
Academic Editor: Peter A. McCullough
Heart failure with preserved ejection fraction (HFpEF) is one of the greatest unmet needs in modern medicine. The lack of an appropriate therapy may reflect the lack of an accurate comprehension of its pathophysiology. Coronary microvascular rarefaction in HFpEF was first hypothesized in an autopsy study that showed how HFpEF patients had lower microvascular density and more myocardial fibrosis than control subjects. This was later confirmed in vivo when it was noted that HFpEF is associated with reduced myocardial flow reserve (MFR) at single photon emission computed tomography (SPECT) and that coronary microvascular dysfunction may play a role in HFpEF disease processes. HFpEF patients were found to have lower coronary flow reserve (CFR) and a higher index of microvascular resistance (IMR). What is the cause of microvascular dysfunction? In 2013, a new paradigm for the pathogenesis of HFpEF has been proposed. It has been postulated that the presence of a proinflammatory state leads to coronary microvascular endothelial inflammation and reduced nitric oxide bioavailability, which ultimately results in heart failure. Recently, it has also been noted that inflammation is the main driver of HFpEF, but via an increase in inducible nitric oxide synthase (iNOS) resulting in a decrease in unfolded protein response. This review summarizes the current evidence on the etiology of coronary microvascular dysfunction in HFpEF, focusing on the role of inflammation and its possible prevention and therapy.
According to the latest ESC guidelines [1], Heart Failure (HF) with
preserved ejection fraction (HFpEF) is defined as signs and/or symptoms
of heart failure with a left ventricular ejection fraction (LVEF)
The prevalence of HF in the general population is around 1-2% [2] and HFpEF is responsible for approximately 50% of HF admissions [3]. Patients with HFpEF are generally older and have a significantly higher incidence of hypertension, diabetes and obesity compared to the sum of HF patients. These characteristics are responsible for the alterations in vascular biology seen in patients with HFpEF [4].
Coronary artery disease is present in 40-55% of patients with diastolic heart failure and impairment of ventricular relaxation represents the first detectable mechanical change during myocardial ischemia. Ischemia depletes myocyte energy stores, leading to an impairment of calcium ion removal in sarcoplasmic reticulum and both acute and chronic ischemia induce, through different mechanisms, diastolic dysfunction. In acute ischemia diastolic dysfunction precedes motion abnormalities and is induced by sudden global hypoxemia and by perfusion ischemia. As regards chronic ischemia, hypoxic myocardium activates an inflammatory profibrotic response that is the major determinant of diastolic dysfunction [5]. However, coronary artery disease means more than just epicardial coronary artery disease. In 1967 Likoff et al. [6] first suggested that coronary microvascular dysfunction (CMD) could play a role in patients with normal coronary angiography. Subsequent studies have shown that CMD is an important contributer not only to ischemic heart disease, but also to a series of systemic conditions that, through CMD, affect the heart, including HFpEF [7].
In 2005, Borbély et al. [8] first analyzed endomyocardial biopsies of patients with diastolic heart failure (DHF). Biopsies were analyzed for collagen volume fraction (CVF) and sarcomeric protein composition and single cardiomyocytes were isolated to assess cellular contractile performance. DHF hearts had higher CVF and resting tension than healthy controls, a characteristic histological feature of HFpEF.
The pathophysiology of HFpEF was further elucidated by
Westermann et al. [9], who demonstrated that inflammation is an
important stimulus for the development of cardiac fibrosis and extracellular
matrix remodeling. He noted that inflammatory cells were increased in the cardiac
tissue of HFpEF patients and that these cells secreted higher levels of
the profibrotic growth factor TGF-
However, it was not clear why these patients had increased cardiac inflammation. The answer to this question was provided by Paulus et al. [10], in their landmark study “A Novel Paradigm for Heart Failure with Preserved Ejection Fraction”. They first proposed that certain comorbidities may lead to a proinflammatory state that induces coronary microvascular endothelial cells to produce reactive oxygen species (ROS) which limits endothelial nitric oxide synthase (eNOS) and nitric oxide (NO) bioavailability in cardiomyocytes. This results in decreased protein kinase G activity, which leads to cardiomyocyte hypertrophy and to titin hypophosphorylation, which causes cardiomyocyte stiffening and diastolic dysfunction. These observations highlighted the role of the coronary microvascular endothelium as the central target for systemic inflammation and its contribution to myocardial dysfunction (Fig. 1).
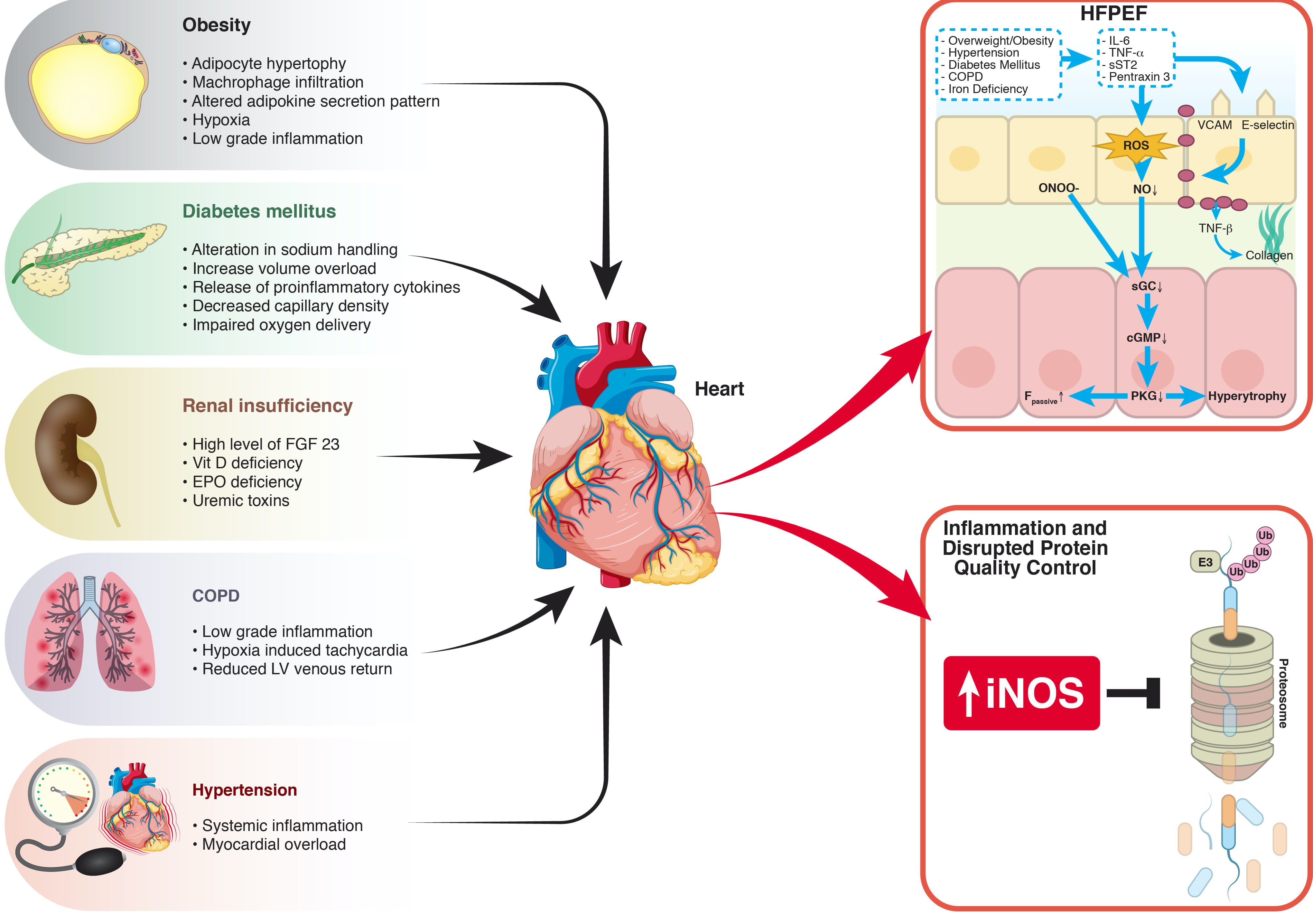 Fig. 1.
Fig. 1.Different Hypothesis of Comorbidities Induced Myocardial Dysfunction. Patients with HFpEF often have multiple concomitant diseases and comorbidities play an important role in HFpEF pathophysiology. Obesity is characterized by an altered adipokine secretion pattern and by a low-grade systemic inflammatory response, that is also present in COPD and hypertension. Diabetes mellitus leads to decreased capillary density, impaired oxygen delivery and release of proinflammatory cytokines, while renal insufficiency is associated with high levels of FGF23 and uremic toxins. Comorbidities induced inflammation represents the main driver of HFpEF in two different pathophysiological models. In the hypothesis of Paulus et al. systemic inflammation leads to production of reactive oxygen species (ROS) and reduced nitric oxide (NO) availability. Schiattarella et al. found that an inflammation induced increase in inducible NO synthase (iNOS) leads to reduce an unfolded protein response. cGMP, cyclic guanosine monophosphate; COPD, chronic obstructive pulmonary disease; EPO, erythropoietin; FGF23, fibroblast growth factor 23; Fpassive, resting tension; HFpEF, heart failure with preserved ejection fraction; IL-6, interleukin 6; iNOS, inducible nitric oxide synthase; NO, nitric oxide; ONOO-, peroxynitrite; PKG, protein kinase G; sCG, soluble guanylate cyclase; sST2, soluble ST2; TNF, tumor necrosis factor; Ub, ubiquitin; VCAM, vascular cell adhesion molecule.
Systemic inflammation may also be the main contributer to HFpEF
as noted in a series of experiments by Schiattarella et al. [11] (Fig. 1).
They exposed mice to HFpEF comorbidities such as obesity and
hypertension, thus simulating the clinical features of human HFpEF. The
unfolded protein response (UPR) was demonstrated to be downregulated in obese and
hypertensive mice through the inactivation of the IRE1
These studies contributed strong evidence that certain co-morbidities induce, through different but complementary mechanisms, a systemic inflammatory response. This so called metainflammation is critical to HFpEF pathophysiology. An unifying theory, that fully explains the complex connection between inflammation and HFpEF, has yet to be delineated and further studies are required in this field.
The importance of inflammation in microvascular dysfunction was also demonstrated in autoimmune rheumatic diseases and psoriasis, which have been associated with a higher risk of HFpEF [12, 13, 14].
The characteristics of microvascular involvement in HFpEF was noted in 2014 by Mohammed et al. [15]. In selected HFpEF patients who underwent an autopsy, they quantified microvascular density (MVD) and myocardial fibrosis. MVD was significantly diminished and it was inversely associated with myocardial fibrosis, suggesting that reduced MVD or processes leading to coronary microvascular alterations may contribute to myocardial fibrosis. Furthermore, group differences in left ventricular fibrosis were attenuated after the adjustment for MVD. This provided further proof that CMD is the result of comorbidity-induced inflammation which leads to myocardial structural changes [16, 17].
One of the first publications [18] regarding the potential role of CMD in HFpEF, measured the peak transcardiac oxygen gradient during exercise, which was found to be lower in HFpEF patients compared to controls. Since myocardial oxygen consumption during increased cardiac work is mainly achieved by an increase in myocardial blood flow, these data suggested an impairment of myocardial oxygen delivery due to microvascular dysfunction.
This same concept was expressed in HFpEF patients undergoing cardiac catheterization, an invasive determination of CFR and IMR (index of microvascular resistance), which reflects coronary blood flow and coronary microvascular resistance [19]. The HFpEF cohort was found to have lower mean CFR and higher mean IMR compared to controls and over one third of these patients exhibited overt CMD (abnormal CFR and IMR). Two other groups of patients were identified: those with abnormal IMR and normal CFR and those with normal IMR and abnormal CFR. Further hemodynamic characterization revealed that in patients with abnormal IMR but normal CFR, precapillary arterioles are able to dilate and provide enough flow when necessary, while patients with normal IMR but abnormal CFR have a normal coronary microvascular bed, whose dilation is elevated at rest due to higher metabolic demands. These two groups identify different patterns of coronary physiology, that could reflect distinct HFpEF entities, which may represent different phases or severity of the disease. Further studies are required to confirm these hypotheses.
Another invasive analysis was performed by Sucato et al. [20], who studied coronary blood flow and myocardial perfusion of the microcirculation in patients with microvascular angina and HFpEF compared to controls. TIMI frame count (TFC) and myocardial blush grade (MBG) were analyzed. HFpEF patients had longer TFC of the three major coronary arteries than non HFpEF patients, thus indicating reduced flow in HFpEF coronary microcirculation. MBG was also lower in HFpEF patients, representing another potential marker of CMD in HFpEF patients.
One study [21] evaluated both endothelium-dependent and independent coronary microvascular function in HFpEF, using acetylcholine and adenosine infusions. Overall, CMD was present in 72% of the patients (isolated endothelium-dependent dysfunction in 29%, isolated endothelium-independent in 33%, combined in 10%). Patients with HFpEF and endothelium-independent microvascular dysfunction had worse diastolic dysfunction and higher overall mortality.
The hypothesis of microvascular dysfunction in HFpEF was also confirmed by Srivaratharajah et al. [22] using cardiac positron emission tomography (PET), to quantify myocardial flow reserve (MFR). MFR was defined as the ratio of myocardial blood flow during exercise and at rest and is considered to be a marker of microvascular function [23]. Resting myocardial blood flow was significantly greater in HFpEF patients compared with normotensive controls (likely reflecting increased resting metabolic demand, as supported by a significantly higher resting rate-pressure product), while stress myocardial blood flow was significantly lower in the HFpEF group. Overall, HFpEF was associated with lower global and regional MFR than non-HF controls and was significantly associated with reduced global MFR independently of potential confounders (such as age, sex and history of hypertension and diabetes mellitus), reflecting the presence of CMD in HFpEF patients. It is unlikely that the reduction of MFR in HFpEF can be explained by a greater resting blood flow, as the percentage difference in MFR for HFpEF versus normotensive controls was greater than the percentage difference between resting flow in these groups. In addition, stress flow was significantly reduced in HFpEF.
Cardiac magnetic resonance (CMR) was used to study CFR in HFpEF patients in 2016 by Kato et al. [24], who measured blood flow in the coronary sinus. Corrected coronary sinus blood flow at rest was found to be substantially higher in both HFpEF and hypertensive patients than in control patients. Interestingly, during ATP infusion, a significant increase in coronary sinus blood flow was observed in all 3 groups, but the corrected differences in sinus blood flow and CFR were lower in HFpEF patients than in hypertensive patients or in controls. A possible explanation for these results is that coronary blood flow is already elevated at rest to account for microvascular dysfunction or for higher metabolic demands in HFpEF patients, whereas their reserve myocardial capacity is decreased in comparison to healthy subjects. Furthermore, decreased CFR was significantly associated with elevated serum BNP concentration, suggesting that microvascular dysfunction may influence disease severity.
A more recent CMR study [25] also confirmed these results: global
myocardial perfusion reserve (MPR) was lower in HFpEF compared to
controls and almost 70% of HFpEF patients had CMD, defined as MPR
Prevalence and correlates of CMD in HFpEF patients were analyzed with echocardiography in the PROMIS-HFpEF study [26]. The prevalence of CMD was reported to be as high as 75% and CFR was associated with markers of heart failure severity (such as NTproBNP and right ventricular dysfunction), but, not to patient quality of life (KCCQ score) nor functional capacity (6 minutes walking test). Furthermore, CMD correlated with urinary albumin to creatinine ratio and with EndoPAT testing (peripheral arterial tonometry), which are markers of systemic endothelial dysfunction [27], suggesting that HFpEF is a systemic disorder associated with endothelial dysfunction and microvascular disease in the heart and in other organs. A follow-up of PROMIS-HFpEF patients has also recently been published [28] and showed a strong association between the presence of CMD and the risk of cardiovascular and HF-related events (CV death and HF hospitalizations), confirming the hypothesis that CMD is related to disease severity.
In 2012 Akiyama et al. [29] investigated whether peripheral endothelial dysfunction could predict the occurrence of cardiovascular events in patients with HFpEF. Peripheral endothelial function was assessed by reactive hyperemia-peripheral arterial tonometry and expressed using the RH-PAT index (RHI), which was significantly lower in patients with HFpEF compared to controls. Furthermore, RHI was an independent predictor of future cardiovascular events in HFpEF patients.
A recent study by Yüksel et al. [30] used nailfold videocapillaroscopy to assess systemic microvasculature involvement in HFpEF patients. The number of patients with abnormal videocapillaroscopic findings was significantly greater in the HFpEF group compared to HF with reduced ejection fraction (HFrEF) and control groups. These results suggest that inflammation causes systemic microvascular dysfunction and is consistent with the results of Zanatta et al. [31], who found that patients with systemic sclerosis (a model of systemic inflammatory disease) had lower CFR than controls and that CFR was correlated with abnormalities found at nailfold videocapillaroscopy.
Two recent studies analyzed the correlation between coronary
microvascular dysfunction and exertional alterations in HFpEF patients.
Ahmad et al. [32] studied patients with unexplained cardiac exertional
symptoms and non-obstructive coronary artery disease who underwent invasive
evaluation of CMD and hemodynamic exercise right heart catheterization. Both
endothelium independent (expressed as CFR) and dependent (expressed as the
percent difference of coronary blood flow after acetylcholine intracoronary
infusion %
The second study [33] assessed the relationship between echocardiography determined CFR and exercise tolerance measured using the 6 minute walking test (6MWT) in HFpEF patients. HFpEF patients had a high prevalence of CMD (66%) and in patients with HFpEF CFR significantly correlated with 6MWT results, being an independent predictor of reduced exercise tolerance.
Myocardial ischemia, assessed with SPECT, is an independent predictor of death and rehospitalization in patients with new onset HFpEF [34]. In the PROMISE-HFpEF follow-up study [28], the presence of CMD correlated with cardiovascular and HF-related events. A similar concept was reported by Taqueti et al. [35], who analyzed patients undergoing evaluation for suspected coronary artery disease but with no evidence of CAD or reduced left ventricular ejection fraction. CFR was assessed with PET imaging and patients were followed for cardiovascular death and hospitalization for non-fatal myocardial infarction or heart failure. Impaired CFR was found to be an independent predictor of adverse cardiovascular outcomes, especially of HFpEF related hospitalizations.
Comparable results were obtained assessing CMD with invasive techniques [36] in a follow up of the PROMISE-HFpEF study by Dryer et al. [19]. HFpEF patients with overt CMD (abnormal CFR and IMR) had lower freedom from heart failure hospitalization compared to other HFpEF patients. This was also noted when comparing HFpEF patients with abnormal CFR to those with normal CFR.
The results of these studies suggest a correlation between CMD and clinical outcomes of HFpEF patients, thus identifying a group of high-risk of HFpEF patients in which therapies targeted at CMD might be clinically significant.
The treatment of HFpEF still represents one of the greatest unmet needs in medicine. Thus far, several trials have failed to show a clear benefit of a specific medical therapy [37]. The PEP-CHF study [38], randomized HFpEF patients to perindopril versus placebo. Inconclusive results were found on long-term morbidity and mortality since there was insufficient power for the primary endpoints. However, improved symptoms, better exercise capacity and fewer hospitalizations for HF in the first year were observed on perindopril, suggesting that it may be of benefit in this patient population. The I-PRESERVE study [39] investigated irbesartan versus placebo in patients with HFpEF, but no definitive conclusions were found on the efficacy of irbesartan. The CHARM-Preserved trial [40], analyzing the role of candesartan, did not find any statistically significant differences in the primary outcomes of composite cardiovascular death and HF hospitalizations.
The TOPCAT trial [41] investigated the potential role of spironolactone and found a nonsignificant trend in favor of the primary outcome. Based on the promising results in HFrEF, the PARAGON-HF trial [42] analyzed the efficacy in HFpEF of sacubitril-valsartan (LCZ696) compared with valsartan alone. PARAGON-HF narrowly missed a significant reduction in its primary endpoint, although patients with an EF below 57% and women derived a significant benefit in the primary outcome, mainly driven in women by a reduction in heart failure hospitalizations [43]. Since women account for over half of HFpEF patients [44, 45], sacubitril-valsartan could be a potentially important therapy in this subgroup of patients.
The role of sodium-glucose cotransporter-2 (SGLT2) inhibitors, whose efficacy has been proven in HFrEF, is still under investigation in two ongoing trials (EMPEROR-PRESERVED trial and DELIVER trial) analyzing the role of empaglifozin and dapaglifozin in HFpEF patients [46, 47].
Based on the pathophysiological hypothesis of Paulus et al. [10], many studies analyzed the potential role of pharmacological intervention in the nitric oxide (NO) pathway. In the Neat-HFpEF trial [48], isosorbide mononitrate treatment resulted in decreased activity levels and in no improvement in the quality of life. The INDIE-HFpEF trial [49], analyzing the treatment with inhaled inorganic nitrite, failed to increase exercise capacity, NYHA functional class and quality of life. The SOCRATES-PRESERVED trial [50], using the soluble guanylyl cyclase stimulator vericiguat, showed improved quality of life but no benefit on NT-pro-BNP levels and the RELAX trial [51], using sildenafil, failed to show an effect on exercise capacity or clinical status. The failure of NO-based therapies could be explained by the more recent hypothesis on HFpEF pathophysiology [11], which focuses on the enhanced pro-inflammatory mediators, iNOS activity and nitrosative stress (Fig. 1). Future therapies for HFpEF may aim at reducing iNOS activity and NO levels, rather than increasing them.
In summary, none of the above cited trials showed a clear benefit of a certain medical therapy on HFpEF. However, none of them specifically sought to address coronary microvascular dysfunction, which is common in these patients.
As we have seen in the previous chapter, there are currently no effective therapies for HFpEF. This highlights the need for novel strategies focused on its prevention [52]. A follow up study from the Framingham Heart Study [53] found that lower physical activity is associated with a higher incidence of HF and that this association was stronger for HFpEF than HFrEF. Similarly, a more recent study [54] showed that improvement in cardiorespiratory fitness over a period of 4.4 years was associated with up to a 17% reduction in HF risk at a later age. These findings could be explained by the beneficial effect of physical activity [55] on HFpEF risk factors, such as obesity, diabetes and hypertension, which could alter the pathophysiological mechanism that leads to HFpEF.
As regards the NO pathway, a study by Mátyás et al. [56] investigated in rats investigated the effect of long-term preventive application of the phosphodiesterase-5A (PDE5A) inhibitor vardenafil in diabetic cardiomyopathy-associated HFpEF. They found that vardenafil effectively prevented the development of HFpEF by maintaining diastolic function, restoring cGMP levels and PKG activation, lowering apoptosis and by alleviating nitro-oxidative stress, myocardial hypertrophy and fibrotic remodeling.
A recent study [57] reviewed the potential role of microRNAs (miRNAs) in the prevention of HFpEF. miR-21 has been proposed as a pleiotropic factor that may regulate several target mRNAs in cardiovascular diseases, leading to a strong relationship between miR-21 upregulation and cardiac hypertrophy and fibrosis [58, 59, 60]. There are ongoing trials analyzing the role of miR-21 inhibitors in patients with kidney fibrosis as a result of Alport syndrome, as previous studies in mice demonstrated benefits related to reduction of pro-inflammatory and pro-fibrotic signaling pathways [61]. Identifying a group of patients at high risk for developing HFpEF, for example using validated biomarkers [62, 63, 64], and miR-21 therapies could be used in very early stages of disease, to alter the pathognomonic mechanisms of inflammation and fibrosis.
This review summarizes the current evidence for the occurrence of coronary microvascular dysfunction in HFpEF. We first reviewed the pathophysiology of CMD in HFpEF. Two different important pathophysiological theories [10, 11] suggested how comorbidities promote an inflammatory state that impairs microvascular function. Even if these theories may differ in some aspects, for example in the role of nitric oxide synthase activation, both agree that comorbidity induced inflammation plays a central role in HFpEF pathogenesis and that CMD is a characteristic of HFpEF.
We then reviewed the evidence of CMD in HFpEF. Starting from the first autopsy study [15] that showed a reduction of microvascular density in HFpEF patients, we moved to in vivo analysis with invasive and non-invasive techniques. Cardiac catheterization [19] showed how, in HFpEF patients, CFR is lower compared to controls and these results were confirmed by nuclear medicine techniques [22], cardiac magnetic resonance [24, 25] and echocardiography [26]. In view of the evidence of CMD in HFpEF, we sought possible clinical correlates and found that CFR directly correlates with exercise capacity, being an independent predictor of reduced exercise tolerance [32, 33]. Furthermore, CMD significantly correlates with cardiovascular and HF-related events, suggesting a possible relation with disease severity [28].
As regards the therapeutic options, we analyzed the most important trials regarding HFpEF and discussed how, most of them, failed to show a clear benefit of a specific medical therapy. Interestingly, none of them specifically aimed at the problem of CMD, which we noted to be so common in these patients.
Given the absence of effective therapies for HFpEF, we analyzed potential preventive strategies. Lower physical activity is associated with a higher incidence of HFpEF [53, 54] and the main reason for this finding could be the beneficial effect of physical activity on HFpEF risk factors. Pharmacologically, the phosphodiesterase-5A (PDE5A) inhibitor vardenafil prevented the development of HFpEF in diabetic rats [56] and the potential role of microRNAs is under investigation [57].
Coronary microvascular dysfunction is a typical feature of HFpEF and this has been proven both histologically and clinically. The accurate comprehension of the mechanisms of microvascular dysfunction could lead to more specific preventive options, whose efficacy would be of paramount importance given the absence of proven therapeutic strategies.
All co-authors have participated in the design, execution or analysis of the study. The submitted version of the manuscript has been read, revised and approved by all co-authors.
We thank the Department of Cardiac, Thoracic, and Vascular Sciences and Public Health (Padua Univerisity Hospital) for the support.
This research received no external funding.
The authors declare no conflict of interest and have no known competing financial interests or personal relationships that may have influenced the work reported in this paper.