
† These authors contributed equally.
§ These authors contributed equally.
Academic Editor: Peter A. McCullough
Global diabetes mellitus prevalence is increasing. Metabolic disorders, such as type 2 diabetes, are associated with abnormal cardiac electrophysiology and increased risk of arrhythmias. Patients with both diabetes types (1 and 2) suffer from sudden cardac death (SCD) as a leading cause of mortality. Cardiovascular death is defined as death attributable to cardiovascular disease (CVD) occurring shortly within the symptom onset. This usually arises from life-threatening ventricular tachyarrhythmias that lead to hemodynamic instability, and subsequent shock and death. A variety of pathways have been suggested that link hypoglycaemia to the development of adverse cardiovascular outcomes, including blood coagulation abnormalities, inflammation, endothelial dysfunction and sympathoadrenal responses. We propose a four-step framework for the optimisation of SCD risk factors in diabetic patients, to include: raising awareness to influence health behaviour, provision of screening programs, use of technology within educational programs to improve patient engagement and effective provision of diabetic community teams.
The global prevalence of diabetes mellitus is increasing, paralleling increasing rates of obesity, dietary changes and sedentary lifestyles. In 1980, just 4.3% of all adults had diabetes (108 million); this later increasing to 9.0% in 2014 (422 million) and is now expected to reach 12.5% (700 million) by 2025 [1]. In addition to this, complications resulting from diabetes accounted for 6% of global mortality in the year 2000 [2].
Diabetes is associated with multiple co-morbidities, including increased risk of cardiovascular disease (CVD), as well as nephropathy (up to a quarter of type-2 diabetic patients present with microalbuminuria [3]), neuropathy (peripheral neuropathy associated in up to half of patients with diabetes [4]), retinopathy (seen in up to 10% of patients in the pre-diabetic stage [2]) and certain types of cancer [5, 6]. Metabolic disorders such as type 2 diabetes and metabolic syndrome result in abnormal cardiac electrophysiology, increasing the risk of arrhythmias that may precipitate SCD [7, 8, 9].
SCD is defined as death attributable to CVD occurring shortly within the symptom onset, usually arising from life-threatening ventricular tachyarrhythmias that cause hemodynamic instability, leading to shock and death [10]. Over 350,000 sudden cardiac deaths occurred in 2014 in the USA [11], making it an important public health issue.
A recent meta-analysis suggests that diabetic patients have twice the risk of SCD in the general population (relative risk: 2.02 (95% CI: 1.81–2.25)) [12]. Given that it occurs shortly after the onset of symptoms, there is little time for effective medical interventions. A majority of SCD occur among the general segments of the population, therefore screening methods applicable to the general population are required. There continues to be interest in identifying clinically useful markers for SCD among the general population, as epidemiological studies have shown that half of SCD victims have no previously diagnosed CVD at the time of death [13, 14].
It is now evident from previous perspective studies that patients with diabetes are at an increased risk of cardiovascult death both in the general population and among different patient groups [12]. This review aims to organise the pathophysiology of SCD in diabetes and outline the potential risk factors. Moreover, this review aims to propose a framework for the optimisation of SCD risk factors in patients with diabetes.
Hypertension and insulin resistance mechanisms are likely to be the most important in the pathophysiology of metabolic syndrome and incident SCD in Diabetes. Indeed, insulin resistance, inflammation and endothelial dysfunction are all interrelated pathophysiological processes contributing to the development of hypertension, metabolic syndrome and cardiovascular disease [15].
Elevated fasting plasma glucose is associated with an increased risk of SCD [16]. Moreover, insulin resistance has been linked with a pro-inflammatory state and the elevation of inflammatory markers [17]. Low-grade inflammation may also increase the risk of metabolic syndrome, although some of this risk is mediated through obesity and factors related to insulin resistance [16], which contributes to an increased risk of SCD.
There are various biochemical mechanisms that may lead to cardiac mortality in diabetic patients. These fall within four distinct categories:
(1) Nervous system involvement, including arrhythmogenic effects caused by cardiac autonomic neuropathy (CAN), repolarization disorders and hypoglycaemia-mediated sympathetic activation.
(2) Disorders involving the coagulation cascade, including atherosclerosis-induced myocardial ischaemia, endothelial dysfunction, platelet aggregation or thrombophilic effects.
(3) Pro-inflammatory tendencies, leading to myocardial alterations, fibrosis, associated hypertension and/or uraemia.
(4) Repolarization failures i.e., disorders of potassium balance (as seen in diabetic nephropathy and hypoglycaemia) [18].
(Note that the following biochemical mechanisms are not necessarily arranged in the above order, yet they will all fall into one if not more of the above categories).
Physiological control of the autonomic nervous system (ANS) function is divided
into the sympathetic nervous system (SNS) and parasympathetic nervous system
(PNS). SNS stimulation traditionally results in “fight or flight” responses,
such as increased heart rate and blood pressure, heightened arousal and
mobilization of energy stores etc. (as shown in Fig. 1) [19]. This is mediated by
adrenaline, noradrenaline and dopamine via interaction with G-protein coupled
adrenergic receptors (
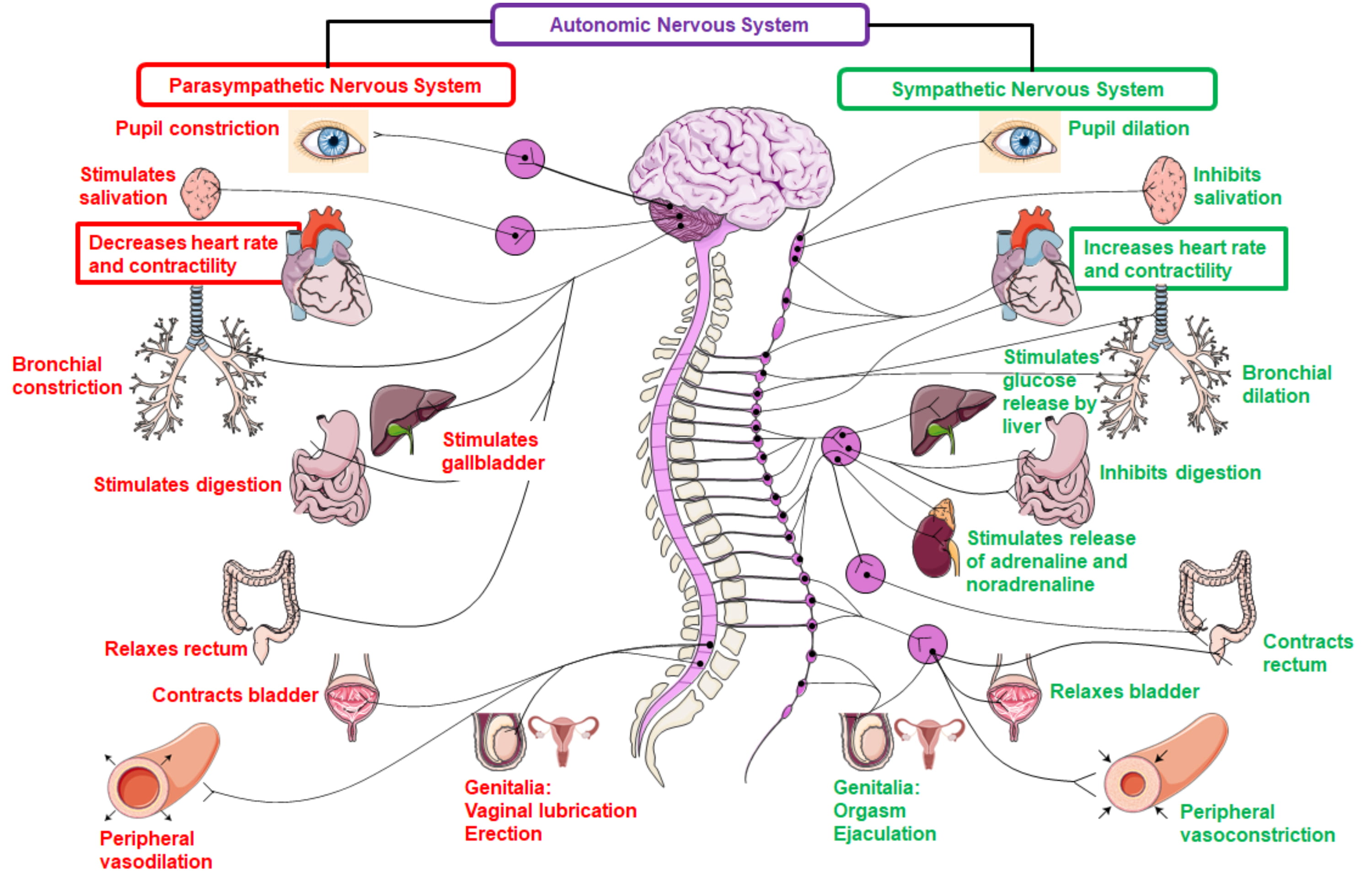 Fig. 1.
Fig. 1.Physiological functions of the parasympathetic and sympathetic
nervous system. The sympathetic nervous system is mediated by adrenaline,
noradrenaline and dopamine via interaction with G-protein coupled adrenergic
receptors (
The ANS is understood to mediate the inflammatory response. It can detect injury and infection, and thus activate a cholinergic anti-inflammatory pathway which modulates the response—this has been cited as a target for future therapies of diabetes [21]. This arc is stimulated by exogenous, Pathogen-Associated Molecular Patterns (PAMPs) or endogenous, Damage-Associated Molecular Patterns (DAMPs), which in turn stimulate receptors in the vagus nerve and associated glomus cells. The innate immune systemic response to PAMP and DAMPs is reduced by activation of the vagus nerve-to-spleen pathway, which in turn leads to reduced inflammation and cytokine release. This is also known as the “inflammatory reflex circuit”. It uses both the vagus nerve (parasympathetic branch) and splenic nerve (sympathetic branch), although this differentiation is still under debate [22]. It was Watkins et al. [23] who discovered that sensory neurons were responsible for detecting inflammation in tissues. Interleukin (IL)-1 is the inflammatory cytokine responsible for stimulating the sensory neurons, which is mediated by the vagus nerve.
Specialized glomus cells, in conjunction with the ANS, sense levels of oxygen,
glucose and other metabolites. Upon activation, these cells release dopamine and
noradrenaline, which depolarise the sensory fibres of the vagus nerve, which
travel to the brainstem and initiate a motor efferent arc [24]. While IL-1
initiates the afferent arc via stimulation of these glomus cells, there are
several other ligands derived from macrophages, monocytes and dendritic cells
which can activate toll-like receptors and result in increased expression of
nuclear factor-kappa B (NF-
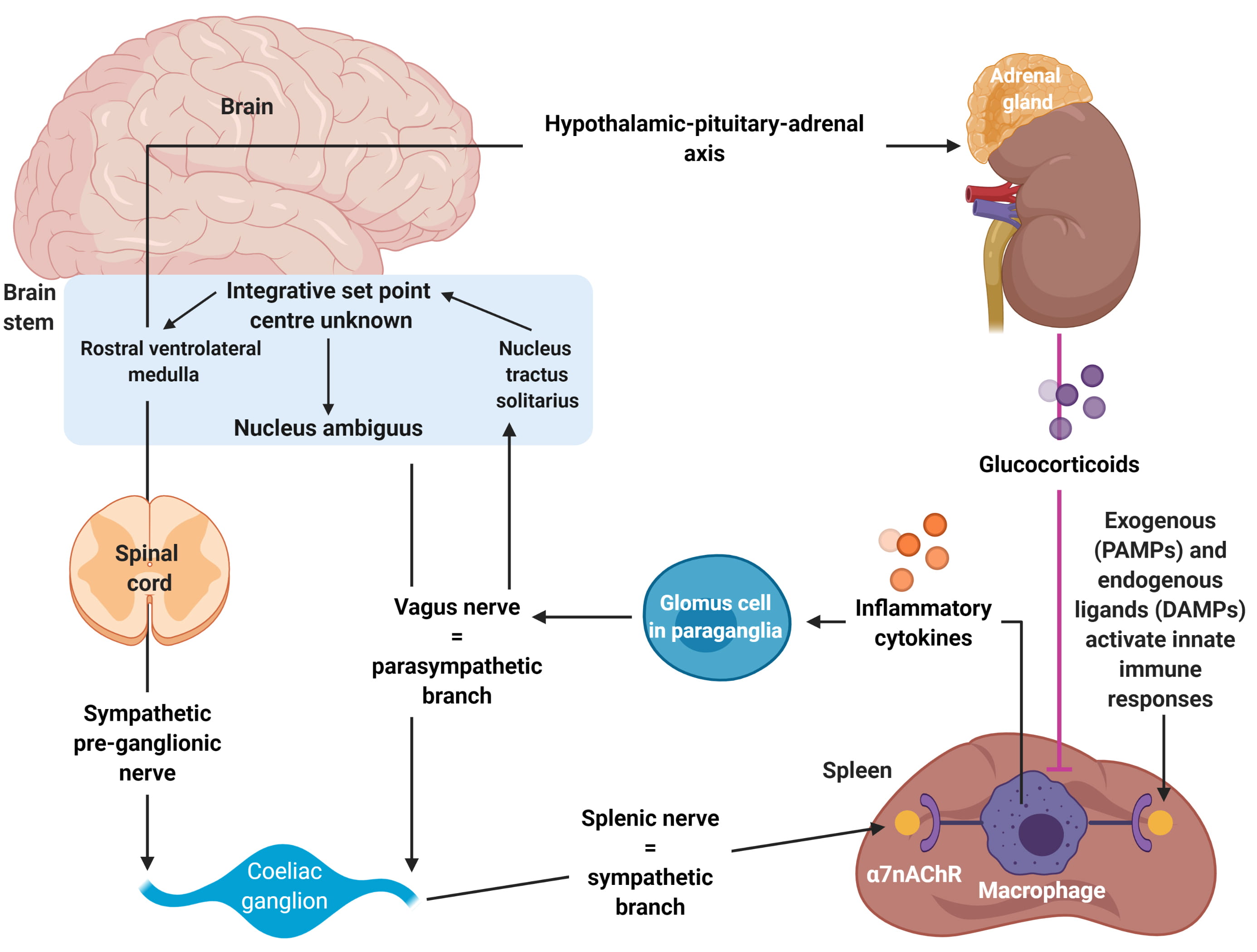 Fig. 2.
Fig. 2.Autonomic modulation of inflammation via altering sympathetic-parasympathetic balance. Adapted from Tracey et al. [24]. The Autonomic nervous system mediates the inflammatory response. It can detect injury and infection, and thus activate a cholinergic anti-inflammatory pathway which modulates the response—this has been cited as a target for future therapies of diabetes. The arc is stimulated by exogenous, Pathogen-Associated Molecular Patterns (PAMPs) or endogenous, Damage-Associated Molecular Patterns (DAMPs), which in turn stimulate receptors in the vagus nerve and associated glomus cells. The innate immune systemic response to PAMP and DAMPs is reduced by activation of the vagus nerve-to-spleen pathway, which in turn leads to reduced inflammation and cytokine release. This is also known as the “inflammatory reflex circuit”.
The efferent arc of this inflammatory response is known as the cholinergic
anti-inflammatory pathway [20]. Acetylcholine interacts with innate immune cells
that express the nicotinic acetylcholine receptor subunit a7 (
The current literature on cardiac autonomic neuropathy (CAN) reflects that there is an early-phase reduction in parasympathetic tone/function associated with increasing resting heart rate and derangement of the expiration/inspiration ratio of heart rate variability [20].
It is not wholly understood if it is caused due to a loss in parasympathetic tone or an early augmentation of sympathetic tone. However, from the outset in diabetes mellitus, there is reduced parasympathetic function and a relative increase in sympathetic function, which imbalances the sympathetic/parasympathetic tone [20].
Later, sympathetic denervation follows from the ventricular apices to the base of the heart. This leads to an imbalance in tone, which exposes the patient to an increased propensity to arrhythmias [25].
Impaired glucose tolerance (also known as the pre-diabetic stage) is also associated with reduced PNS modulation of the heart and a shift towards increased sympathetic tone. We thus infer that parasympathetic tone may decline over time due to autonomic imbalances that shift towards increased sympathetic tone during the transition from regular to impaired glucose tolerance, before finally resulting in DM [26].
Chronic hyperglycaemia has also been linked to inflammation via increases in protein glycation causing accumulation of advanced glycation end products (AGEs) in body tissues [20]. Clinical evidence shows that an accumulation of AGEs in collagen tissue shows a correlation with the severity of peripheral and autonomic nerve abnormalities in diabetes, which occurs prior to clinical manifestation [27]. These AGEs form intra-, and extracellularly via complex protein, lipid, and nucleic acid arrangements, which lead to cross-linking (Fig. 3) [28]. This process is controlled through RAGE, a pattern recognition receptor, commonly expressed, but induced by inflammation-initiating reactions. Carboxymethyl lysine (CML) is another AGE, which patients with diabetes often have higher levels of. It also activates RAGE. The AGE-RAGE interaction causes a chronic cascade of inflammation. Diabetic mice that had their RAGE removed during a study were shown to have a reduced propensity towards developing neuropathy [29]. Furthermore, a soluble AGE receptor (sRAGE) can be used as a decoy, and therefore can further reduce binding of ligands to RAGE and thus, further inhibit the inflammation cascade. Severe and autonomic neuropathy presence has shown to have a correlation to reduced levels of sRAGE in patients with Charcot neuropathy [30]. There are low levels of RAGE expression in regular homeostasis, however during biological stress, AGE accumulation increases, leading to increased RAGE expression.
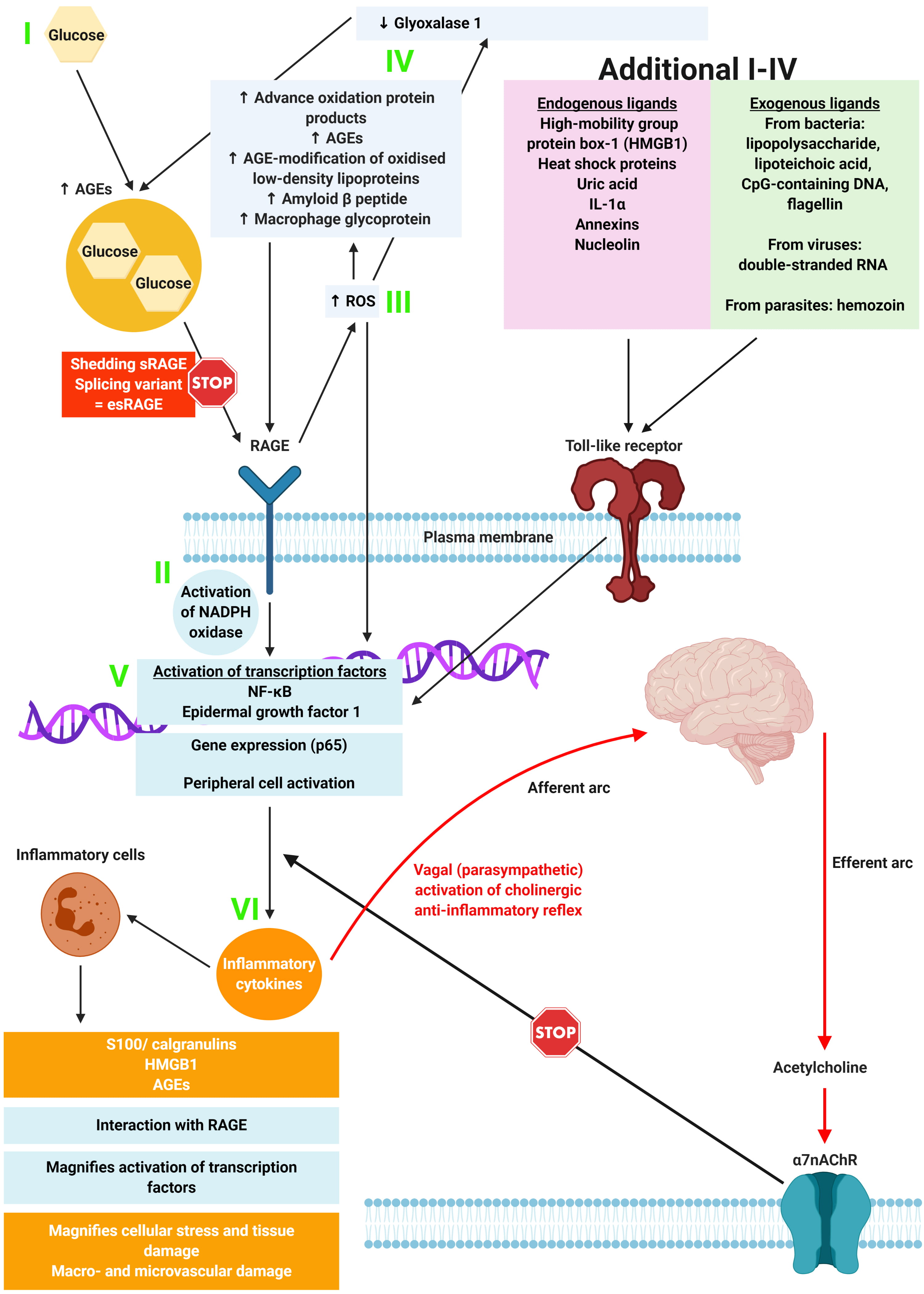 Fig. 3.
Fig. 3.The relationship between the binding of ligands onto RAGE, and associated inflammation, gene expression, oxidative and nitrosative stress, and damage to the macro- and microvasculature. Adapted from Yan et al. [28]. Constant increases in low-level inflammation in diabetes is mediated by a large group of exogenous and endogenous ligands along with the CNS and ANS. Thus, the loss of autonomic control, along with reduced parasympathetic function (which is a common feature of loss of autonomic balance in DM), initiates an inflammatory response which, if not controlled, results in considerable morbidity and mortality.
As shown in Fig. 3, the hypothesis shows that in metabolic syndromes and diabetes, “constant increases in low-level inflammation is mediated by a large group of exogenous and endogenous ligands along with the CNS and ANS” [20]. Thus, the loss of autonomic control, along with reduced parasympathetic function (which is a common feature of loss of autonomic balance in DM), initiates an inflammatory response which, if not controlled, results in considerable morbidity and mortality.
Interestingly, reduced heart rate variability correlates to an increase in circulating inflammatory markers C-Reactive Peptide (CRP), IL-6 etc. The Adiponectin-to-leptin-ratio, which is a marker of adipose tissue-derived inflammation, is correlated with cardiac autonomic imbalance, and can be seen early in T2DM patients [31]. Another study found a link between loss in HRV (heart rate variability) and IL-6 levels [32], with another study finding an inverse association between HRV and CRP levels [33]. The authors of the previous study concluded that this effect could be due to low-grade inflammation from a reduction of anti-inflammatory cascades in the cholinergic pathway, which seems to be consistent with the role of the cholinergic anti-inflammatory pathway being to exert a tonic inhibitory influence on immune responses, which shows potential as a target for future intervention.
Perhaps the major risk factor developed in patients with diabetes is a propensity to incur thrombotic events. According to one source, up to eighty percent of patients with diabetes mellitus die from thrombotic events [34]. Of these, seventy-five percent are due to cardiovascular complications, with the remainder being split between cerebrovascular and peripheral vascular complications. Thus, we can assume that the vascular endothelium is dysfunctional in diabetes mellitus. It has been observed that coagulation activation markers (i.e., prothrombin activation fragments 1 & 2) and thrombin—anti-thrombin complexes are elevated in diabetes. Furthermore, plasma levels of clotting factors including fibrinogen, kallikrein, factors VII, VIII, XI and von Willebrand Factor (vWF) are also elevated in diabetes [34]. The levels of anticoagulant protein C (PC) is also decreased. This distorts the coagulative balance of blood towards the pro-thrombotic side. Furthermore, the fibrinolytic system (the primary mechanism of clot removal) is relatively inhibited in diabetes. This is due to abnormal clot structure formation which are more resistant to degradation, as well as an increase in plasminogen activator inhibitor type 1 (PAI-1) [34]. In addition to this, there is an increase in circulating platelet agonists, along with an increased platelet contractile force (PCF), and the presence of higher levels of platelet release products (B-thromboglobulin [35], platelet factor 4 [36], thromboxane B2 [37]). It is thus inferred that platelet activity is increased drastically in diabetes mellitus.
Insulin increases cell permeability to potassium via activation of the sodium-potassium ATPases on cell membranes. Patients with Type 1 Diabetes (more so than type 2 diabetes) suffer from insulin deficiency, which contributes to net efflux of potassium from cells. Thus, hyperkalaemia can be induced through the net efflux of potassium into the extracellular space [38]. This efflux of potassium can be caused by intracellular dehydration, which leads to sodium and water entering the cell at the expense of potassium [39]. The administration of dextrose in water as a short-term therapy for hyperkalaemia may be counterproductive in patients with type 1 diabetes mellitus due to insufficient or unpredictable endogenous secretion of insulin.
Diabetes can also induce hypokalaemia if circulating insulin levels remain too high for too long, leading to a redistribution of potassium ions from the extracellular to intracellular fluid compartment [18]. Gastrointestinal loss of potassium ions may also occur in patient with diabetes with malabsorption syndromes such as diabetic-induced motility disorders, bacterial overgrowth and chronic diarrheal states. There may also be renal potassium loss due to osmotic diuresis and/or coexistent hypomagnesemia, which causes the renal outer medullary potassium channel to secrete more potassium to correct the imbalance [40]. Insulin-induced hypoglycaemia may also encourage adrenaline and aldosterone secretion from the renal medulla, which can further exacerbate mild hypokalaemia [41].
As shown in Fig. 4, there are three main metabolic disturbances responsible for
diabetic cardiomyopathy [42]. Diabetes typically starts as hyperlipidaemia (via
increases in triglycerides (TGs) and non-esterified fatty acids (NEFAs) along
with early hyperinsulinemia, which develops into pancreatic
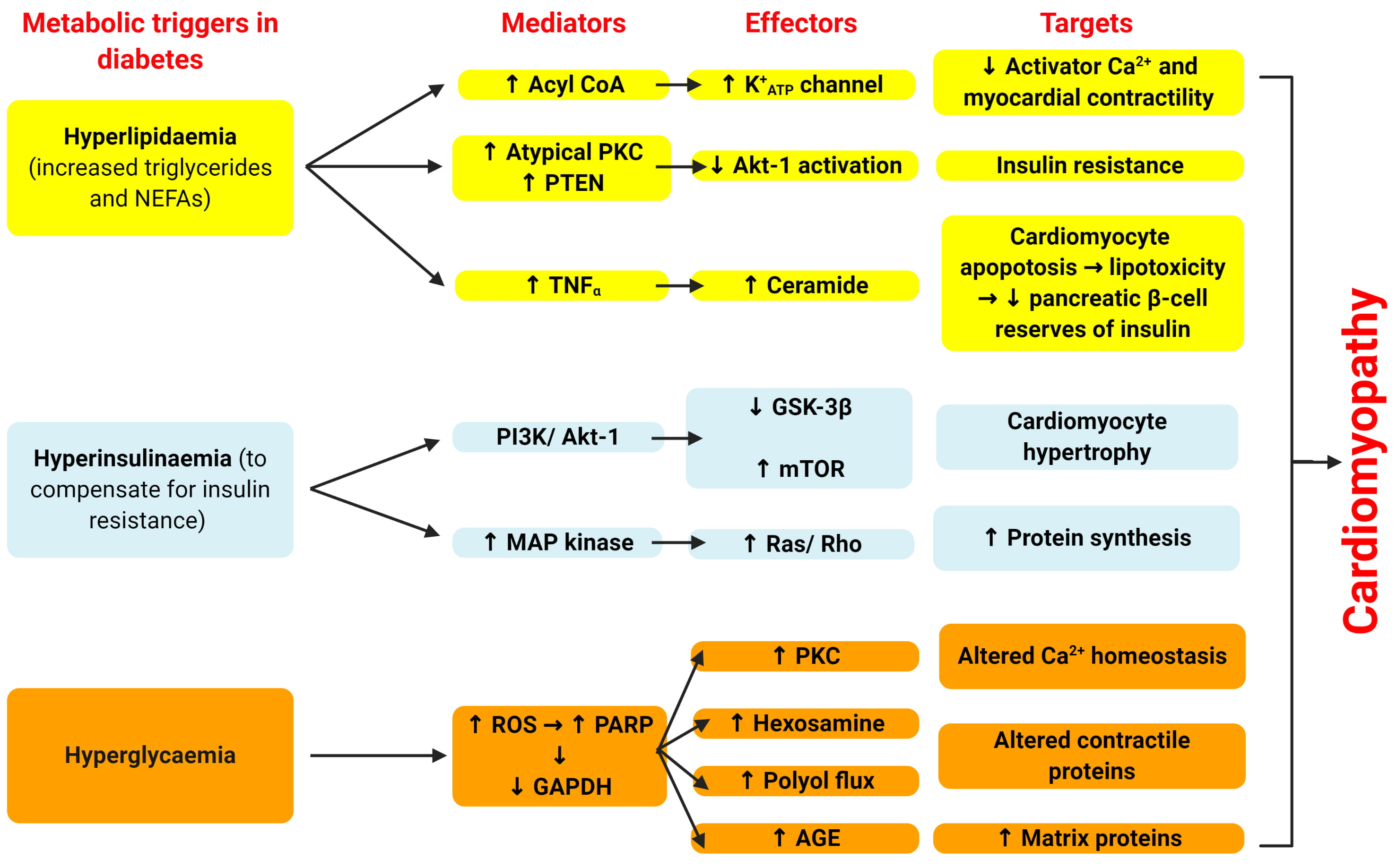 Fig. 4.
Fig. 4.Cellular mechanisms by which diabetes leads to cardiomyopathy.
Adapted from Poornima et al. [42]. There are three main
metabolic disturbances responsible for diabetic cardiomyopathy. Diabetes
typically starts as hyperlipidaemia (via increases in triglycerides (TGs) and
non-esterified fatty acids (NEFAs) along with early hyperinsulinemia, which
develops into pancreatic
NEFAs induce atypical protein kinase C activation, which phosphorylates and thus
activates I
NEFAs can directly alter myocardial contractility independent of altered insulin action via increasing NEFA flux into the myocardium [49]. According to a study by Liu et al. [50], increases in fatty acyl Coenzyme A (CoA) esters within cardiac myocytes may modulate myocardial contraction by opening of the Potassium ATP channel, thus reducing the action potential, also reducing transsarcolemmal calcium flux, leading to reduced myocardial contractility.
NEFA accumulation intracellularly can also directly lead to cell death under
circumstances in which the accumulating NEFAs fail to undergo
Cellular insulin resistance typically presages “mature” diabetes by a decade or more and, as cellular insulin resistance increases, the body needs to compensate by increasing circulating plasma insulin levels, leading to chronic hyperinsulinaemia. The characteristics of the insulin resistance may vary in organ systems, and in terms of its metabolic, mitogenic, pro-survival and vascular impact. However, not all tissues develop cellular insulin resistance (the myocardium is one such tissue). Thus, the mitogenic actions of insulin on myocardium during chronic systemic hyperinsulinaemia commonly manifest itself as cardiac hypertrophy in diabetic cardiomyopathy [52, 53, 54].
There are at least three cellular mechanisms by which hyperinsulinaemia can
mediate cardiomyocyte hypertrophy (Fig. 5). One is via the PI3K
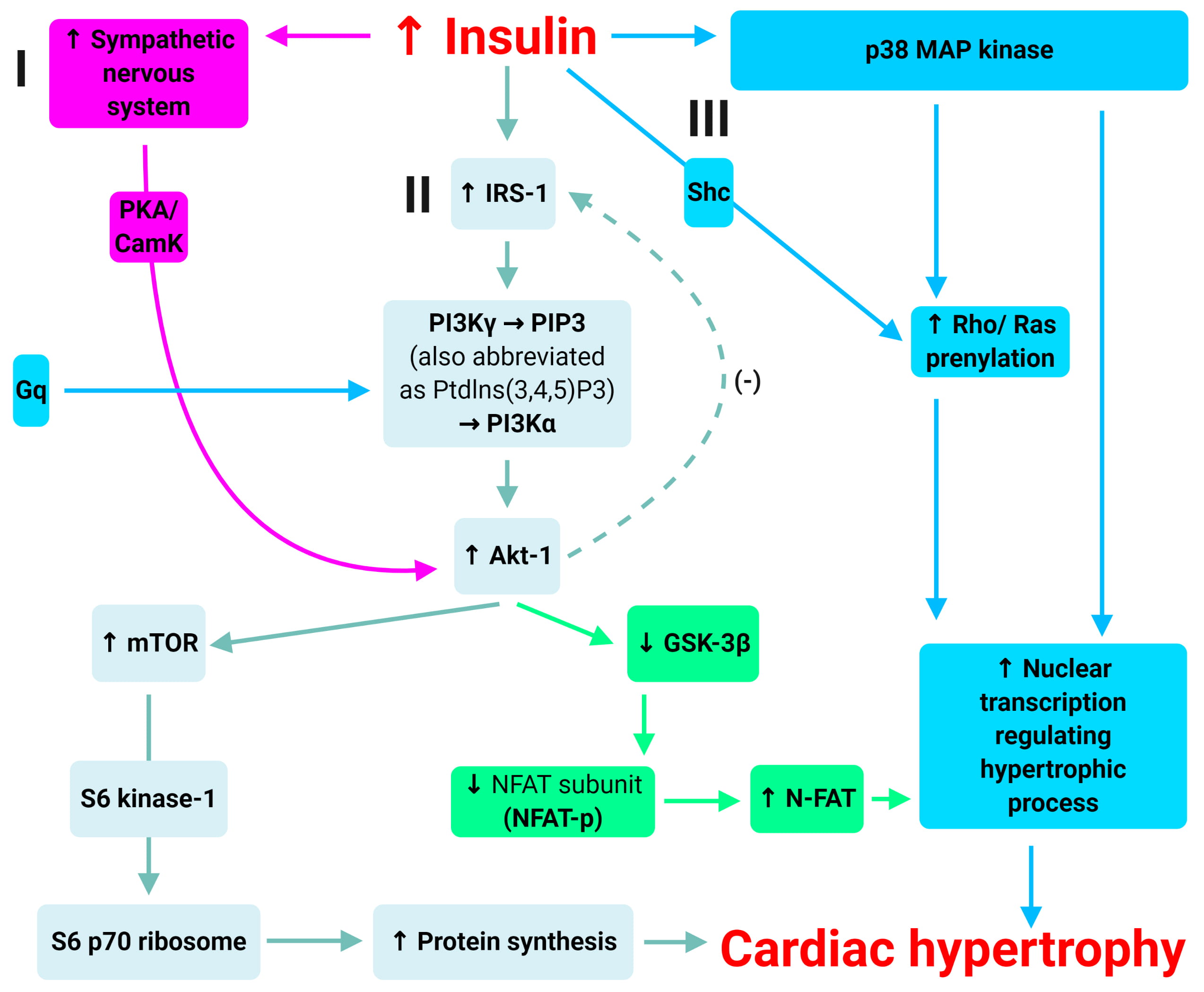 Fig. 5.
Fig. 5.Mechanisms of insulin-induced hypertrophy. Adapted from Poornima
et al. [42]. There are at least three cellular mechanisms by which
hyperinsulinaemia can mediate cardiomyocyte hypertrophy: (1) via the
PI3K
There is another pathway that augments myocardial Akt-1 activation. This is via
increased nervous system activation [58]. There is evidence that chronic Akt-1
activation in cardiac myocytes is mediated through
Finally, there are other Akt-1-independent, but insulin-mediated pathways that also promote cardiac hypertrophy. The most notable example is the extracellular signal-regulated kinase (ERK)/mitogen-activated protein (MAP) kinase pathways [59, 60]. There is strong evidence that insulin induces the activation of the p38 MAP kinase pathway [59], as well as the prenylation of both Rho and Ras in the setting of hyperinsulinaemia, which leads to cardiomyocyte hypertrophy and extracellular matrix expression. Combined, these pathways provide a foundation for the development of cardiac hypertrophy associated with chronic hyperinsulinaemia (associated with type 2 diabetes).
The mechanism by which hyperglycaemia mediates tissue injury through the generation of reactive oxygen species has been largely elucidated through the work of the Brownlee and colleagues (Fig. 4) [61, 62, 63]. Hyperglycaemia results in increased glucose oxidation and mitochondrial generation of superoxide [62]. Excess superoxide leads to DNA damage and the activation of poly (ADP ribose) polymerase (PARP) as a reparative enzyme [61]. PARP also mediates the ribosylation and inhibition of glyceraldehyde phosphate dehydrogenase (GAPDH), diverting glucose from its glycolytic pathway into alternative pathways that are considered the mediators of hyperglycaemia induced cellular injury. Table 1 (Ref. [64, 65, 66, 67, 68, 69, 70, 71, 72, 73]) shows the end products from these pathways including AGEs, increased hexosamine and polyol flux and activation of classical isoforms of protein kinase C. The consequences of the interactions with these mediators lead to multiple adverse consequences inside the cardiac tissue.
| Mediators | Mechanisms of action | Consequences |
| Increased AGE [64, 65, 66] | Crosslink RyRs [64]; crosslink type III collagen [65, 66] | Decreased SR calcium release and myocyte contractility; increased ventricular stiffness; impaired ventricular filling |
| Increased hexosamine flux [67, 68] | Sp1-O-GluN acylation of transcription factors decreasing SERCA2a expression [64] | Prolonged calcium transients; impaired relaxation |
| Increased polyol flux [69, 70] | Decreased regeneration of reduced glutathione leading to oxidative stress; increased DNA fragmentation [69]; sorbitol-induced AGEs [70] | Increased myocyte apoptosis; increased ventricular stiffness |
| Increased protein kinase C activation [71, 72, 73] | Increased cardiac hypertrophy; increased extracellular matrix; decreased SERCA 2a function [73] | Impaired relaxation; increased ventricular stiffness |
| NB: AGE, Advanced Glycation End Products; SR, Sarcoplasmic Reticulum. | ||
Hypoglycaemia is a common homeostatic imbalance, primarily affecting patients with type 1 diabetes [74], yet is frequent those with type 2 diabetes. Hypoglycaemia occurs primarily as a side-effect of insulin treatment, and also due to treatment with sulfonylureas (to a lesser degree) [75]. The associations between cardiovascular outcomes and episodes of severe hypoglycaemia are poorly understood. However, there are a variety of plausible pathways linking hypoglycaemia to the development of adverse cardiovascular outcomes, including, but not limited to: blood coagulation abnormalities, inflammation, endothelial dysfunction (previously covered) and sympathoadrenal responses [76].
Hypoglycaemia is known to influence platelet aggregability and alter several components of the inflammatory cascade. Hypoglycaemia drastically increases P-selectin expression (a marker of platelet activation), as well as fibrinogen and factor VIII levels [76]. Plasminogen activator inhibitor-1 (PAI-1) is also reduced in studies of subjects with type 1 diabetes mellitus [76]. The combination of these two factors encourage acute ischaemic events which are seen in patients with diabetes mellitus and uncontrolled hypoglycaemia. In another study, subjects with type 2 diabetes mellitus showed greater platelet aggregation in spite of treatment with aspirin and adenosine diphosphate antagonists compared with control subjects without diabetes [77].
Hypoglycaemia also increases important circulating inflammatory markers such as
CD40, CD40 ligand, interleukin-6 (IL-6), CRP, oxidative stress, along with other
proinflammatory and atherothrombotic biomarkers such as vascular adhesion
molecules vascular cell adhesion molecule 1, intercellular adhesion molecule 1,
E-selectin, vascular endothelial growth factor, and TNF-
The normal physiological response to hypoglycaemia typically involves an endogenous shutdown of insulin secretion along with release of counterregulatory hormones such as glucagon and adrenaline, in an attempt to increase glucose availability for tissues. Adrenaline causes vasoconstriction and platelet aggregation, and thus in patients with significant coronary artery disease (CAD) their chances of myocardial ischaemic events may increase [79]. We know that during acute hypoglycaemia, supply of glucose to the myocardium, splanchnic circulation and brain are prioritised. Significant autonomic activation also occurs, principally of the sympatho-adrenal system, resulting in large amounts of adrenaline released which results in significant haemodynamic changes, such as tachycardia, increased peripheral systolic blood pressure (SBP), decreased central blood pressure, increased myocardial contractility and increased ejection fraction [80, 81]. This increase in sympathetic activity, coupled with the secretion of other hormones and peptides (in particular endothelin, a powerful vasoconstrictor) have marked effects on intravascular coagulability and viscosity [82]. Increases in erythrocyte concentration increases blood viscosity during hypoglycaemia, whilst platelet activation and increments in factor VIII and vWF encourages coagulation. Endothelial dysfunction can occur during hypoglycaemia due to an increase in CRP, in addition to increased mobilization and activation of neutrophils and platelets.
The catecholamine excess also directly affects platelet reactivity and may
potentially also be proarrhythmic. For example, hypoglycaemia significantly
prolongs the QT interval, which was an independent predictor of mortality in the
MONICA/KORA Augsburg study [83]. Stahn et al. [84] also highlighted a
significant relationship between asymptomatic hypoglycaemic episodes and
ventricular asystoles/nonsustained ventricular tachycardia. Hypoglycaemia may
also impair autonomic function through various autonomic neuropathic mechanisms
discussed earlier, which can lead to reduced heart rate variability. This has
been shown to be an independent predictor of poor outcomes in the population with
diabetes mellitus [83]. Catecholamine excess may also induce hypokalaemia and
increase intracellular Ca
The high cardiovascular risk in asymptomatic diabetic patients has warranted significant interest in the early detection of silent CAD through screening [85]. As well as exercise electrocardiogram test (EET), recent technological advances have been useful to non-invasively assess the presence and severity of CAD. These include stress echocardiography (SE), stress radionuclide myocardial perfusion imaging (MPI), coronary artery calcium scoring (CACS) and computed tomography coronary angiography (CTCA) [86].
There is considerable debate regarding whether pre-emptive coronary revascularization and intensification of medical therapy based on routine CAD screening may result in improved outcomes for asymptomatic patients with diabetes [87, 88]. Those who favour screening maintain that invasive treatment leads to reduction of scintigraphic CAD progression [89] and improvement of risk classification [90]. On the other hand, opponents argue for optimal medical treatment without screening, as revascularization has not been shown convincingly to decrease cardiovascular events in patient with diabetes [91, 92]. Furthermore, randomised controlled trials (RCT’s) have failed to show any prognostic benefit of CAD screening. It must be noted that these trials may have been negatively affected by a low sample size and statistical Type II error due to lower event rates than expected [93, 94].
A systematic review and meta-analysis of published RCT’s concluded, with higher statistical power, that there was a reduction of cardiac events when using a systematic non-invasive CAD screening strategy in asymptomatic diabetes patients [95]. Further research is required to demonstrate the precise magnitude of the effect in specific subgroups. This would require larger, appropriately sized randomized trials. It may well be that larger benefits from screening are found in specific subgroups, for example, the very high-risk patients where medical therapy has failed to normalize blood glucose levels. Other subgroups may include the elder patient with diabetes [96] or those with high levels of exercise. The effects of screening patients with Type 1 Diabetes also needs to be studied more extensively. Only the FACTOR-64 randomized controlled trial included 12% of patients with Type 1 diabetes [97]. This corresponds to 3% of the total sample in the systematic review by Clerc OF et al. [98] with 97% of patients having Type 2 diabetes.
The cost of diabetes to the National Health Service (NHS) in the United Kingdom exceeds £9 billion per annum (approximately a tenth of the NHS budget) [99]. A systematic review, involving 26 different types of articles, was conducted to assess the cost-effectiveness of diabetes education. It was found that over half of the studies reviewed indicated that diabetes education was associated with reduced cost [100]. Moreover, a cost-utility analysis using data from a 12-month, multicentre randomised controlled trial concluded that diabetes education and self-management for ongoing and newly diagnosed (DESMOND) is cost-effective compared with usual care. There were also reductions in CVD risk, particularly a decrease in weight and smoking [101]. Various diabetes education courses are currently being carried out in the United Kingdom, including Dose-Adjustment for Normal Eating (DAFNE), DESMOND and X-PERT. These help to improve awareness and knowledge of diabetes among patients, and empower them to manage their own condition effectively. Various factors hinder people to gain access to diabetes knowledge, including cost, distance and a lack of educators or centres [100]. Any diabetes service should offer a well-structured diabetes education programme, as well as being cost-effective, attractive to the public and easily accessible.
To encourage patients with diabetes to participate in self-management behaviours, they should have access to diabetes self-management education (DSME). This is especially difficult in primary care and rural areas. Technology has been proposed as a method to enhance delivery of patient information [102, 103, 104]. Education has been previously provided through smartphone applications, laptops and tablets [105]. Patients with diabetes are highly receptive to technology-based education that allows them to control the pace of learning [106]. Studies have shown that the use of individualized education (based on the educational needs of each person) and behavioural change strategies results in improved blood glucose control [97, 98]. Computerised education using touchscreens or alternative technological applications may provide a potential solution to reduced diabetes education in rural areas [107].
Patients with diabetes experience difficulty initiating and sustaining healthy eating habits [100, 108]. An educational application, such as the one piloted by Hunt CW et al. [101], could be used to provide dietary information. This can include information on portion control, healthy food shopping and monitoring carbohydrate intake. Hunt CW et al. [101] delivered ten diabetes self-management educational modules electronically via iPads to thirty adults living with Type 2 diabetes who attended health promotion clinics in rural communities. The authors found that there was a statistically significant increase in diabetes knowledge scores from pre- to post- educational intervention. Diabetes education delivered electronically can provide the necessary information about diabetes self-management. This can especially help to improve education delivery to those living in areas where access to healthcare resources is limited.
SCD risk factors in patients with diabetes can be optimised by targeting four key domains (Fig. 6). Firstly, patients with diabetes must be aware of the potential risk of SCD so that become in position to influence their health behaviour. We recommend that health provide basic and accurate information in a clear and unambiguous way via multiple channels, such as leaflets and online patient information platforms. Secondly, we recommend that healthcare providers promote uptake of screening for SCD risk factors in patients with diabetes, for instance by implementing financial incentives on attendance and employing text message reminders. Thirdly, we recommend that healthcare providers optimise patient engagement with online information. Finally, we recommend that services are integrated with diabetic community teams to ensure greater accessibility to healthcare provisions and engagement with technology that can remote monitor blood glucose levels, heart rate and rhythm.
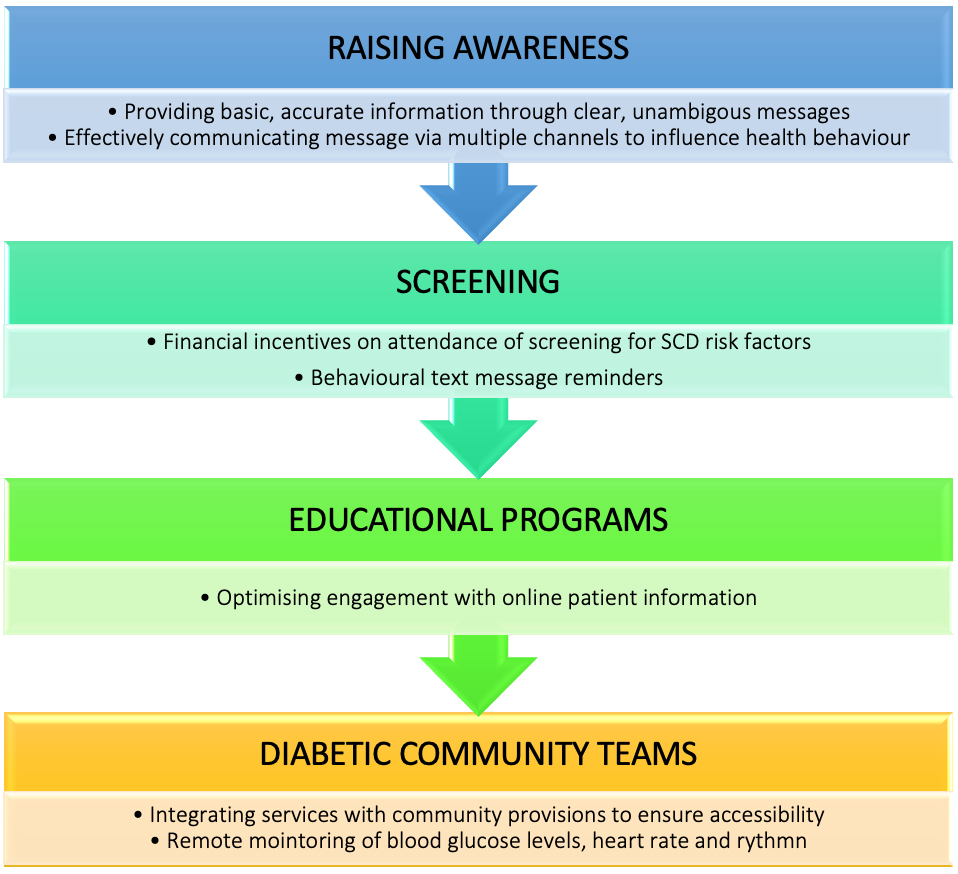 Fig. 6.
Fig. 6.Recommendations for optimising SCD risk factors in diabetic patients. SCD risk factors in patients with diabetes can be optimised by targeting four key domains: raising awareness to influence health behaviour, provision of screening programs and providing invectives to encourage uptake, use of technology within educational programs to improve patient engagement and effective provision of diabetic community teams.
Patients with diabetes mellitus suffer from cardiovascular death as a leading cause of mortality. Mechanisms related to hypertension and insulin resistance are likely to be the most important in the pathophysiology underlying the metabolic syndrome and incident SCD.
A variety of pathways have been suggested as linking hypoglycaemia to the development of adverse cardiovascular outcomes, including, but not limited to blood coagulation abnormalities, inflammation, endothelial dysfunction and sympathoadrenal responses.
Optimisation of SCD risk factors is recommended via: raising awareness to influence health behaviour, provision of screening programs and providing invective to encourage uptake, use of technology within educational programs to improve patient engagement and effective provision of diabetic community teams.
EA and SS conceived and designed the paper. All authors wrote and revised the paper critically for important intellectual content. All authors approved the final manuscript.
Not applicable.
We thank https://biorender.com/ and smart.servier.com with which we created all figures (Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License).
This research received no external funding.
The authors declare no conflict of interest.