MACE compared with the low-VSTM1 group at the 1 year follow
up stage (P = 0.04). In accordance with the presence or absence of MACE,
the patients were divided into two groups as follows, MACE group and non-MACE
group. Table 5 shows the baseline characteristics of the two groups. Compared
with the non-MACE group, the proportion of smokers, and the levels of LDL,
observed in the MACE group were significantly higher in comparison to the
non-MACE group (P
| Non-ACS | ACS | |||
| SA (N = 154) | Asyp (N = 51) | UA (N = 49) | AMI (N = 56) | |
| Cardiac death | 0 | 0 | 0 | 0 |
| Non-fatal MI | 0 | 0 | 0 | 1 |
| TVR | 0 | 0 | 0 | 0 |
| TLR | 0 | 2 | 3 | 5 |
| MACE, major adverse cardiac event; MI, myocardial infarction; TVR, target vessel revascularization; TLR, target lesion revascularization. | ||||
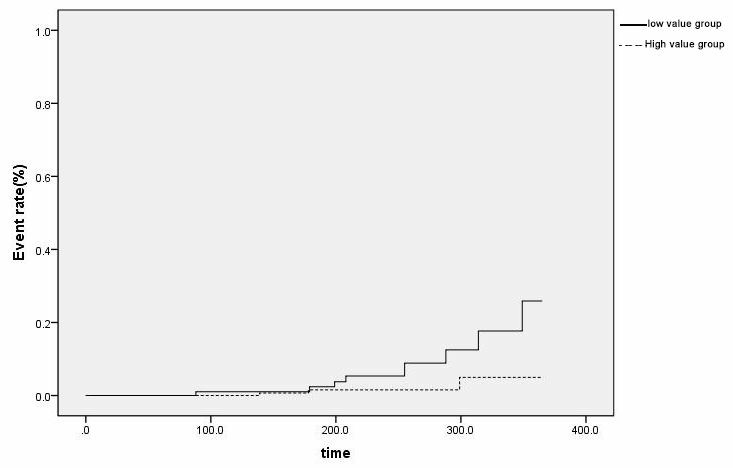 Fig. 5.
Fig. 5.Kaplan–Meier graphs comparing event rate between the study groups. The Kaplan-Meier curves for the high VSTM1 and low VSTM1 groups (P = 0.04).
| Total | MACE group | Non-MACE group | P | |
| (310 patients) | (11 patient) | (299 patients) | ||
| Age | 68.04 |
69.30 |
67.73 |
0.687 |
| Man | 191 | 8 | 183 | 0.440 |
| Smoke | 95 | 8 | 87 | 0.006 |
| Hypertension | 184 | 7 | 177 | 0.768 |
| Diabetes | 81 | 6 | 75 | 0.067 |
| Total cholesterol, mg/dL | 177.2 |
199.4 |
175.5 |
0.072 |
| HDL cholesterol, mg/dL | 39.0 |
37.5 |
39.1 |
0.728 |
| LDL cholesterol, mg/dL | 107.1 |
119.7 |
106.2 |
0.008 |
| Triglycerides, mg/dL | 134.0 |
144.2 |
133.2 |
0.654 |
| HbA1c, % | 6.5 |
6.7 |
6.5 |
0.663 |
| Creatinine, mg/dL | 1.1 |
1.11 |
1.1 |
0.965 |
| LVEF, % | 59.9 |
58.4 |
60.1 |
0.625 |
| Prior MI | 15 | 0 | 15 | 0.446 |
| Prior PCI | 56 | 4 | 52 | 0.108 |
| Prior CABG | 4 | 0 | 4 | 0.699 |
| VSTM1 | 61093.51 |
53989.84 |
75599.42 |
0.002 |
In the present study, the independent correlation between monocyte expression of VSTM1 and the severity of the lesion natures in different stages of CHD, that is, the predictive value of VSTM1, were explored. VSTM1 provided an objective and effective method by which prognosis could be estimated in CHD patients undergoing PCI, and could therefore act as a potential biomarker in detecting plaque vulnerability and assessing the incidence of MACE events in CHD patients.
It is known that inflammation contributes greatly to the formation of atherosclerotic plaques [20, 21]. AS is amonocyte/macrophage inflammatory response to the “pathogenic” lipoproteins invading the arterial wall, and these cells promote the initiation and progression of AS, and dysfunction of monocytes/macrophages act as a critical factor in the pathological progression and vulnerability of atherosclerotic plaques [6, 7, 22, 23, 24]. Therefore, the migration and invasiveness of macrophages both play critical roles in the occurrence and development of AS [25, 26]. Furthermore, it is now generally believed that the enhancement of intimal monocytes/macrophages accumulation can deteriorate the vulnerability of the plaques, or make the plaques prone to rupture, leading to thrombosis in atherosclerosis [27, 28]. Based on these factors, vulnerable atherosclerotic plaques in humans present with a thin-cap fibroatheroma (TCFA) and a large lipid-rich core with monocyte/macrophage infiltration [29, 30], and the hyperfunction of phagocytosis, migration, chemotactic responses of monocytes/macrophages are prone to enlarging the lipid core and weaken the fibrous cap. Mechanically, the formation of TCFA and a large lipid-rich core are the most important determinants of plaque rupture in the occurrence of ACS, which may be interpreted as a process of plaque stabilization [8, 31, 32, 33].
Importantly, VSTM1 expression in the ACS group was significantly lower when
compared with the non-ACS patients (Table 2). A total of 310 CAD patients were
followed up for 1 year, and they were sub-divided into a low-VSTM1 group (VSTM1
Our study unveiled that the objective assessment of reduced VSTM1 expression was independently associated with an increased incidence of AMI after the other cardiovascular risk factors had been considered. At the 1 year follow-up stage, the ratio of revascularization and combined adverse events in the ACS group was higher in comparison to the non-ACS group. This might be attributed to VSTM1 acting as a negative index of inflammation. In other words, the more unstable the plaque, the lower the level of VSTM1 expression. Studies have previously found that VSTM1 expression in ACS patients was significantly lower than that in patients with stable angina pectoris. The pathological basis of ACS is atherosclerotic plaque rupture and thrombosis, which may not occur even in severe cases of patients with stable CAD. Our study showed that VSTM1 acts as a protective factor and is independently associated with the prognosis of CHD even after other cardiovascular risk factors were adjusted and accounted for. This novel study reported the impacts of VSTM1 expression on AS and its association with prognosis. Our findings suggested that VSTM1 expression was an important marker, which reflected the development of unstable plaques in CAD.
Limitations existed within our study: (1) In our heart center, due to strict follow-up and medication management (including DAPT, statin, ezetimibe, PCSK9 inhibitors and other interventions), the incidence of MACE was kept to a relatively low degree, which led to the sample size being relatively small in this study. But the sample of MACEs derived from the CHD patient population, which was overall relatively large, ensures this information was relatively complete. (2) It is not clear whether monocytic VSTM1 expression or monocytic dysfunction induced by abnormal expression of VSTM1 may interact with other CHD risk factors such as smoking, diabetes mellitus, CKD and so on. Therefore, the interaction between VSTM1 and related risk factors of CHD require further research.
Our study revealed that monocytic VSTM1 expression may reflect the severity of CHD, therefore it may be possible to partly predict the occurrence of ACS based on CCS. In addition, monocytic VSTM1 expression levels could act as predictors of MACE events, which may be an effective method of preventing and treating CHD.
JFZ, CQW, YQF and XQQ conceived, designed and funded the study. XFW, MCX, CYM performed the experiments and wrote the paper. EZ and QH reviewed and edited the manuscript. All authors read and approved the manuscript.
The clinical study was approved by the hospital ethics review board of Shanghai Ninth People’s Hospital, Shanghai Jiao Tong University School of Medicine (SH9H-2019-H21-2). All experimental contents were in accordance with principles outlined in the Declaration of Helsinki.
We thank public laboratory platform of School of Medicine, Shanghai Jiaotong University for the flow cytometry experiment.
This study was supported by grants from the National Natural Science Foundation of China (No. 81470546.), Natural Science Foundation of China (Grant No. 81870264), Natural Science Foundation of Shanghai Committee of Science and Technology (Grant No. 19ZR1429000), Clinical research project of the Ninth people’s Hospital Affiliated to Medical School of Shanghai Jiaotong University (Grant No. JYLJ017), Shanghai Committee of Science and Technology, China (Grant No. 18411950500), and a 3-year action plan to promote clinical skills and innovation in municipal hospitals (Grant No. 16CR20348).
The authors declare no conflict of interest.