







1 Princess Máxima Center for Pediatric Oncology, Utrecht 3584 CS, The Netherlands
2 Department of Pediatric Oncology/Hematology, Cancer Center Amsterdam, Amsterdam UMC, VU University, Amsterdam 1081 HV, The Netherlands
Abstract
Tumors of the central nervous system (CNS) are the main cause of cancer-related death in children. While improvements in survival rates for various childhood cancers have been obtained over the last decades, little progress has been made for pediatric brain tumors. In addition, current conventional treatment gives rise to severe long term toxicity, which underpins the burning need for the development of novel therapeutic modalities. Immunotherapy was shown to be successful in both adult solid tumors and pediatric hemato-oncology, and may be an option for pediatric CNS malignancies. However, pediatric brain tumors have a strong immunosuppressive microenvironment, which is considered a major hurdle for effective immunotherapy. The low mutational burden of these tumors may compromise immunotherapy for this patient group even further. The possibility to directly apply the current immune modulating therapies directly into the tumor, however, opens new options for immunotherapy in this population. This review covers immunotherapeutic approaches including immune checkpoint inhibition, chimeric antigen receptor T (CAR-T) cell therapy, therapeutic cancer vaccines, and oncolytic virotherapy. We review their effect on the immunosuppressive tumor microenvironment, summarize current trials, and discuss future directions. We conclude that immunotherapy holds promise for children with CNS malignancies, especially when combined with different (immune) therapeutic strategies.
Keywords
- Brain tumors
- pediatric
- immunotherapy
- immune checkpoint inhibitors
- oncolytic virotherapy
- CAR-T
- dendritic cell
- cancer vaccines
Tumors of the central nervous system (CNS) are the main cause of cancer-related death in children [1]. Overall, five-year survival rates for pediatric brain and CNS tumors is approximately 72% [2]. Survival rates vary substantially depending on type, grade, location, size and age. Five-year survival rates range from around 90% for low-grade gliomas (LGGs) down to 25% for high-grade gliomas (HGGs) [2]. Pediatric cancer mortality rates have been declining continuously over the last decades. However, little progress has been made in the treatment of pediatric brain tumors compared to other pediatric tumors [3]. In addition, survivors are often left with neurologic and neurocognitive morbidities, caused by the treatment with chemotherapy, radiotherapy and surgery. The limited progress in achieving decreased mortality rates among children with brain tumors and the severe side effects that survivors endure indicate that novel therapies are highly needed.
Immunotherapy is a rapidly evolving new treatment modality in cancer treatment. One of the advantages of immunotherapy is the ability to specifically target tumor cells while leaving healthy tissue undamaged. Immunotherapy has been successfully used in the treatment of several hematologic malignancies and solid tumors and has been able to achieve increased survival. This clinical benefit of immunotherapy raised interest for application in children with brain tumors, for whom it might be a promising treatment option as well. Immunotherapy harnesses the body’s own immune system to fight cancer cells. An immune response is elicited when the immune system recognizes a non-self pathogen or immunogenic non-self components. The first phase of the immune response is the innate immune response. When phagocytes and natural killer (NK) cells bind to a pathogen via their receptors, cytokines and chemokines are produced and secreted which induce inflammation. These molecules can attract naive T-cells to respond to and eliminate the pathogen [4]. Adaptive immunity is the response of a lymphocyte towards a specific antigen, and is recruited when the innate immune system is unable to resolve the inflammation by itself. Dendritic cells (DCs) are the most potent antigen-presenting cells (APCs). DCs migrate to the lymph nodes where antigens can be presented to pathogen-specific T-lymphocytes by displaying the antigens on a major histocompatibility complex (MHC) [5, 6, 7, 8]. Therefore, DCs provide an essential link between the innate- and adaptive immune system. Upon binding to an antigen, CD4+ T-cells, CD8+ T-cells, and B-cells undergo clonal expansion [7, 9, 10]. The activated cell produces identical clones that can secrete monoclonal antibodies. An overview of the immune response is depicted in Fig. 1.
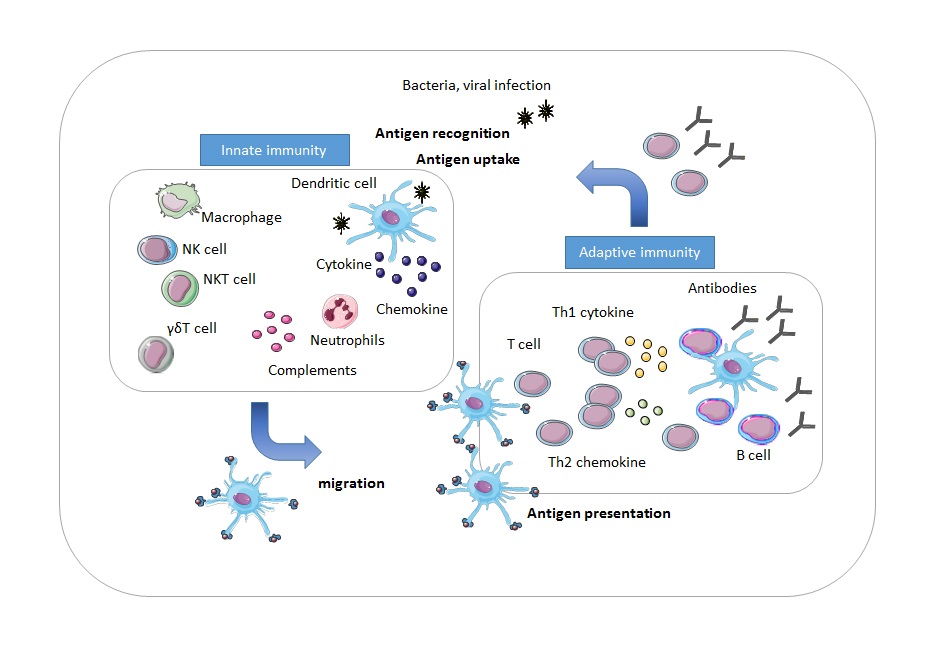 Figure 1.
Figure 1.Overview of the immune response. During the innate immune response, phagocytes and NK cells secrete cytokines and chemokines to mediate destruction of the pathogen. Dendritic cells travel to the lymph nodes, where they present the antigen to pathogen-specific T-cells. In response, T-cells and B-cells expand and B-cells produce antibodies to eliminate the pathogen.
The combination of these processes usually eliminates the pathogen and resolves the inflammation. In cancer however, one or more steps are undermined; the inflammation fails to resolve, giving rise to a chronic inflammatory tumor microenvironment (TME) [11], which stimulates immunosuppression. Tumors exploit various mechanisms for immune evasion. This escape from the immune attack is linked to the TME, as the tumor stromal cells are responsible for the production of immunosuppressive factors. The mechanisms exploited by the tumor are the secretion of immunosuppressive cytokines (such as transforming growth factor beta (TGF-β) and interleukin 10 (IL-10)), depletion of essential nutrients (by indoleamine dioxygenase), recruitment of immunosuppressive cells (regulatory T-cells, tumor associated macrophages, and myeloid-derived suppressor cells) and expression of inhibitory molecules like Fas ligand (FasL), or programmed death ligand 1 (PD-L1) [12]. In addition, the tumor can downregulate molecules required for T-cell recognition as well, such as MHC, the antigen itself, or molecules implicated in processing of the antigen [11]. Regulatory T-cells inhibit effective antitumor immunity through various suppressive mechanisms, which include modulation of the cytokine microenvironment, metabolic disruption of the target cell and alteration of DC function [13]. For example, regulatory T-cells produce the immunosuppressive cytokines TGF-β and IL-10 [13] and induce a functional hyporesponsive state of tumor infiltrating cytotoxic T-lymphocytes, characterized by impaired cytokine secretion and cytotoxic granule release as well as co-expression of the inhibitory molecules PD-L1 and T-cell immunoglobulin and mucin domain 3 (TIM-3) [14]. Immunosuppression consists a complex set of processes, of which the individual mechanisms interact, as well as maintain each other’s function.
Pediatric brain tumors are known to be immunologically ‘cold’, indicating the lack of tumor-infiltrating T-cells [15]. This deficiency in T-cell infiltration might be due to a lack of tumor antigens, a deficit in antigen presentation, an absence of T-cell activation, or a deficit of homing into the tumor bed [16]. In turn, the lack of tumor antigens can be associated with the low mutational burden that characterizes pediatric brain tumors, resulting in fewer neoantigens to activate T-cells [17]. The absence of infiltrating T-cells and the immunosuppressive TME are intrinsically linked, illustrated by the example of TME induced downregulation of molecules required for T-cell recognition, resulting in subsequently impaired T-cell infiltration.
In contrast to the ‘cold’ phenotype that characterizes both adult and pediatric brain tumors, pediatric brain tumors might exhibit distinct immunophenotypes when compared to adult brain tumors, in terms of inflammatory and immunosuppressive TME. Adult HGGs are known to have an immunosuppressive microenvironment and extensive immune cell infiltration [18]. In contrast, various immunophenotypes have been identified for pediatric pilocytic astrocytoma (PA), ependymoma (EPN), glioblastoma and medulloblastoma. Myeloid and lymphocyte infiltration levels were significantly lower in pediatric glioblastoma and medulloblastoma compared to PA and EPN [19]. Remarkably, diffuse midline glioma (DMG) was neither shown to have an inflammatory nor an immunosuppressive TME [20, 21]. In addition, medulloblastoma was found to have low expression of immune markers, and immune-stromal microenvironmental patterns even differ between subtypes of medulloblastoma, indicating distinct types of local immunosuppression [22]. These outcomes point to a possible different focus in designing immunotherapy for pediatric brain tumor compared to adult brain tumors.
The next part of the current review discusses the different immunotherapeutic approaches for pediatric brain tumors, as well as the advantages, considerations and challenges that are associated with the application of these immunotherapeutic strategies for children with brain tumors. The following research question will be addressed: Does immunotherapy have the potential to become a new modality in pediatric brain tumor treatment?
Tumors utilize various molecular mechanisms to evade an immune attack. One such mechanism is the so-called immune checkpoint proteins. An example of an immune checkpoint protein is programmed cell death protein 1 (PD-1). Binding of PD-1, a receptor expressed on T-cells, to its ligand PD-L1 expressed on tumor cells, results in inhibition of T-cell responses [23]. Blocking the PD-1/PD-L1 pathway using anti-PD-1/PD-L1 antibodies that serve as immune checkpoint inhibitors (ICIs) can restore T-cell activation towards tumor cells and markedly enhance antitumor responses (Fig. 2).
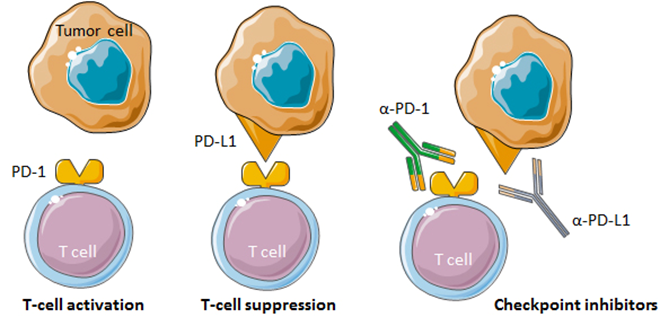 Figure 2.
Figure 2.Immune checkpoint inhibition by anti-PD-1 and anti-PD-L1 monoclonal antibodies. T-cell activation is inhibited upon binding of PD-1 to its ligand PD-L1 on tumor cells. Monoclonal antibodies that bind to PD-1 or PD-L1 can block the PD-1/PD-L1 interaction and promote antitumor responses.
ICIs have proven efficacy in the treatment of different adult malignancies, such as non-small cell lung cancer [24] and melanoma [25]. In a study investigating the response to pembrolizumab, an anti PD-1 monoclonal antibody, in both adults and children with recurrent primary brain tumors, no clinical or histological efficacy was demonstrated [26]. This might be attributed to the low mutational burden that is found in CNS tumors. A higher tumor mutational burden is proposed to predict responsiveness to ICIs across various tumors [27], because a higher mutational burden results in an increased amount of neoantigens that can be recognized by the immune system which triggers an antitumor response. This finding was confirmed in studies that investigated the association between tumor mutational burden and efficacy of ICIs in children with brain tumors. Hypermutant biallelic mismatch repair deficient (bMMRD) glioblastomas were found to have a significantly higher mutational burden than sporadic adult and pediatric gliomas and other brain tumors. Consequently, bMMRD glioblastomas were found to have a mean neoantigen load of 7-16-fold higher than other immunoresponsive cancers including melanoma. Based on this observation, two siblings with bMMRD-induced glioblastoma were treated with the anti-PD-1 inhibitor nivolumab and showed significant clinical responses [28]. In addition, positive PD-L1 expression of the tumor might serve as a biomarker for response to ICIs. Responses to immune checkpoint inhibition with nivolumab in children with brain tumors were observed in patients with PD-L1 expression and a high mutational burden [29]. Responsiveness to ICIs may depend on the tumor type, and might even differ in individual patients. PD-L1 expression was positive in a part of atypical teratoid rhabdoid (ATRT) tumors, EPNs and HGGs [30, 31]. Given the selective efficacy of ICIs targeting the PD-1/PD-L1 pathway in pediatric brain tumors, the application of this ICI in this patient population will be rather limited.
Another therapeutic target that might be more relevant for pediatric brain tumors is cluster of differentiation 47 (CD47), a highly expressed protein on the surface of tumor cells involved in immune evasion. When CD47 binds to signal-regulatory protein alpha (SIRPα) on myeloid cells, macrophage-induced phagocytosis is inhibited. Hu5F9-G4, a humanized anti-CD47 antibody, blocked the CD47-SIRPα pathway, resulting in macrophage-mediated phagocytosis. Hu5F9-G4 induced phagocytosis and inhibited tumor growth in patient-derived xenografts of group 3 medulloblastoma, ATRT, primitive neuroectodermal tumor (PNET), glioblastoma and diffuse midline glioma (DMG) [32]. For all the tumor types, Hu5F9-G4-treated mice displayed increased survival compared to controls. Intraventricular administration of Hu5F9-G4 inhibited leptomeningeal metastases in medulloblastoma xenografts. Finally, Hu5F9-G4 proved to be safe. Collectively, these findings point to the promise of this immune checkpoint to be successfully targeted in pediatric brain tumors.
Chimeric Antigen Receptor T-cell (CAR-T) therapy is a form of adoptive immunotherapy that does not require pre-existing immunity to be present, but instead can activate immune responses. In CAR-T therapy, autologous cytotoxic T-cells are genetically modified to express a chimeric antigen receptor that binds a tumor specific antigen [33]. CAR T-cells are injected back into the patient to destroy the cancer cells (Fig. 3). CAR T-cell therapy provides an abundance of tumor-specific T-cells that bypasses pre-existing T-cell requirements for antigen presentation and processing. Therefore, CAR T-cell therapy might be more useful for pediatric brain tumors that lack a high mutational burden or positive immune checkpoint expression and do not respond to treatment with ICIs.
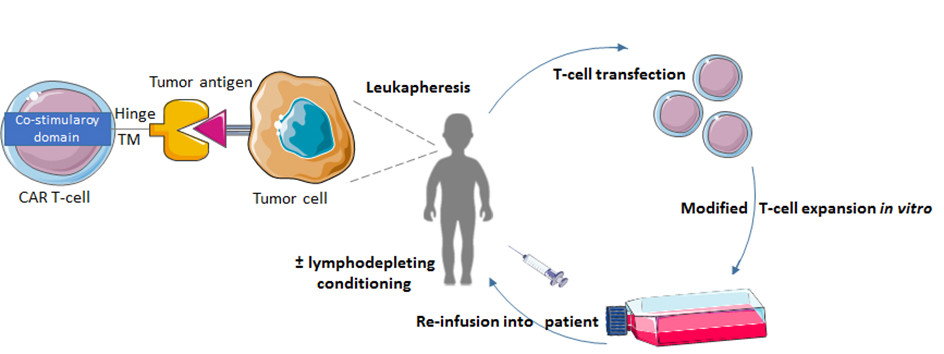 Figure 3.
Figure 3.The process of CAR T-cell therapy. Patient T-cells are collected via leukapheresis. These T-cells are then genetically engineered with a viral expression vector to express the CAR protein. The CAR T-cells are expanded and re-infused into the patient, where the small chain fragment variable of the CAR T-cell can bind to the tumor antigen.
Clinical studies on CAR T-cell therapy focusing on pediatric brain tumors are scarce. Some target antigens have been identified in preclinical studies, one of which is the disialoganglioside GD2. Patient-derived DMGs with Histone 3-Lysine-27-Methionine (H3K27M) mutations were found to have high expression of GD2. Use of GD2 CAR-T therapy resulted in potent antitumor efficacy in DMG xenografts of the H3K27M glioma subtypes of the pons, thalamus and spinal cord [34]. Treatment related toxicity occurred in a few mice. Histologic analysis revealed that this was caused by GD2 CAR-T induced hydrocephalus and a widespread inflammation of the brain parenchyma, meninges, ventricles and especially the brainstem. In neuroblastoma patients, GD2 CAR T-cell therapy is considered safe and demonstrated clinical efficacy [35, 36]. The clinical efficacy of this therapy in children with DMG has yet to be assessed. The susceptible location of DMGs and the severe toxicities observed in animals as a consequence of the GD2 CAR-T treatment emphasize the importance of carefully designed trials with extensive monitoring of possible neurotoxic effects and protocols for potential tackling of neurotoxicities for translation to humans.
Recently, cluster of differentiation 276 (B7-H3) as a new CAR-T therapeutic target for pediatric brain tumors was discovered in a preclinical trial [37]. B7-H3 was found to be homogenously overexpressed on various pediatric solid tumors and brain tumors, among which medulloblastoma, HGG and DMG. B7-H3 CAR T-cells were co-cultured in vitro with medulloblastoma cell lines. It was shown that B7-H3 CAR T-cells cleared medulloblastoma xenografts and significantly increased survival. In addition, it was shown that a high target antigen expression on the surface of tumor cells is needed for B7-H3 CAR-T efficacy, and that efficacy is limited when target cells express low antigen levels. Many targeted tumor antigens, such as B7-H3, are expressed by healthy tissues as well, which can result in on-target, off-tumor toxicities through antigen binding on non-tumor tissues. In some studies, this has resulted in severe on-target off-tumor toxicities, including treatment-related mortalities [38, 39]. Therefore, an integral part of application of CAR T-cell therapy is the management of these severe toxicities. However, MGA271, the antibody on which B7-H3 CAR T-cell therapy is based, exhibits minimal binding to healthy tissues [40], providing a favorable therapeutic window, and displaying promise for translation to the clinic.
Clinical trials on the efficacy of CAR-T therapy in brain tumors have mainly been performed in adult glioblastoma patients. Brown et al. [41] reported on a patient with recurrent multifocal glioblastoma who showed regression of the intracranial and spinal tumors after CAR T-cell treatment targeting the tumor associated antigen interleukin-13 receptor alpha 2 (IL-13α2), a remarkable response that continued for almost eight months after the initiation of the treatment. Although a promising result, the application in a single patient makes it difficult to draw conclusions about the true impact of this therapy. In another study, 16 patients with progressive human epidermal growth factor receptor 2 (HER2) positive glioblastoma received HER2 specific CAR-modified virus specific T-cells [42]. Among the 17 patients were seven children. One patient, a 17-year-old male, had a partial response, which was defined as a 30% decrease in the longest diameter of the tumor, which lasted nine months. Seven patients showed stable disease for 2 to 29 months, and the other eight had progressive disease from the CAR T-cell infusion onwards.
CAR-T therapy is associated with some challenges regarding safety and feasibility, among which are the on-target, off-tumor toxicities and neurotoxicities, as indicated in the preclinical studies mentioned above. Cytokine releasing syndrome is the most common adverse event following CAR-T therapy, characterized as an elevation in inflammatory cytokines resulting from immune activation [43]. The potential risks regarding safety are demonstrated in a dose escalation study in which two patients with recurrent glioblastoma experienced severe hypoxia, including one treatment related mortality, after infusion of epidermal growth factor receptor variant III (EGFRvIII) CAR T-cells at the highest dose [44]. Heterogeneity and loss of antigen expression are feasibility impediments identified for efficient CAR T-cell therapy in glioblastoma [45, 46]. Moreover, CAR T-cell therapy induced compensatory immunosuppressive responses. Compared to pre-CAR T-cell infusion, in situ evaluation of the TME post-CAR T-cell infusion, revealed upregulation of expression of various regulatory T-cells and immunosuppressive molecules [46]. Future studies should include preclinical trials focusing on overcoming these barriers of heterogeneity of expression, loss of antigen expression and adaptive resistance. It should be noted that molecular features underlying pediatric brain tumors are substantially distinct from those in adult brain tumors. For example, epidermal growth factor receptor (EGFR) gene amplifications, which are common in adult HGGs, were shown to be rare in pediatric HGGs [47, 48]. Furthermore, EGFR is underexpressed in pediatric glioblastomas compared to their adult equivalent [47]. As such, EGFR seems a less likely target for CAR-T therapy in children. However, the platelet-derived growth factor receptor A (PDGFRA) gene was found to be amplified in pediatric glioblastoma, and was overexpressed compared to adult glioblastoma [47]. This indicates that PDGFRA may be a more useful target in pediatric brain tumors. Thus, although promising targets have been found in preclinical studies of CAR T-cell therapy in pediatric brain tumors, further investigations of these and other targets in clinical trials are warranted. Some safety risks and feasibility limitations are brought in association with CAR T-cell therapy. For translation to clinical trials in children, these require close monitoring and management strategies to be set up.
Another new immunotherapeutic modality is therapeutic cancer vaccines. The latter are designed to induce an immune response against antigens overexpressed by tumor cells. Dendritic cell (DC) vaccines and peptide vaccines as a treatment for pediatric brain tumors will be discussed below.
Peptides representing a specific tumor antigen can be administered as a vaccine (Fig. 4). In accordance with other immunotherapeutic modalities, only a few clinical trials on peptide vaccines in brain tumor treatment have focused on the pediatric population. Vaccinations with peptide epitopes derived from three glioma-associated antigens (GAA) were well-tolerated and showed some evidence of immunological and clinical responses in children with newly diagnosed malignant brainstem and non-brainstem gliomas [49], recurrent LGGs [50] and recurrent HGGs [51]. The three GAAs, interleukin-13 receptor alpha 2 (IL-13Rα2), ephrin type-A receptor 2 (EphA2) and survivin, were shown to be highly expressed in pediatric gliomas [52]. Vaccination induced immune responses to at least one of the vaccine-targeted GAA epitopes in the majority of the patients [49, 50, 51]. No dose-limiting toxicities were encountered and occurring toxicities were generally mild, supporting the conclusion that peptide vaccines from GAA epitopes are well-tolerated by the pediatric population with various gliomas. The peptide vaccines are suggested to have modest clinical efficacy, although the true efficacy is difficult to ascertain based on a small number of patients and in the absence of controls. In the three studies, a small proportion of the children displayed a partial (> 50% decrease in maximal tumor area) or minor response (25% to 50% decrease in maximal tumor area), with some partial responses persisting over an extraordinary long period. In children with recurrent HGGs, a six-month progression free survival (PFS) rate of 33% and a six-month overall survival (OS) rate of 73% was noted, a result that the authors propose being favorable compared to other studies in the same population.
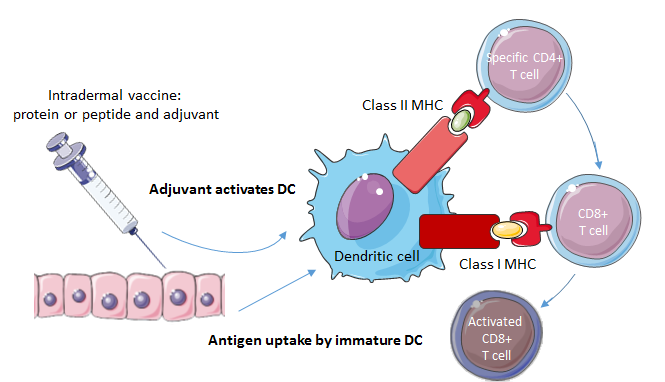 Figure 4.
Figure 4.Mechanism of action of cancer vaccines. In case of a peptide vaccine, a peptide and an adjuvant are injected into the patient. The adjuvant activates the DC, which presents the antigen to T-cells by displaying the antigen on MHC class 1 and class 2 molecules. CD4+ T-cells help the CD8+ T-cells to mature. An activated CD8+ T-cell can lyse the tumor cell bearing the tumor antigen.
Pseudoprogression, which is defined as a new or an expanded area of contrast enhancement in the absence of true tumor growth [53], occurred in five of the 26 patients with a newly diagnosed brainstem or non-brainstem glioma [49]. Patients exhibited transient increases in tumor size or enhancement in combination with new or worsening neurologic deficits, but upon Magnetic Resonance Imaging (MRI), they showed stabilization and/or clinical improvement after administration of dexamethasone to diminish the pseudoprogression. Interestingly, pseudoprogression was identified to be associated with prolonged survival. Four of the patients with pseudoprogression had a brainstem glioma, and their OS was 19.5 months compared to the 10.9 months in brainstem glioma patients without pseudoprogression. Therefore, pseudoprogression is proposed to be a possible sign of efficacy.
In the three studies investigating peptide vaccines in children with brain tumors, the relatively limited clinical response might be attributed to a few factors. Firstly, a clinical response might be hampered as a consequence of lacking antigen processing components such as MHC, immunosuppressive regulatory T-cells, or inhibitory checkpoint pathways. Secondly, effectiveness may be limited by an absence of any of the target antigens on a subset of the tumors as noted in various studies [49, 52, 54]. Moreover, target antigen reactivity may be lost over time [54], as was observed in a substantial number of patients, especially in the group with newly diagnosed brainstem and non-brainstem gliomas [49]. Therefore, a current clinical trial for HLA-A2+ children and young adults with newly diagnosed DIPG (NCT02960230) targeted the H3.3K27M mutation that is thought to be present in all subclones, as it is an early event in tumorigenesis [55]. In preclinical experiments, stimulation of HLA-A2+ CD8+ T cells with a synthetic peptide encompassing the H3.3K27M mutation effectively suppressed the progression of glioma xenografts in mice [56].
Since DCs can induce an antigen specific T-cell response by presenting antigens to T-cells, DCs present a promising target for immunotherapy. Isolated DCs can be loaded with various antigenic agents, such as tumor lysate, peptides, proteins, RNA and DNA, which the DCs process and display as epitopes on their MHC class 1 and 2 molecules [57]. The loaded DCs can be administered to the patient as a vaccine to induce an antigen-specific T-cell response to tumor antigens (Fig. 5).
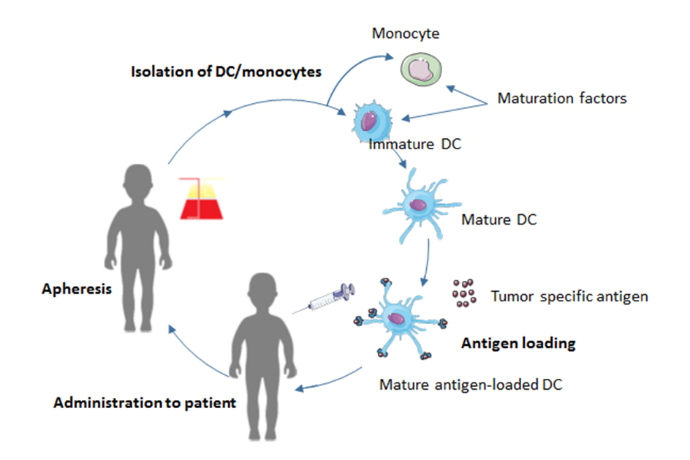 Figure 5.
Figure 5.The process of DC vaccination. Monocytes or autologous DCs are obtained from the patient’s blood by apheresis. They are differentiated into DCs and matured. Subsequently, the DCs are loaded with tumor antigens, such as tumor lysate, and injected back into the patient.
Several clinical trials on DC-based vaccine therapy have been conducted in brain tumors, but only a few focused on pediatric patients. One study assessed the feasibility of tumor-lysed DC vaccines in pediatric patients with newly diagnosed or recurrent HGG [58]. Three patients were eligible to receive vaccinations, of which two showed no evidence of disease after the treatment. However, these two were newly diagnosed glioma patients that underwent a gross total resection before the DC vaccination. The authors speculated that DC-based immunotherapy may offer most benefit in those patients, which is supported by another study in which total resection of HGG before vaccination was associated with a trend for prolonged OS and PFS [59]. However, gross total resection is a positive prognostic factor in itself, which may complicate the ability to determine the true effect of the DC vaccine. In the study of Ardon et al. [60], 45 children with various brain tumors received tumor-lysed DC vaccinations. Of the different brain tumors included, the therapy seems to be especially beneficial for HGG and ATRT. For those tumors, remarkable responses were observed in terms of OS and PFS. However, for some brain tumor subgroups sample size was very small, with only one or two patients included, which makes it difficult to draw conclusions about the clinical benefit in these brain tumor subtypes. In addition, children included in this study received different chemotherapeutic agents prior to, during, or after the vaccine treatment and were on different radiotherapy schedules. Thus, other therapy modalities may have had some effect as well. This may hamper the evaluation of the DC-based vaccination; alternatively, it has been suggested that immunotherapy in combination with chemotherapy or radiotherapy could potentiate antitumor activity [61, 62]. In another study, seven children with recurrent brain tumors received DCs pulsed with tumor RNA [63]. Despite clinical responses in three out of seven patients, no cell-mediated antitumor responses were detected in vitro. Moreover, patients appeared to be relatively immunocompromised before receiving the DC vaccines, as they had below-normal cellular proliferative responses compared to normal controls. An absence of T-cell tumor infiltration pre- and post-DC vaccination has been noted as well [58]. In the three clinical trials in children with pediatric brain tumors discussed, only mild adverse events were present as a consequence of the DC vaccinations. DC-based vaccine therapy was shown to be feasible, well-tolerated and safe. Whether DC-based vaccines are clinically effective in the pediatric population is difficult to interpret due to the small and heterogeneous sample sizes.
Clinical trials regarding DC-based vaccinations in adults are somewhat more sophisticated. The efficacy of a glioblastoma DC vaccine was evaluated in a study by comparing various functional immune parameters between a control group, receiving surgery and radio-chemotherapy post-surgery and a treatment group, who in addition, also received the DC vaccine [64]. Tumor control rate, survival rate and time to relapse, were significantly increased in the treatment group compared to the control group nine months after the vaccination. The same holds for Karnofsky performance status (KPS). The KPS scale assesses functional status of a patient and the significantly higher score in the treatment group reflects an improvement in the quality of life for patients that received the DC vaccination. Furthermore, CD3+, CD3+/CD4+, CD4+/CD8+, NK cells, interleukin 2 (IL-2), interleukin 12 (IL-12) and interferon gamma (IFN-γ) were significantly higher in the treatment group compared to the control group, indicating that DC vaccinations were associated with an improved antitumor immune response. In a study with a similar design, one-, two-, three-year survival rates, median OS and median PFS were significantly higher in the treatment group that received the DC vaccination compared to the control group [65]. Recently, the first results of a large phase three clinical trial evaluating the addition of tumor lysate-pulsed DC vaccines to conventional therapy for newly diagnosed glioblastoma were published [66]. After surgery and chemoradiotherapy, patients were randomized to receive either the DC vaccine or a placebo. Kaplan-Meier (KM) curves were generated to yield estimates of median survival times. Based on these curves, approximately 30% of the patients treated with the DC vaccine showed extended survival with a KM derived median OS estimate of 40.5 months, which could not be explained by known prognostic favorable factors. Although follow-up is still ongoing, it has been suggested that the patients live longer than expected.
Despite the more extensive research into the immunologic factors accompanying DC vaccine efficacy in the non-pediatric glioblastoma population, immune response monitoring has not yielded a clear association with clinical efficacy. Ardon et al. [59] did not find any positive correlation between immune reactivity and clinical outcome. In five of the eight patients, an increase in IFN-γ producing tumor antigen reacting T-cells between the first and the fourth DC vaccination was observed, which is assumed to reflect an induction in the tumor antigen directed immune response. The discordance between clinical and immunological responses is illustrated by the fact that the two patients with the longest survival did not show an increased IFN-γ secretion. Decreased regulatory T-cell populations and decreased expression of the negative costimulatory cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) on peripheral T-cells were found to be associated with increased survival in glioblastoma patients receiving DC vaccinations [67]. Various microenvironmental related factors have been proposed to be associated with clinical success of DC vaccine therapy [68, 69]. High PD-1+ tumor infiltrating lymphocyte counts were found to be negatively associated with OS and PFS in a glioblastoma patient group receiving DC vaccinations [61]. This is not surprising, given the immunosuppressive nature of PD-1, but it is even more supported by the observation that PD-1 and PD-L1 can mediate adaptive immune resistance in response to DC vaccine therapy [70]. DC vaccination was associated with upregulation of PD-1 expression in vivo. Treatment with both DC vaccination and PD-1 blockers drastically improved survival in glioma-bearing mice, whereas neither agents alone improved survival. In turn, this was associated with a second finding of the study of Jan et al. [61], in which a lower PD-1+/CD8+ ratio was also shown to constitute a prognostic factor for improved OS and PFS. This lower ratio is interpreted as being similar to PD-1 blockade, reflecting a decreased immunosuppression, and improved efficacy of the DC vaccination. These findings suggest that a combinational therapy of DC vaccinations and PD-1 blockade may improve clinical outcomes, especially in tumors that recruit immunosuppressive agents.
The immune factors underlying DC vaccine efficacy remain to be elucidated in further research. DC vaccine therapy may not work for every tumor, but instead may depend on tumor subtypes or TME factors. For optimization of the DC vaccine therapy, identification of biomarkers of antitumor immune responses elicited by the therapy are needed. Biomarkers can offer more insights regarding the added value of this therapy in the clinical setting. The promising results in clinical trials in adult glioblastoma encourages clinical trials, preferably randomized, to be set up in the pediatric population, since many different types of brain tumors exist in children for which extrapolation of findings in glioblastoma may not be possible.
Oncolytic virotherapy is another immunotherapeutic approach that utilizes viruses to selectively target and eradicate cancer cells, while leaving normal tissues unharmed. After intratumoral or intravenous administration, the virus enters tumor cells where it replicates and causes destruction via cell lysis (Fig. 6). In addition, oncolytic viruses (OVs) can generate an antitumor immune response, as a consequence of enhanced immunogenicity of the TME and activation of innate and adaptive immune responses [71].
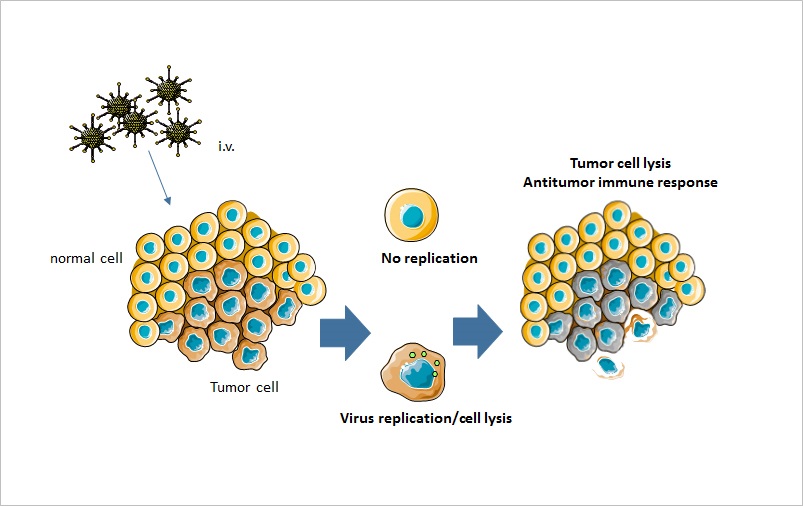 Figure 6.
Figure 6.Mechanism of action of oncolytic virotherapy. Oncolytic viruses can be administered intravenously or intratumorally. The virus only replicates in tumor cells, where it causes tumor cell lysis. In addition, oncolytic viruses lead to an increased antitumor immune response.
Measles virus showed efficacy in orthotopic xenograft models of medulloblastoma [72] and ATRT [73]. Patient derived cell lines exhibited significant cell killing and viral replication after infection with the measles virus. In an orthotopic xenograft model, survival was significantly increased in animals treated with the active virus compared to animals treated with the inactivated virus. Herpes simplex virus 1 (rRp450) demonstrated efficacy in orthotopic xenograft models of medulloblastoma and ATRT as well [74]. Apart from the survival benefit observed for mice that received the herpes virus, addition of the chemotherapeutic agent cyclophosphamide was shown to enhance rRp450-mediated cytotoxicity and survival. The animals’ median survival was increased compared to animals treated with rRp450 or cyclophosphamide only. Fifty percent of the animals that received the combination therapy, were tumor free at the end of the study, whereas the animals in the individual treatment groups all showed recurrence of disease, an impressive result that highlights the potential of this combination therapy.
More recently, the possible use of neuro-attenuated recombinant poliovirus, (PVSRIPO) was evaluated. Tumor specimens were analyzed for polio virus receptor positivity (CD155) by immunohistochemistry (IHC). The 12 samples contained medulloblastoma, ATRT, anaplastic ependymoma and (low grade) xanthoastrocytoma (PXA). All revealed a robust expression of CD155. As a proof of concept, PXA and medulloblastoma cell lines were exposed to PVSRIPO. The virus was able to efficiently infect and propagate in both entities and a significantly diminished tumor cell growth inhibition in the different tumor cell lines was observed [75].
The antitumor effect of OVs has also been explored in preclinical studies of pediatric malignant gliomas. The oncolytic herpes simplex virus 1716 (HSV1716) inhibited the invasion and migration of pediatric HGG and DMG cells in vitro [76]. Accordingly, the HSV1716 might have a role as anti-invasive therapeutic, which is promising considering the invasive phenotype that malignant gliomas display [77]. The Seneca Valley virus (SVV-001) was shown to be able to traverse the blood-brain barrier (BBB) after systemic administration, hence causing cell killing in vitro, and infecting glioma cells without harming normal cells and significantly prolong survival in orthotopic xenografts of pediatric malignant gliomas [78]. Furthermore, the binding of α2,3- and α2,6-linked sialic acids were identified to mediate SVV-001 infectivity and are suggested to be a diagnostic marker that can help discern between tumors permissive to SVV-001 killing and tumors resistant to SVV-001 killing.
These results provide strong indications for the efficacy of OVs in pediatric brain tumors, but clinical testing of oncolytic virotherapy in this patient group is still in its infancy. Clinical trials are ongoing and results are yet to be published. A case series included three pediatric patients with malignant HGGs that received the Newcastle disease virus [79]. At the time of publication, the patients were still receiving the virus as maintenance therapy, already showed survival ranging from seven till nine years and had a good quality of life. Various OVs have demonstrated safety and antitumor activity in clinical trials of adult malignant HGGs [80, 81]. In a dose-determining and toxicity study evaluating intratumoral delivery of a recombinant poliovirus in patients with recurrent glioblastoma, 19% of the patients exhibited virus-related adverse events [82]. Nevertheless, the authors were able to determine a safe dose. Additionally, patients treated with the poliovirus were shown to have a survival benefit compared to historical controls. A case report of a phase I study with oncolytic adenovirus Delta-24-RGD (DNX24-01) for newly diagnosed DIPG patients (NCT03178032) [83] reported safe delivery of the virus by intratumoral administration, and no virus toxicity during the first 4 weeks after infusion in one patient [84], but the final results remain to be published. Administration of this virus was found safe and therapeutically effective, however, in a subset of adult patients with recurrent glioblastoma [85], and preclinical data of DNX2401 showed a robust antitumor effect in pediatric HGG and DIPG models [86], which was even enhanced by subsequent radiotherapy [87]. As such, the results of the NCT0317803 trial are eagerly awaited.
The major hurdle for successful use of oncolytic virotherapy represents the ability of a virus to induce an antiviral immune response, which may consequently inhibit an antitumor response through the development of neutralizing antibodies and/or the induction of cytotoxic T-cells to mediated immune responses [88]. Therefore, the main challenge for oncolytic virotherapy is the development of strategies aimed at overcoming these obstacles. Various strategies are proposed to overcome these hurdles including the hiding of the virus inside carrier cells (such as mesenchymal stem cells), utilization of varying virus serotypes in order to enable serotype switching to evade antibody neutralization, PEGylation of the viral coat, polymer coating, and modification of the virus by bispecific fusion proteins or antibodies [71, 88].
Oncolytic virotherapy might show optimal efficacy when combined with other therapies. OVs can upregulate PD-1 and PD-L1 expression, and addition of PD-1/PD-L1 axis blocking agents to the OV was associated with improved survival in mice [89]. Likewise, the antitumor response of oncolytic virotherapy could be enhanced when the virus was combined with an immunosuppressive agent such as cyclophosphamide, since the immunosuppressive agent might avoid clearance of the virus and prolong persistence of the virus in the TME [90].
Taken together, oncolytic virotherapy represents a promising immunotherapeutic modality because it can specifically target tumor cells and is able to induce an antitumor response by stimulating the innate and adaptive immune systems. No severe adverse events seem to be associated with oncolytic virotherapy. Indications for synergistic interactions between oncolytic virotherapy and other therapies exist, and these deserve further investigation to optimize the antitumor response.
Pediatric brain tumors are the main cause of cancer related death in children and improvements in survival over the last decades for these tumors have drastically lagged behind when compared to those observed for other pediatric cancers. This emphasizes the burning need for new therapies to be developed for this pediatric brain tumors. Accordingly, the current review took into consideration whether immunotherapy could be a potential new treatment modality for pediatric brain tumors. Many immunotherapeutic treatment modalities have shown efficacy in various malignancies. In contrast, little is known about the possibilities to use immunotherapy for the treatment of pediatric brain tumors.
Multiple relevant targets for immunotherapy have been identified in preclinical studies, which require testing in the clinical setting to elucidate their full potential. Results regarding safety, feasibility and efficacy in the first clinical studies are encouraging. Immunotherapy particularly shows promise due to its ability to specifically target tumor cells, while sparing normal tissue cells. The ability to target cancer cells that are slowly dividing or quiescent, such as cancer stem cells, and the ability to suppress the re-emergence of cancer as a consequence of immunological memory, represent other advantages of immunotherapy [91]. Overall, immunotherapy has been shown to be a potential treatment option for pediatric brain tumors.
Yet, various challenges need to be overcome in order to optimize immunotherapeutic applicability and efficacy in the clinical setting. Firstly, the immunosuppressive microenvironment of pediatric brain tumors can limit the effectiveness of immunotherapy and an increased understanding of the immunosuppressive microenvironment of pediatric brain tumors is warranted. This knowledge can aid in the development of therapies targeting this immunosuppressive microenvironment to enhance antitumor efficacy. Cancer cells interact with the extracellular matrix and stromal cells of the TME in order to stimulate tumor growth [92]. Various targets interfering with the recruitment of stromal cells into the TME, tumor-stromal interactions or specific pathways activated by the TME have been identified for adult tumors [93]. The TME of pediatric brain tumors is less studied than that of adult tumors. Targeting angiogenesis using anti-angiogenic agents might be a possible strategy for the targeting of the immunosuppressive TME in pediatric brain tumors, since angiogenesis contributes to tumor growth. However, thus far, anti-angiogenic agents targeting vascular endothelial growth factor (VEGF) have shown minimal efficacy in pediatric EPN, malignant glioma and DMG in terms of increased survival [94, 95]. In pediatric LGGs, short-term disease control was observed until cessation of the treatment [96]. Other targets associated with the immunosuppressive TME may be worth investigating in pediatric brain tumors, such as the regulatory T-cells that were shown to be critical in the suppression of the anti-glioma response [97].
The chronic inflammatory and immunosuppressive TME induces T-cell exhaustion as a result of prolonged antigen stimulation, characterized as a poor effector function, expression of co-inhibitory receptors and reduced production of immunostimulatory cytokines [98]. Breaking this self-sustaining cycle of immunosuppression and impaired cytotoxic T-cell function collaborating in repressed immune responses is critical for enhancing antitumor efficacy. Combination therapies, aimed at activating an immune response or enhancing effector T-cell function in conjunction with immunomodulatory agents, might be a powerful solution for this and might increase antitumor responses. ICIs have shown limited efficacy in pediatric brain tumors and are unlikely to be impactful by themselves, but can be used in combination with other therapies. Activation-induced cell death of GD2-specific CAR T-cells was observed after repeated antigen stimulation; however, addition of PD-1 blockade could enhance GD2-directed CAR T-cell survival and promoted killing of PD-L1+ tumor cells [99]. In addition, neoadjuvant PD-1 blockade was found to confer a survival benefit in patients with recurrent glioblastoma [100]. Synergistic interactions between therapies should be unraveled in future research, in order to optimally utilize the full potential of immunotherapy in pediatric brain tumors.
A key question in the development of immunotherapy for pediatric brain tumors is how to reliably monitor antitumor response. Immune monitoring strategies have not yielded clear associations with clinical efficacy. Immunotherapy might not work for every patient. Treatment response may depend on the TME or the tumor subtype. Biomarkers for treatment response should be identified, that can distinguish between patients that will benefit from the therapy from those that won’t benefit from the therapy. The identification of biomarkers will guide immunotherapy towards a therapy of precision medicine, in which individual treatment regimens are tuned to tumor characteristics.
Future research should consider the long-lasting effects of inflammation on the developing brain. So far, this has been an understudied area, as research about immunotherapeutic feasibility and efficacy gained priority. The nervous system is particularly vulnerable to structural and functional damage in response to systemic inflammation [101]. Peripheral immune cells and inflammatory molecules can cross the BBB and exert cytotoxic effects. The resulting cognitive and behavioral changes can become permanent during persistent systemic inflammation [101]. Given the remarkable advances in the immunotherapeutic field, increased attention to the long-term effects of inflammation on the brain is warranted.
Understanding the role of the BBB drug delivery of immunotherapeutics is relevant when applying these agents to pediatric brain tumors as well. The BBB might be a hurdle for effective immunotherapeutic drug delivery, due to its selective permeability. However, immunotherapeutic agents might cross the BBB because the BBB is frequently disrupted in tumors of the CNS [102]. The degree of BBB permeability can vary between brain tumors. The wingless (WNT)-medulloblastoma subtype was shown to lack a functional BBB, whereas the sonic hedgehog (SHH)-medulloblastoma subtype was found to contain an intact BBB [103]. Wnt-inhibitors partially disrupted the BBB in mouse Shh-medulloblastomas, indicating that this inhibitor might be a target to enhance drug delivery across the BBB. Identifying BBB integrity in a given brain tumor subtype might provide insights towards increasing immunotherapeutic efficacy by improving delivery of immunotherapeutic agents across the BBB. At the same time, inflammation as a result of increasing BBB permeability should be limited in order to prevent neurotoxicity.
In conclusion, immunotherapy is a promising new treatment modality for pediatric brain tumors. Immunotherapy could significantly improve survival of children with brain tumors, although it has yet to be defined whether immunotherapy can lead to long-term remissions. Despite the remarkable advances in immunotherapy over the last decades, much work lies ahead in order to overcome obstacles and optimize treatment for pediatric brain tumors. The immunosuppressive TME represents the major hurdle in immunotherapy, and strategies for targeting the immunosuppressive TME are needed to increase immunotherapeutic efficacy. Randomized controlled trials of immunotherapeutic approaches in pediatric brain tumors are warranted. Clinical trials in pediatric brain tumors are still few for some immunotherapeutic modalities, and randomized controlled trials could provide insights about the true clinical benefit. Maximal antitumor efficacy could be achieved when different immunotherapeutic strategies are combined, or when immunotherapy is combined with conventional treatment modalities. These synergistic effects from combination therapies warrant investigation in future research.
APC: antigen-presenting cell; ATRT: atypical teratoid rhabdoid tumor; BBB: blood brain barrier; bMMRD: baillelic mismatch repair deficiency; CAR-T: chimeric antigen receptor T; CD47: cluster of differentiation 47; CNS: central nervous system; CTLA-4: cytotoxic T-lymphocyte-associated antigen 4; DC: dendritic cell; DMG: diffuse midline glioma; DNX24-01: adenovirus Delta-24-RGD; EGFR: epidermal growth factor receptor; EGFRvIII: epidermal growth factor receptor variant III; EphA2: ephrin type-A receptor 2; EPN: ependymoma; FasL: Fas ligand; GAA: glioma-associated antigens; GD2: disialogangliosid; H3K27M: Histone 3-Lysine-27-Methionine; HER2: human epidermal growth factor receptor 2; HGG: high-grade glioma; HSV1716: herpes simplex virus 1716; ICI: immune checkpoint inhibitor; IFN-γ: interferon gamma; IHC: immunohistochemistry; IL-2: interleukin 2; IL-10: interleukin 10; IL-12: interleukin 12; IL-13Rα2: interleukin-13 receptor alpha 2; KM: Kaplan-Meier; KPS: Karnofsky performance status; LGG: low-grade glioma; MHC: major histocompatibility complex; MRI: magnetic resonance imaging; NK cell: natural killer cell; OS: overall survival; OV: oncolytic virus; PA: pilocytic astrocytoma; PD-1: programmed cell death protein 1; PDGFRA: platelet-derived growth factor receptor A; PD-L1: programmed death ligand 1; PFS: progression free survival; PNET: primitive neuroectodermal tumor; PXA: pleomorphic xantoastrocytoma; SHH: sonic hedgehog; SIRPα: signal-regulatory protein alpha; SVV-001: Seneca Valley virus; TGF-β: transforming growth factor beta; TIM-3: T-cell immunoglobulin and mucin domain 3; TME: tumor microenvironment; VEGF: vascular endothelial growth factor.
EK wrote the first draft of the manuscript, while JLU and EH revised the manuscript. All authors contributed to the conception and design of the paper. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
EHU is funded by the Children Cancer-free foundation (KiKa).
The authors declare no competing interests.