
Heart failure with preserved ejection fraction is a very common clinical problem. Its prevalence is increasing with aging of the population. A diverse group of risk factors and etiologies comprise the HFpEF syndrome. No specific therapies have been shown to improve survival for the vast majority of HFpEF cases. Restrictive cardiomyopathies account for a significant portion of HFpEF patients and are characterized by diastolic dysfunction due to infiltration of the myocardium or ventricular hypertrophy. Many of these restrictive diseases occur in the context of myocardial infiltration by other substances such as amyloid, iron or glycogen or endomyocardial fibrosis. These infiltrative diseases usually have important clues in the clinical picture and on cardiac imaging that may allow differentiation from the usual HFpEF phenotype (that is commonly seen in the older, hypertensive patient). Noninvasive diagnosis has replaced endomyocardial biopsy for most instances in the workup of these conditions. Early recognition is important to institute specific therapies and to improve prognosis. In this review, we describe 4 major infiltrative cardiomyopathies (Cardiac Amyloidosis, Sarcoidosis, Hemochromatosis and Fabry disease), and their key imaging features.
Heart failure with preserved ejection fraction (HFpEF) refers to heart failure symptoms with normal or near-normal cardiac function on echocardiography. It is characterized by diastolic dysfunction and reduced ventricular compliance. Patients with HFpEF typically have symptoms such as shortness of breath, excessive fatigue, and exercise intolerance. HFpEF causes significant morbidity and mortality with a 1-year mortality of ~25% (Bhatia et al., 2006; Dunlay et al., 2017; Owan et al., 2006; Perez de Isla et al., 2009). While beta-blockers and renin-angiotensin-aldosterone system inhibitors are known to improve survival in heart failure with reduced ejection fraction (HFrEF), no therapies show similar effectiveness in HFpEF. The prevalence of HFpEF has increased in the last two decades. About half of all new HF patients are now diagnosed with HFpEF (Dunlay et al., 2017). The risk factors for developing HFpEF include age, hypertension, obesity, diabetes mellitus (Mollee et al., 2014), atrial fibrillation, and dyslipidemia. Additionally, several studies consistently report that HFpEF is more common in female patients (Bursi et al., 2006; Gurwitz et al., 2013; Owan et al., 2006). Most HFpEF cases are associated with common cardiac risk factors such as hypertension, diabetes, renal disease, sleep apnea, obesity and aging, but a few cases are due to specific infiltrative disorders wherein the myocardial architecture and function is altered by excess accumulation of either abnormal proteins, glycosphingolipids, glycogen or other substances (Table 1). It is essential to distinguish between these primary and secondary forms of HFpEF since the latter group of infiltrative cardiomyopathies usually have distinctive clinical features and specific therapies. For example, hypertensive heart disease causing HFpEF is common in older individuals, especially in hypertensive women and is treated with control of blood pressure and diuretics, whereas patients with Fabry cardiomyopathy are usually younger men who also have other characteristic clinical features of that disease (e.g., cornea verticillata) and are treated with enzyme replacement therapy.
| Restrictive Cardiomyopathies | |
|---|---|
| Infiltrative | Non-Infiltrative |
| · Protein - amyloidosis | · Idiopathic, also includes familial |
| · Iron - cardiac siderosis: blood transfusions, hemochromatosis | · Scleroderma |
| · Oxalate - oxalosis | · Pseudoxanthoma elasticum |
| · Sarcoidosis | · PRKAG2 cardiomyopathy |
| · Lysosomal storage diseases - | · Hypertrophic cardiomyopathy |
| a) Sphingolipidoses - Fabry, Niemann-Pick, Gaucher | |
| b) Glycogen storage - Pompe, Danon | |
| c) Mucopolysaccharidoses - Hurler, Schie, Hunter, Sanfilippo, Morquio, Maroteaux-Lamy, Sly, Natowicz | |
| · Endomyocardial - EMF (Loeffler), Hypereosinophilic syndromes | |
| · Radiation | |
| · Drugs (ergotamine, anthracyclines, chloroquine) | |
| · Carcinoid | |
| · Metastases | |
The majority of patients with HFpEF will have some element of interstitial myocardial fibrosis especially in advanced stages of the disease which is a common myocardial response to the initial insult or injury and contributes to the abnormal diastolic function. Many older individuals will also have patchy deposits of ATTRwt amyloid which are seen in over 20% of autopsies in octogenarians dying of any cause. (Maurer et al., 2016) The mere finding of amyloid fibrils in the hearts of older patients does not automatically establish that they had amyloid cardiomyopathy as it may have been silent or subclinical. The degree of infiltration and other poorly understood environmental and genetic factors undoubtedly affect the phenotypic expression in any given individual.
In this review, we discuss the imaging features of the common infiltrative cardiomyopathies that cause HFpEF and outline their key diagnostic features, testing and treatment.
Restrictive cardiomyopathy is characterized by impaired myocardial compliance, diastolic dysfunction and preserved EF (Pereira et al., 2018). In younger individuals (< 30 years of age), it is mostly due to genetic disorders that cause increased fibrosis and/or abnormal deposition of iron, proteins, or glycogen. In older adults, common causes include cardiac amyloidosis (CA), iron overload (cardiac siderosis), radiation-induced heart disease and sarcoidosis (Table 1). It is important to recognize these etiologies as some of these now have disease-specific therapies. The major clinical presentation of RCM is HFpEF with dyspnea on exertion and fatigue being very common symptoms; however, HFrEF may occur in the later or terminal stages of the disease and is not uncommonly seen in advanced CA and hemochromatosis. Symptoms such as dyspnea are mainly due to increased LV filling pattern secondary to reduced viscoelastic properties and increased parietal stiffness. On echocardiography a common finding is that of grade 2 or 3 diastolic abnormality. One also encounters conduction abnormalities, arrhythmias and sudden cardiac death as well as thromboembolic complications in RCM (Muchtar et al., 2017). RCM is a disease with a 5-year mortality exceeding 40-50%. In one study of RCM in children, about 53% experienced SCD shortly after diagnosis; 75% of the remaining patients had HF, of whom all died or underwent heart transplantation within a few years of diagnosis (Rivenes et al., 2000). In adults, RCM has a 5-year survival rate of 55%, with the major mortality being from HF in 42% of patients (Kubo et al., 2007). The prototypical infiltrative RCM is cardiac amyloidosis. Other causes include: sarcoidosis, hemochromatosis, and storage disorders. RCMs are diagnosed by a combination of clinical, laboratory and imaging findings. On echocardiography, RCMs have normal or near-normal systolic function, at least in the early-to-mid stages of the disease and prominent diastolic dysfunction (with grade II to IV diastolic abnormality). Often, echocardiography and Cardiac magnetic resonance (CMR) imaging will allow one to hone in on a specific diagnosis or narrow it down to 2-3 major contenders based on the clinical context. Endomyocardial biopsy (EMB) is helpful to affirm the diagnosis but suffers from sampling bias in that if the RCM is not diffuse or extensive, the tissue sample may show normal myocardium if the involvement by the disease process is patchy which often occurs in the earlier stages. Furthermore, it has the disadvantage of being an invasive tool; this may lead to a delay in the diagnosis as it is used only when clinical suspicion is high or noninvasive tests have yielded inconclusive results.
It is now well-established that RCM due to cardiac amyloidosis is an underrecognized entity. Amyloidosis is an infiltrative disorder occurring due to extracellular deposition of a fibrillar material derived from various precursor proteins, called amyloid, that self-assemble with highly ordered abnormal cross β-sheet conformation (Hawkins, 1995). The two most common types of cardiac amyloidosis are transthyretin (Srimahachota et al., 2013), amyloidosis and the immunoglobulin light chain amyloidosis (AL); the former has two types: ‘wild-type' transthyretin form (ATTR-wt) and mutant transthyretin form (ATTR-m) (Mollee et al., 2014). The wild type, previously called senile cardiac amyloid is more common. Patients with the same type of amyloidosis, whether AL or ATTR, can have different patterns of organ involvement and different amounts of amyloid deposition in the involved organs, which determines the phenotype and course of the disease. Cardiac involvement drives mortality in AL amyloidosis; AL-CA is a rare condition with an estimated prevalence of 8 to 12 per million (Kyle et al., 1992). There are about 3,000 newly diagnosed cases of AL amyloid per year in the United States (US). 10-15% of patients with multiple myeloma develop AL amyloidosis. Of these, 30-50% have symptomatic cardiac involvement (Muchtar et al., 2016). The prognosis of untreated AL amyloidosis is very poor with median survival being around 6 months, and 5-year mortality exceeding 90%. ATTR-wt is now increasingly recognized as a cause of unexplained HFpEF in elderly patients. It is frequently accompanied by carpal tunnel syndrome and autonomic neuropathy. Patients with ATTR amyloidosis have a somewhat longer median survival ranging from 24 to 66 months; nevertheless, the overall prognosis is still poor in part due to the advanced age of these patients (Maurer et al., 2016).
As the presentation of amyloidosis can be nonspecific, diagnosing CA can sometimes be quite challenging. Patients may have been previously subjected to extensive evaluation but because of lack of awareness of the disease up until recently, many don't have a firm diagnosis other than “heart failure” which is often ascribed to other causes or nonspecific (Maurer et al., 2016). Many are diagnosed very late in the disease such that their survival is poor due to advanced HF. Nevertheless, a thorough history and exam, EKG and tailored cardiac imaging can often lead to a straightforward diagnosis CA provided it is being sought after (Fig. 1). Left ventricular thickening (often mislabeled “hypertrophy”) in the absence of a diagnosis of hypertension or valvular heart disease and low voltages on ECG will suggest CA (“EKG-Echocardiography mismatch”, also called “low volts-low BP-thick heart”) (Mohty et al., 2013). However, cardiac involvement can also occasionally occur without LV (left ventricular) thickening as in few cases of AL amyloidosis. Low voltage on ECG may be absent in 50% of AL-CA and 75% of TTR-CA. In ATTRm and in AL amyloidosis, extracardiac organ occurs more often with the most common being nephropathy, autonomic or peripheral neuropathy and gastrointestinal involvement (Gonzalez-Lopez et al., 2017). About 20% of AL amyloidosis patients have macroglossia. In ATTRwt, such extracardiac involvement is rather uncommon aside from orthostatic hypotension and carpal tunnel involvement - the latter may precede the diagnosis by 7-10 years. Vitreous involvement may rarely occur. Family history of cardiomyopathy and neuropathy should be inquired about in all patients.
 Figure 1.
Figure 1.Clinical diagnostic algorithm for evaluation of a patient suspected of having amyloidosis. PYP-SPECT: Pyrophosphate single photon emission computed tomography. EMB: endomyocardial biopsy. TTR-wt: wild-type transthyretin. TTR-m: mutant transthyretin
Echocardiography is the first and universally applied diagnostic imaging modality for CA. It provides both diagnostic and prognostic information. The characteristic features of CA on echocardiography includes left ventricular thickening - often in a concentric manner, enlarged atria, advanced diastolic dysfunction, thickened valves, pulmonary hypertension and the frequent presence of a pericardial effusion (Fig. 2) (Mohty et al., 2013). Right ventricular and interatrial septum thickening is also seen in CA. Sparkling or ‘speckled' appearance (Mohty et al., 2013) of the myocardium is common but not specific to CA, and its absence should not be used to rule it out. It occurs due to increased backscatter of ultrasound signal due to amyloid fibrils in the myocardium and is also termed a frosted glass appearance. Doppler imaging reveals diastolic abnormalities - there is often increased E/A and E/e' ratios. However, these findings may also be seen in other disorders with increased afterload, such as aortic stenosis, hypertrophic cardiomyopathy, hypertensive heart disease, renal disease and Fabry disease. Therefore, it is important to correlate the echocardiography findings with the patient's history and overall clinical picture.
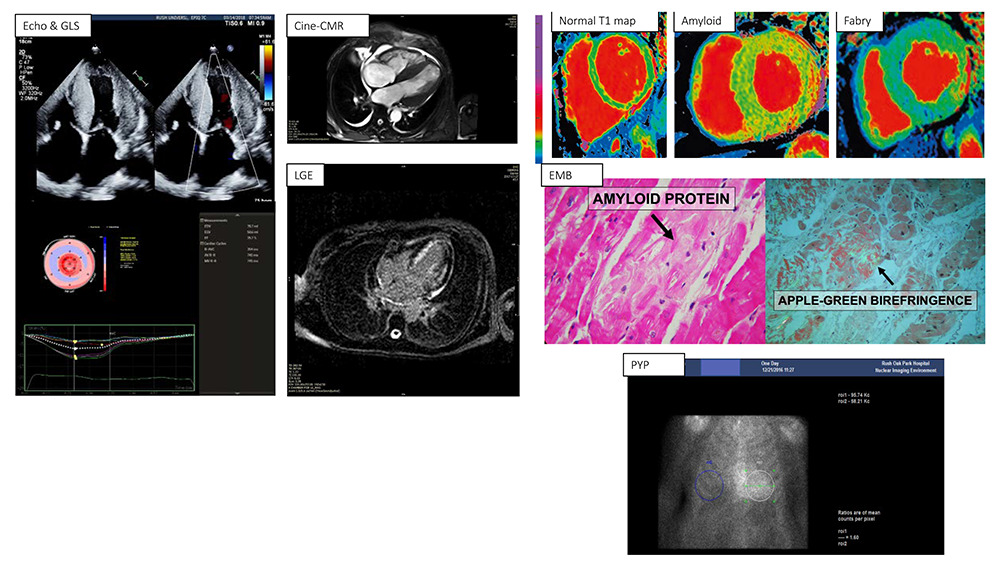 Figure 2.
Figure 2.Imaging in cardiac amyloid. Echo (Apical 4 chamber view) showing severe hypertrophy with granular sparkly myocardium, atrial enlargement, valve thickening. Bull-eye plot of regional longitudinal strain shows apical sparing (Cherry on top) pattern. Cine-CMR (Apical 4 chamber view) showing severe hypertrophy again with small pericardial and pleural effusion, preserved ejection fraction. There is subendocardial diffuse late gadolinium enhancement (LGE) on phase sensitive inversion recovery imaging. T1 maps (short axis view, 1.5T, MOLLI sequence) in a normal subject show homogeneous normal T1 values (970 ms, coded as green color). Amyloid patient with hypertrophy and patchy red color denoting increased T1 value (1190 ms). In contrast, the Fabry patient has low T1 value (bluish color, 850 ms, due to GB3 deposition) except in lateral wall where focal fibrosis is seen (red color, focally increased T1). Endomyocardial biopsy (EMB) showing amorphous appearing amyloid deposits in myocardial interstitial space on hematoxylin-eosin staining (left) and classic apple-green birefringence on polarized light microscopy when stained with Congo red stain. PYP-SPECT scan showing Heart:Lung ratio of 1.6 in a 83 year old man with HFpEF from ATTR-wt
Regional longitudinal strain is typically more impaired in the basal left ventricular segments than in the apical segments (i.e. an apical sparing pattern). When seen on a bull's eye plot, this has the appearance of a “cherry on top” (Fig. 2). Strain imaging also has prognostic utility (Buss et al., 2012) and is useful in detecting early CA, even before symptoms of hypertrophy are present (Buss et al., 2012). Additionally, it may help distinguish CA from other causes of LVH such as athlete's heart or hypertensive heart disease (Phelan et al., 2012).
CMR is an ancillary imaging technique useful in diagnosing CA, particularly, when echocardiographic findings are suggestive or uncertain and clinical suspicion for CA exists. CMR classic findings include an inability to null the myocardium on TI scout and diffuse gadolinium enhancement (LGE) in the ventricles and atria. Amyloidosis results in increase in the extracellular volume/space due to excessive interstitial amyloid deposition. Gadolinium is an interstitial agent, therefore, such CMR patterns are very suggestive of amyloidosis, and can help differentiate it from other causes of unexplained hypertrophy.
Amyloidosis typically produces a unique pattern of subendocardial myocardial late gadolinium enhancement (LGE) on CMR (Fig. 2). However, the pattern of LGE can be patchy, diffuse or transmural (Selvanayagam et al., 2007). LGE is associated with poorer prognosis, with transmural enhancement having worse survival compared to other patterns of enhancement (Fontana et al., 2015). The use of gadolinium contrast agent is contraindicated in patients with a glomerular filtration rate of less than 30 mL/min/1.73 m2 because of the increased risk of developing nephrogenic systemic fibrosis, therefore, its application to patients with amyloid who also have advanced kidney disease is limited (Yang et al., 2012).
T1 mapping and extracellular volume fraction are 2 techniques in CMR that can further support the diagnosis of CA and also have prognostic value. T1 mapping helps in further characterization of the tissues and is particularly useful in patients with advanced renal disease when gadolinium cannot be given (called native T1 mapping which does not require gadolinium contrast). Myocardial amyloid infiltration leads to elevated non-contrast or native T1 relaxation times (Fig. 2) (Karamitsos et al., 2013). Extracellular volume (ECV) fraction is an additional method in CMR imaging that is useful in diagnosing CA and can be calculated by obtaining pre- and post-contrast T1 values for myocardium and blood pool. ECV fraction progressively increases in patients with CA fibril deposition and thus, can be used as a surrogate to quantify amyloid burden (Banypersad et al., 2013).
Many nuclear bone tracers such as technetium-labeled bisphosphonates (99mTc-DPD, 99mTc-HMDP, and 99mTc-PYP) have been studied in CA. These tracers bind ATTR more than AL amyloid fibrils. Tc99m-PYP SPECT imaging can detect ATTR amyloidosis (Castano et al., 2016) with a high sensitivity and specificity, but not AL amyloidosis. Castano et al. showed sensitivity of 99% in ATTR-CA with 86% specificity (Castano et al., 2016). Therefore, it can be useful in distinguishing the two major types of CA. This differentiation of the amyloid type, including definition of ATTRm and ATTRwt, detected via bone scintigraphy and evaluation for extracardiac involvement, including neuropathy in ATTRm, is crucial for specific treatment (Gonzalez-Lopez et al., 2017). Additionally, Tc99m-PYP SPECT can help in determining prognosis. The ratio of a region of interest in the myocardium relative to a region of interest in the contralateral lung provides what is known as the ‘H:CL ratio' (Fig. 2). A H:CL ratio equal or greater than 1.6 predicts accurate diagnosis as well as increased mortality in ATTR amyloidosis (Castano et al., 2016). The Perugini score (grade 0-3) is also useful in characterizing the intensity of myocardial uptake and may help differentiate ATTR-wt from AL-CA. The latter typically has a low score of 0-1 while ATTR-wt has a score of 2-3. Nonetheless, up to 20% of patients with AL-CA may have higher degrees of uptake. Thus, bone scintigraphy alone should not be used to exclude the diagnosis of AL-CA and screening for monoclonal protein must always be performed also (Fig. 1).
Cardiac troponin and B-type natriuretic peptide (BNP) or NT-proBNP (N-terminal pro-B-type natriuretic peptide) are useful markers for diagnosis and prognosis. Elevation in BNP/NT-proBNP levels occurs due to myocardial stretch. Elevation in cardiac troponin is due to myocyte injury. Higher levels of NT-ProBNP and Tn are seen in AL-CA as compared to TTR-CA due to the direct toxicity of immunoglobulin light chains on myocytes. These biomarkers are often elevated in other cardiac conditions causing hemodynamic overload and also in renal disease, making its use to detect CA challenging due to low specificity. In AL-CA, high sensitivity troponin values higher than 50-54 ng/L are associated with higher mortality and correlate with New York Heart Association functional class, left ventricular ejection fraction and wall thickness (Dispenzieri et al., 2014; Kristen et al., 2010). Novel markers such as soluble ST2 and galectin-3 further improve prognostication in patients with AL amyloidosis (Dispenzieri et al., 2015).
EMB is the gold standard for diagnosis of CA. However, it is an invasive procedure; it may rarely result in complications and sometimes may not provide good yield depending on the specimen obtained; fibrillar deposition tends to be patchy and thus a negative EMB (usually obtained from a random portion of the right ventricular septal myocardium) does not rule out CA with certainty (Mollee et al., 2014). Owing to multimodality imaging techniques and the ancillary use of cardiac biomarkers, the need for EMB to diagnose CA has decreased significantly. A positive Congo red staining of the biopsy coupled with accurate typing of fibril protein content (tissue proteomics, usually by mass spectrometry) are used to diagnose CA.
50% of patients have a positive fat pad biopsy and the yield is higher in AL than in ATTRm or ATTRwt (80%, 67% & 14%) (Fine et al., 2014). While a fat pad biopsy is a preferred initial site due to ease of access, a negative result is insufficient to exclude the diagnosis and an endomyocardial biopsy should be performed when suspicion persists. When AL amyloidosis is suspected, one should obtain serum free light chains and serum and urine immunofixation electrophoresis (SPEP and UPEP miss ~20-25% of cases of AL - thus immunofixation is more sensitive). If a monoclonal protein is present, a bone marrow biopsy should be done to evaluate for a plasma cell clonal disorder (myeloma or MGUS). Bone marrow aspirate can also be stained for Congo red and when coupled with the serum/urine tests, can provide a diagnosis in over 90% of AL cases (Fig. 1). Sometimes, TTR-CA can coincidentally coexist with a monoclonal gammopathy in the absence of AL, and this makes the diagnosis even more challenging.
Treatment of cardiac amyloidosis includes symptomatic therapy of heart failure as well as the underlying disease. Treatment of AL amyloidosis is according to regimens used for multiple myeloma. Treatment will halt production of the pathogenic paraprotein, aid organ recovery and improve quality of life and survival. Recently, however, targeting the abnormal amyloid deposits is also being studied as another potential option. Patients with ATTR and AL are often elderly and frail due to advanced disease, their age and comorbidities - thus treatment selection should include patient wishes and minimize treatment-related toxicity.
Until few years ago, diagnosis of ATTR was uncommon and histological confirmation was necessary, making its diagnosis a real challenge in daily clinical practice. Moreover, specific therapeutic options were very limited. However, advances in cardiac imaging and diagnostic strategies have improved recognition of ATTR. Given that effective therapies are now available, cardiologists should be familiar with diagnosis and treatment of CA (Fig. 3).
 Figure 3.
Figure 3.Formation of TTR-amyloid fibrils and treatments. TTR is normally produced in the liver and circulates as a tetramer. In TTR-CA, the tetramer dissociates to form monomers which are unstable and aggregate to form amyloid fibrils that deposit in the heart. Knock-down of TTR synthesis in the liver can be achieved by RNA therapies or liver transplant. A second treatment approach is to use stabilizers that prevent tetramer dissociation. A third strategy employs drugs that degrade amyloid fibrils. mTTR: mutant thransthyretin; siRNA: small interfering ribonucleic acid; ASO: antisense oligonucleotide; TUDCA: Tauroursodeoxycholic acid; SAP: serum amyloid P component
Tafamidis is a selective small molecule that stabilizes the transthyretin homotetramer and subsequently avoids the rate limiting step of tetramer dissociation. It has been demonstrated to be useful in decreasing neurological deterioration in patients with early-stage hATTR polyneuropathy and has been used in Japan and Europe for that purpose for over 2 decades (Barroso et al., 2017). Recently, the efficacy and safety of tafamidis were also shown in CA (both ATTRwt and ATTRm). In the seminal ATTR-ACT trial which randomized more than 400 patients to either tafamidis or placebo, after 30 months there was a significant impressive reduction in all-cause mortality by 30% and cardiovascular-related hospitalizations by 32% for tafamidis (pooled data for both the 20 and 80 mg once daily groups) compared to placebo. Patients receiving tafamidis also experienced a significant reduction in the decline of functional capacity assessed via the 6-min walk test and quality of life compared. Therefore, tafamidis is now approved for the treatment of ATTR cardiomyopathy by the US Food and Drug Administration (Maurer et al., 2018).
Patisiran is an RNAi therapeutic agent composed of a small interfering RNA formulated as a lipid nanoparticle that enables delivery to hepatocytes (Fig. 3). After intracellular release, the small interfering RNA blocks production of mutant and wild-type TTR protein by inducing cleavage of TTR messenger RNA resulting in lowering of TTR blood levels up to 80% when administered intravenously every 3 weeks (Coelho et al., 2013). This drug has recently been approved for hereditary TTR-amyloid polyneuropathy in adults based on favorable data from the APOLLO trial (Adams et al., 2018). Another RNA-based therapy is Inotersen which is a double stranded antisense oligonucleotide and works by binding to cognate TTR mRNA and subsequent RNAse H1 mediated degradation of cellular TTR mRNA, thereby reducing serum TTR protein and tissue deposits. This drug is administered by subcutaneous injection by the patient once weekly and is also approved for hereditary TTR-amyloid polyneuropathy in adults (Benson et al., 2018). Side effects include thrombocytopenia and glomerulonephritis. 3rd generation formulations of these drugs are being studied to mitigate the adverse effect profile and allow for monthly or less frequent home subcutaneous administration for convenience.
All the aforementioned drugs either prevent new deposition of fibrils or stabilize the amyloidogenic TTR tetramer but don't actually remove the deposits in the organs. Trials are ongoing with the RNA-based therapies in CA also - with promising data available looking at echocardiography endpoints (wall thickness, strain) (Solomon et al., 2019). However, to date there is no treatment available to remove the amyloid already deposited in the tissue. Thus, early diagnosis is still critical.
Sarcoidosis is a multisystem granulomatous disease. Noncaseating granulomas are the pathological hallmark of the disease and are most often associated with pulmonary and lymph node involvement; however, the heart is also frequently involved (Iannuzzi et al., 2007). Clinically evident cardiac involvement is seen in 5% of patients and has a male predominance. However cardiac involvement is seen in a higher percentage of patients on autopsy (25%) indicating that many cases can go undetected due to lack of cardiac symptoms. HF symptoms are seen in 30% of patients. In patients with manifest CS, there is a higher risk of bundle branch block (especially RBBB), high-grade atrioventricular block requiring pacemaker and ventricular tachycardia and sudden cardiac death requiring implantable cardioverter-defibrillator therapy. Biomarkers such as angiotensin-converting enzyme, lysozyme, urinary calcium, interleukin, interferon, neopterin, B-type natriuretic peptide, or high-sensitivity troponin levels are often elevated in patients with CS but unfortunately these tests have limited clinical utility because of low sensitivity and specificity (Iannuzzi et al., 2007). Chest X-ray is the initial imaging study used to identify lung and lymph node involvement and is abnormal in > 90% of patients with extracardiac sarcoidosis (Criado et al., 2010). High-resolution CT of the lung enables better characterization of lung parenchymal and interstitial involvement. ECG abnormalities are present in 3.2% to 8.6% of patients with clinically silent CS (Birnie et al., 2016). Right bundle branch block and atrioventricular block and are the most common ECG findings (Birnie et al., 2014). Echocardiography typically shows wall motion abnormalities in a noncoronary distribution, and basal anteroseptal or inferolateral thinning with increased echogenicity (Uemura et al., 2005). Other abnormalities include reduced left and right ventricular systolic and diastolic function, ventricular dilatation or aneurysms, valvular abnormalities, and pericardial effusion (Blankstein and Waller, 2016). Imaging modalities such as CMR and positron emission tomography may provide valuable information for diagnosis and treatment response in CS. CMR findings in the acute phase of the disease include increased intramyocardial signal intensity on T2-weighted images due to granulomatous lesions and edema associated with inflammation (Kouranos et al., 2017). In the chronic phase of CS, myocardial wall thinning or aneurysms and a low signal on T2-weighted images but high LGE signal intensity may be seen, indicating scarring or fibrosis (Fig. 4A). Cardiac PET imaging for CS involving the use of FDG to evaluate for myocardial inflammation is very useful. Special patient preparation is required before 18F-FDG-PET/CT imaging to adequately suppress normal myocardial FDG uptake and increase the utilization of free fatty acids to identify focal inflammation within sarcoid granulomas. The classical patterns of myocardial FDG uptake in patient with active CS are either diffuse, focal, and focal-on-diffuse patterns (Youssef et al., 2012).
 Figure 4.
Figure 4.CMR in cardiac sarcoidosis (A) Apical-3 chamber view demonstrating multiple patchy areas of LGE in the epicardium and mid-myocardium that reflect sites of granulomatous inflammation and scarring. Typically involvement occurs in the lateral and septal myocardium but can be quite variable such that cardiac sarcoid can mimic the LGE imaging patterns of many other diseases. CMR T2* imaging in cardiac iron overload (B). This 23-year-old man had thalassemia and multiple blood transfusions leading to cardiac hemochromatosis. Since iron is paramagnetic, it decreases the T2* relaxation time of the adjacent water protons in the myocardium. Using a multi-echo sequence, one can plot the sequential decay of T2* on a curve to compute the iron content within myocardium. This patient had severe iron overload (T2* time <10 ms, normal is > 40 ms) and was started on chelation therapy.
Increased gastrointestinal iron absorption in hemochromatosis results in iron overload. This condition is then characterized by increased cellular uptake of non-transferrin bound iron. In the myocardium, excess iron results in transmembrane Ca2+ flux abnormalities and diastolic dysfunction. Eventually this leads to myocyte loss, replacement fibrosis and cardiac dilatation (Pereira et al., 2018). There is an early phase of RCM with a HFpEF phenotype that progresses to a later phase characterized by HFrEF with dilated cardiomyopathy. Once HF sets in, the prognosis is poor, even with chelation therapy. Early identification is thus very important for the management of patients with cardiac siderosis and can be done by CMR imaging. CMR T2* imaging is very sensitive to the presence of cardiac iron and has been well validated in quantifying it noninvasively and has been shown to improve patient prognosis by early institution of therapy (Fig. 4B). Other tools to follow patients are biomarkers such as NT-proBNP levels and echocardiography (especially tissue Doppler and strain imaging).
Fabry disease is an X-linked recessive lysosomal storage disorder. Mutations in the GLA gene encoding the lysosomal enzyme α-galactosidase result in impairment of metabolism of glycosphingolipids and accumulation of globotriaosylceramide (GB3) in various tissues, particularly in the endothelium of the heart, kidney, brain and skin. Males present in childhood with acroparesthesias (pain and tingling in extremities, often precipitated by heat or exercise or dehydration), hypohidrosis, cornea verticillata (whorling appearance on slit-lamp), angiokeratomas of the skin in the lower abdomen and groin, and gastrointestinal disturbances. Females are usually affected 10-15 years later and a may develop a milder phenotype. Cardiac manifestations include stroke and myocardial infarction at a young age and hypertension that is often worse than would be expected for the degree of renal dysfunction or proteinuria. GB3 accumulates in all cardiac cell types, including cardiomyocytes, microvascular endothelial and smooth muscle cells, fibroblasts, and the valves, producing myocardial hypertrophy that mimics the morphological and clinical picture of hypertrophic cardiomyopathy or unexplained left ventricular hypertrophy. The 2 main types of disease manifestations include the classic variant and the cardiac variant. In the classic variant, males have very low or absent α-Gal A activity, which results in severe systemic manifestations that typically begin in childhood or adolescence. Cardiac involvement typically becomes noticeable between 20 and 40 years of age, and renal involvement usually progresses to the point of requiring dialysis or renal transplantation. Heart failure, arrhythmias, and myocardial infarction are usually late manifestations. Patients with the “cardiac variant” of Fabry disease have residual enzyme activity (approximately 1%-5% of normal values). They present in the fifth and sixth decades of life with unexplained hypertrophy and conduction disease, without other classic manifestations of Fabry disease. In male patients who have classic Fabry disease, the diagnosis is straightforward to establish by a blood test for assaying α-Gal A activity in leukocytes. In those with atypical presentations or in heterozygous females, α-Gal A activity may be within the normal range (especially during early stages of the disease) and genetic testing is recommended for identifying a pathogenic mutation in the GLA gene.
Echocardiography reveals increased wall thickness, often in a symmetric fashion and in ~25% of cases, a striped endomyocardial pattern (Fig. 5) (binary sign) (Mundigler et al., 2011). Additional features include mild valvular regurgitation, diastolic dysfunction, abnormal longitudinal strain and in the later stages, decline in ejection fraction. CMR may show low T1 values in areas of myocardium with GB3 accumulation, T2 hyperintensity and abnormalities in perfusion in addition to late gadolinium enhancement (most often in the basal inferolateral segment) (Fig. 5).
 Figure 5.
Figure 5.Cardiac imaging in Fabry disease. Echo (parasternal long axis view) showing severe left ventricular hypertrophy. CMR LGE image showing severe LVH and LGE in basal inferolateral wall in mid-myocardium (arrow). b) T1 mapping shows low T1 in most of the myocardium due to GB3 deposition except in area of fibrosis in the same wall where it is elevated (arrow). c) T2 mapping shows high T2 in same wall signifying ongoing inflammation and edema (arrow) Reproduced with permission from Nordin et al. (2016). Electron microscopy (EM) of cardiac biopsy showing GB3 deposits inside the lysosomes appearing as concentric lamellae spaced ~5nm apart. These are not pathognomonic and may also be seen with drugs such as chloroquine, amiodarone and few other lysosomal storage diseases.
Treatment includes recombinant enzyme replacement therapy (agalsidase-ß) that is given intravenously every 2 weeks and restores α-galactosidase A function exogenously. Some data shows that this may delay the progression to renal failure and heart failure if instituted early. Migalastat is a pharmacological chaperone that facilitates trafficking of amenable mutant forms of α-gal A enzyme from the endoplasmic reticulum to lysosomes and increases its lysosomal activity (McCafferty and Scott, 2019). Oral migalastat is the first approved medication for treating patients with Fabry disease who have an amenable GLA mutation.
Our understanding of the epidemiology and natural history of HFpEF is fairly robust, but knowledge gaps exist. Greater study of HFpEF subgroups to understand the underpinnings of the prognostic, pathophysiological, and therapeutic differences in this population are needed. In infiltrative cardiomyopathies, knowledge of the unique clinical features of the various major cardiomyopathies can help the clinician decide which laboratory and imaging tools to use to confirm the diagnosis and to decide which specific therapies to use. Multimodality imaging is often employed and can avoid invasive endomyocardial biopsy in many cases to arrive at the correct diagnosis.
AL-CA, Light chain cardiac amyloidosis; BNP, B-type natriuretic peptide; CA, Cardiac amyloidosis; CMR, Cardiovascular magnetic resonance; CS, Cardiac sarcoidosis; DM, Diabetes mellitus; EMB, Endomyocardial biopsy; FDG, 18-fluoro-deoxyglucose; GB3, Globotriaosylceramide; HF, Heart failure; GLS, Global longitudinal strain; HCM, Hypertrophic cardiomyopathy; HFpEF, Heart failure with preserved ejection fraction; HFrEF, Heart failure with reduced ejection fraction; Tn, Troponin; TTR, Transthyretin; TTR-m, Mutant transthyretin; TTR-wt, Wild type transthyretin; RCM, Restrictive cardiomyopathy
Both the authors contributed to the manuscript
The authors declare no funding, no contribution from other persons.
The authors declare no conflicts of interest.