

 , Cecchin Erika 1
, Cecchin Erika 11 Experimental and Clinical Pharmacology Unit, Centro di Riferimento Oncologico di Aviano (IRCCS), via F. Gallini 2, 33081 Aviano (PN), Italy
2 Cancer Epidemiology Unit, Centro di Riferimento Oncologico di Aviano (IRCCS), via F. Gallini 2, 33081 Aviano (PN), Italy
3 Radiation Oncology Unit, Centro di Riferimento Oncologico di Aviano (IRCCS), via F. Gallini 2, 33081 Aviano (PN), Italy
4 Department of MedicalOncology, Centro di Riferimento Oncologico di Aviano (IRCCS), via F. Gallini 2, 33081 Aviano (PN), Italy
5 Department of Oncology, Azienda Ospedaliera Santa Maria degli Angeli, 33170 Pordenone (PN), Italy
6 Medical Oncology Unit, San Filippo Neri Hospital, Piazza Di S. Maria Della Pietà, CAP Rome (RO), Italy
7 Medical Oncology Unit, Ospedale Cà Foncello, Piazzale Ospedale 1, 31100 Treviso (TV), Italy
8 Department of Experimental and Clinical Medicine, University of Florence, Piazza di San Marco 4, 50121 Florence (FI), Italy
9 Department of Health Sciences, University of Florence, Piazza di San Marco 4, 50121 Florence (FI), Italy
Abstract
Pre-treatment DPYD genotyping of a panel of 4-single nucleotide polymorphism (SNP) including DPYD*2A, DPYD*13, c.2846A$>$T and c.1236A$>$G-HapB3, has been strongly recommended by the current pharmacogenetics guidelines in order to avoid severe fluoropyrimidine (FL)-related toxicity. However, translation to clinical practice is still lagging behind. This requires a better definition of the relationship between genetic variants of DPYD and dose-limiting toxicities (DLTs) and development of new methods to investigate the effect of DPYD variants, such as the DPYD activity score. The aim of the current study was to support the clinical implementation of DPYD genetic testing, by assessing the relationship between DPYD variants in the 4-SNP panel and the risk to develop DLTs and by stratifying patients according to the DPYD gene activity score (GAS) model. (GAS $=$ 1.0 if carriers of one DPYD*2A or DPYD*13 alleles and GAS $=$ 1.5, if carriers of one c.2846A$>$T or c.1236G$>$A-HapB3 allele. Non-carriers GAS $=$ 2.0). A retrospective population of 763 colorectal cancer patients treated with FL-based chemotherapy, was selected and genotyped$.$ Patients carrying at least one decreased function DPYD variant in the 4-SNP panel, displayed a significant association with the risk of developing DLT ($i.e.$ grade $\geq $ 3 non-hematological toxicity or grade $\geq $ 4 hematological toxicity) either within the first three cycles of chemotherapy (OR$=$ 2.7, 95% CI $=$ 1.33-5.41) or during the entire course of treatment (OR $=$ 2.7, 95% CI $=$ 1.42-5.04). Patients' GAS was found to better define the risk of DLT for both acute (GAS $=$ 1.5, OR $=$ 1.80, 95% CI $=$ 0.78-4.15 and GAS $=$ 1.0, OR $=$ 10.12, 95% CI $=$ 2.55-40.20) and total toxicity (GAS $=$ 1.5, OR $=$ 2.08, 95% CI $=$ 1.02-4.27 and GAS $=$ 1, OR $=$ 7.09, 95% CI $=$ 1.69-29.65). In conclusion, the present study demonstrates that the DPYD 4-SNP panel and the associated GAS can predict the occurrence of DLT related to treatment with FL. These findings further support the implementation of pre-emptive DPYD genotyping in the routine clinical practice.
Keywords
- Fluoropyrimidine
- Dihydropyrimidine dehydrogenase
- Drug-related toxicity
- Chemotherapy
- Pharmacogenetics
- Colorectal cancer
Abbreviations
5-FU: 5-Fluorouracil
CI: Confidence Interval
CPIC: Clinical Pharmaceutical Implementation Consortium
DLT: Dose Limiting Toxicity
CTCAE: Common Terminology Criteria for Adverse Events
DPD: Dihydropyrimidine Dehydrogenase, enzyme
DPWG: Royal Dutch Association for the Advancement of Pharmacy
DPYD: Dihydropyrimidine Dehydrogenase, gene
ESMO: European Society for Medical Oncology
FDA: Food and Drug Administration
FL: Fluoropyrimidine
NCI-CTC: National Cancer Institute -- Common Toxicity Criteria
OR: Odds Ratio
SNP: Single Nucleotide Polymorphism
TYMS: Thymidylate Synthase
Since the beginning of 1950, 5-fluorouracil (5-FU) represents one of the most commonly prescribed anticancer drugs for the treatment of various solid tumors. Belonging to the antimetabolite fluoropyrimidine (FL) class, 5-FU and its oral prodrug capecitabine (Xeloda) inhibit pyrimidine biosynthesis in cells with high rates of proliferation by acting as a false high-affinity substrate for thymidylate synthase (TYMS). Both the biosynthetic depletion of endogenous thymidine and the direct damage to DNA and RNA, result in a potent cytotoxic activity of 5-FU metabolites, leading to cell death and tumor growth suppression. Although the pharmacological efficacy of FLs is well-established, the narrow therapeutic index of the drug represents one of the main issues in the management of chemotherapy. In fact, despite the fact that the vast majority of patients can be safely treated with FLs, it is well established that a substantial fraction of these patients comprising 20 -- 30%, are likely to develop severe (grade $\geq $ 3) or even life-threatening toxicity during the course of chemotherapy, thus producing treatment delays and discomfort for the patients[1].
Since one of the leading causes of FL-related toxicity is detectable in an inefficient catabolism of the drug, that is mainly mediated by the dihydropyrimidine dehydrogenase (DPD) enzyme, many efforts have been made in order to better characterize these genetic variants potentially impairing DPD activity[2,3,4,5,6,7]. Currently, although more than 160 single nucleotide polymorphisms (SNPs) have been identified in the DPYD gene encoding for the enzyme DPD, only four of them are classified as clinically relevant and listed among the international pharmacogenetics guidelines for drug dose recommendations, such as those provided by the Clinical Pharmacogenetics Implementation Consortium (CPIC)[8] and the Royal Dutch Association for the Advancement of Pharmacy (DPWG)[9].
Dysfunctional variants, known for their role in impairing DPD activity comprise: DPYD*2A (rs3918290) and DPYD*13 (rs55886062) associated with an almost complete protein deficiency in homozygotes[10] whereas c.2846A$>$T (rs67376798) and c.1236G$>$A-HapB3 (rs56038477) are associated with a moderate loss of protein function[7,8,9,10,11]. Recently, CPIC and DPWG regulatory organizations have moved toward the harmonization of drug dosing adjustments according to genotype by referring to the GAS model originally described by Henricks et al., in 2015 that is aimed to translate the results of the genetic test into the most likely phenotypic outcome[12]. Briefly, DPYD alleles are classified as completely non-functional (GAS $=$ 0), intermediate functional (GAS $=$ 0.5) or normally functional (GAS $=$ 1.0), according to the observed enzyme activity. By summing single alleles score, individual patient estimation of the protein functionality status is obtained, hence allowing to tailor the starting 5-FU dose based on the patients' genotype. The association between DPYD variants DPYD*2A, DPYD*13, c.1679 T$>$G and c.1236G$>$A-HapB3 and the increased risk of severe toxicity is widely supported by numerous retrospective studies, thus providing increasing support for the translation of pre-emptive DPYD genotyping in clinical practice[13,14]. However, a pre-emptive pharmacogenetic testing for the above mentioned DPYD variants is not reported in the drug label yet. Moreover, while the administration of 5-FU and capecitabine in colorectal cancer patients with low or absent DPD activity is contraindicated by FDA, no dose adjustment is recommended for the intermediate metabolizers, leaving the decision up to the clinicians. Moreover, the recent publication of ESMO guidelines for colorectal cancer management did not include any recommendation for pre-emptive DPYD genotyping rekindling the debate about the validity and utility of this test in the clinical practice[15].
Although several studies have previously addressed the effect of genetic DPYD variants on toxicity occurrence[16,17,18], to the best of our knowledge, toxicity is rarely looked at, from the point of view of its effect on the patients' quality of life. In fact, although a toxicity level graded as equal to or higher than NCI-Common Toxicity Criteria grade 3 is commonly considered severe in the pharmacogenetic association studies, in the clinical practice, grade 3 hematological or non-hematological toxicities have different clinical consequences as well as they have a different impact on patients' quality of life. A grade 3 neutropenia is hardly noticed by the patients and did not usually drive therapeutic decisions as treatment interruption or dose modification, thus not representing a DLT. On the other hand, a grade 3 gastrointestinal or other non-hematological toxicities, are more likely to lead to therapy discontinuation and serious daily disadvantages. For this reason, a more complete definition of the clinical impact of DPYD variants and a better definition of the functional relevance of the SNPs should be mandatory.
Hence, the aim of the current study was to evaluate the relationship between DPYD pharmacogenetics variants that belong to the 4-SNP panel (DPYD*2A, DPYD*13, c.2846A$>$T and c.1236A$>$G-HapB3) and the onset of DLT. To address this point, patients were stratified according to the DPYD activity score model.
Between 1999 and 2015 a systematic collection of clinical data and biological samples (blood) of consenting patients receiving FL-based chemotherapy, was carried out within the Clinical and Experimental Pharmacology Unit of Centro di Riferimento Oncologico (CRO), Aviano (PN).
Patients were selected from a database of 1,122 clinical cases based on the following inclusion criteria: (i) diagnosis of colorectal cancer, (ii) peripheral blood samples availability, (iii) signed written informed consent approved by the local Ethical Committee, (iv) assumption of FL-based chemotherapy, and (v) detailed clinical data availability.
Patients' medical records were examined to collect the following clinical information: (i) baseline patient assessment, (ii) chemotherapy information, (iii) toxicity data related to each of the chemotherapy cycles (classified according to NCI- Common Toxicity Criteria version 3).
Adverse events that occurred during chemotherapy were classified according to the Common Terminology Criteria for Adverse Events (CTCAE) v.3.0. (https://ctep.cancer.gov/protocolDevelopm-ent/electronic_applications/docs/ctcaev3.pdf). For our purposes, only DLT, defined as grade $\geq$ 3 non-hematological toxicities and grade $\geq$ 4 hematological toxicities were considered.
For the total toxicity estimation, the maximum grade developed during whole chemotherapy was evaluated, while only adverse events reported within the first three cycles of chemotherapy were considered for the assessment of acute toxicity.
The study was performed in accordance with the Declaration of Helsinki and was approved by the Local Ethical Committee. All recruited patients provided informed consent for data and blood collection.
Genomic DNA was extracted from peripheral blood collected in EDTA containing tubes using the EZ1 DNA Blood 200 $\mu $L kit (Qiagen) and the BioRobot EZ1 (Qiagen). DNA samples were stored at $+$ 4$^{\circ}$C.
DPYD*2A and DPYD*13 alleles were examined through pyrosequencing analysis on PSQ96MA system (Qiagen) according to the manufacturer's instructions.
DPYD c.2846A$>$T and c.1236G$>$A-HapB3 were analyzed by pre-designed TaqMan SNP genotyping assays with the Applera TaqMan Universal Master Mix used on ABI 7900HT (AB Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. Previously genotyped samples were used as positive and negative controls in each analysis. The results were validated using Sanger sequencing in ABI PRISM 3130xl Genetic Analyzer (Applied Biosystem) and the results were analyzed with Chromas Lite software v. 2.01. Primer sequences and reaction conditions are available under request.
The association between the DPYD genotype and the risk to develop DLT within the first three cycles or during the entire course of FL-based chemotherapy was evaluated. Patients were grouped according to the presence of at least one variant allele in the 4-SNP panel (DPYD*2A, DPYD*13, c.2846A$>$T or c.1236G$>$A-HapB3) and defined accordingly as `carriers' or `non-carriers'. Carriers of one DPYD*2A or DPYD*13 alleles were assigned a GAS of 1.0, carriers of one c.2846A$>$T or c.1236G$>$A-HapB3 were assigned a 1.5, whereas non-carriers were assigned a GAS of 2.0[12].
Genotype frequencies were compared with those reported for Caucasian population in dbSNP (https://www.ncbi.nlm.nih.gov/SNP/) and tested for deviation from Hardy-Weinberg equilibrium[19].
The association between DPYD variant patient status and toxicity occurrence was estimated through an unconditional logistic regression model, adjusted for gender, age, chemotherapy scheme and radiotherapy exposure. Odds ratio (OR) and 95% confidence interval (CI) were calculated. Statistical significance was set at $p <$ 0.05 (two-sided).
A final set of 763 patients were selected for the current study. Patient details concerning age, gender, chemotherapy regimen and radiotherapy exposure are listed in Table 1. Genetic results for DPYD polymorphisms are also depicted (Table 1). Concerning pathological characteristics of the population, 423/763 (55.4%) patients had colon as primary tumor site, whereas 340/763 (44.5%) had rectal cancer. With respect to treatment setting, 196/763 (25.7%) patients were treated in neo-adjuvant setting, 282/763 (37.0%) in adjuvant setting, whereas 279/763 (36.6%) were treated on the first line or 6/763 (0.8%) on the second line for a metastatic disease.
| Characteristic | N | % |
|---|---|---|
| Sex |
479 | 62.8 |
| Female | 284 | 37.2 |
| Age, years |
61 (20-85) | |
| FL- association therapy Monotherapy | 229 | 30.0 |
| Oxaliplatin | 265 | 34.7 |
| Irinotecan | 269 | 35.3 |
| Radiotherapy |
197 | 25.8 |
| No | 566 | 74.2 |
| DPYD genotype |
9 | 1.2 |
| *13 | 0 | 0.0 |
| c.2846 A > T | 5 | 0.7 |
| c.1236G > A | 31 | 4.1 |
Abbreviations: FL, Fluoropyrimidines.
All 763 enrolled patients were successfully genotyped for the selected DPYD variants. All polymorphisms were detected in heterozygosity and no compound heterozygosity was detected. Forty-five patients (5.9%) carried at least one variant allele for any DPYD polymorphism, whereas the remaining 718 (94.1%) resulted wild-type for all the four variants. In particular, 9 patients (1.2%) carried the DPYD*2A allele, 5 patients (0.7%) carried the c.2846A$>$T allele and 31 patients (4.0%) carried the c.1236G$>$A-HapB3 allele. No patient carried any polymorphic allele for the DPYD*13 variant. The variants followed the Hardy-Weinberg equilibrium, and the detected allele frequencies were consistent with those reported in previous studies[11,20].
By classifying the patients according to the GAS model, 9 patients (1.2%), all of which harbored the DPYD*2A allele, were associated to a DPYD activity score of 1.0, whereas the remaining 36 DPYD mutated patients (4.71%) were associated to a 1.5. The 718 non-carrier patients (94.1%) were considered as full-metabolizers, so the related GAS was 2.0.
When looking at the toxicity experienced by the patients, 178/763 (23.3%) developed at least one kind of DLT during the entire course of chemotherapy, whereas 585/763 (76.7%) did not. In particular, among the first group, 38/178 patients (21.3%) developed grade 4 hematological toxicity as the most severe toxicity, 154/178 (86.5%) grade 3 or 4 non-hematological, and 14/178 (7.9%) both.
Considering the early onset toxicities, 655/763 patients (85.8%) did not develop any DLT within the first three cycles of chemotherapy, and 108/763 (14.2%) did. Among this group, 22/108 patients (20.4%) developed hematological toxicities, 94/108 (87.0%) non-hematological and 8/108 (7.4%) both (Table 2). Among the 178 patients experiencing DLT during the entire course of treatment, the most common toxic events were hand-foot syndrome, neurological disorders, neutropenia, leukopenia and gastrointestinal toxicities, mainly represented by diarrhea and vomiting.
| Acute1 | Total2 | |||
|---|---|---|---|---|
| DLT** | N | % | N | % |
| All | ||||
| No | 655 | 85.8 | 585 | 76.7 |
| Yes | 108 | 14.2 | 178 | 23.3 |
| Haematological | ||||
| No | 741 | 97.1 | 725 | 95.0 |
| Yes | 22 | 2.9 | 38 | 5.0 |
| Non-haematological | ||||
| No | 669 | 87.7 | 609 | 79.8 |
| Yes | 94 | 12.3 | 154 | 20.2 |
** Hematological toxicity ≥grade 4 or non-hematological toxicity≥grade 3
1 DLT developed within the first three cycles of chemotherapy.
2 DLT developed during the entire course of treatment.
The onset of any kind of DLT resulted in a significant association with the presence of at least one DPYD variant within the analyzed 4-SNP panel. When considering both acute or total toxicity, carriers of at least one variant allele had about a 2.7-fold risk to develop at least one event of DLT (Table 3). Specifically, 28.9% and 44.4% of patients carrying at least one DPYD variant developed a DLT within the first 3 cycles (acute toxicity), or within the entire course of chemotherapy (overall toxicity), respectively. In WT DPYD patients, DLTs were observed in 13.2% patients within the first 3 cycles and in 22.0% of patients when considering the whole course of chemotherapy.
| N | % | Acute1 | Total2 | |||
|---|---|---|---|---|---|---|
| OR3 | 95% CI | OR3 | 95% CI | |||
| SNP panel | 45 | 5.9 | 2.69 | 1.33-5.41 | 2.67 | 1.42-5.04 |
| DPYD activity score | ||||||
| 2 | 718 | 94.1 | 14 | 14 | ||
| 1.5 | 36 | 4.7 | 1.80 | 0.78-4.15 | 2.08 | 1.02-4.27 |
| 1 | 9 | 1.2 | 10.12 | 2.55-40.2 | 7.09 | 1.69-29.65 |
| X2 for trend | P | = 0.0007 | P | = 0.0009 |
1DLT developed within the first three cycles of therapy were considered.
2DLT developed during the whole treatment course were considered.
3Estimated from unconditional logistic regression model including terms for gender, age, chemotherapy scheme and radiotherapy.
4Reference category.
4-SNPs Panel, DPYD*2A DPYD*13, c.2846A>T and c.1236G>A-HapB3.
GAS better defined the risk to develop DLTs than the single DPYD variant. The risk (OR) to develop acute toxicity was 1.80 (95% CI $=$ 0.78--4.15) and 10.12 (95% CI $=$ 2.55--40.20) in patients with GAS $=$ 1.5 and 1.0, respectively. For total toxicity, the risk was 2.08 (95% CI $=$ 1.02--4.27) and 7.09 (95% CI $=$ 1.69--29.65) in patients with GAS $=$ 1.5 and 1.0, respectively (Table 3). Among patients with a GAS of 1.0, the onset of DLT during the entire course of chemotherapy was reported in 6 out of 9 patients (66.7%), 5 of which (55.6%) developed toxicity within the first three cycles of treatment. Patients with a GAS of 1.5 showed DLT in 14 out 36 cases (38.9%) when considering the entire treatment course, 8 of which (22.2%) reported DLT within the first three cycles of chemotherapy. On the other hand, patients with a GAS of 2.0 (WT DPYD) presented DLT in 158 out 718 cases (22.0%) when considering the whole therapy and in 95 cases (13.2%) when considering the first three cycles of treatment. Fig. 1 shows the toxicity grades developed within the first three cycles of chemotherapy (A) and during the whole treatment (B) in relationship with the GAS. Toxicities were classified as mild (grade from 0 to 2), severe (grade 3) and very severe (grade 4). Patients with a DPYD activity score of 1.0 were more likely to develop grade 4 toxicity rather than other patients. Patients with a DPYD activity score of 1.5 showed an increased risk to develop grade 3 toxicity rather than WT DPYD patients (Fig. 1).
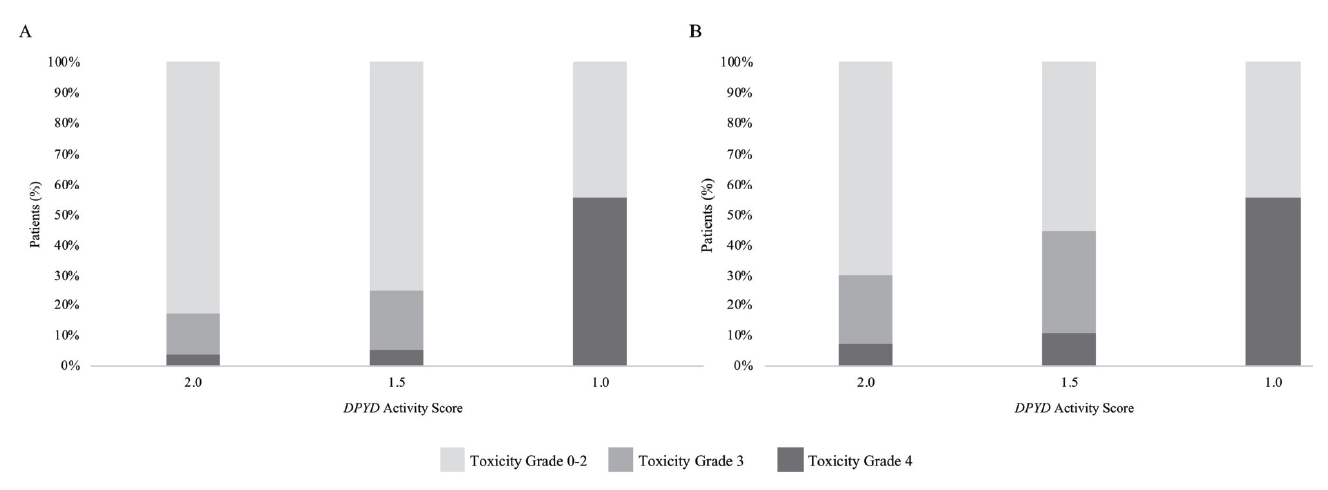 Fig. 1.
Fig. 1.Toxicity grade (by NCI CTC scale) distribution according to the DPYD activity score model within the first three cycles of chemotherapy (A) and during the whole treatment (B).
Although the pharmacogenetics guidelines for the management of FL-based chemotherapy toxicity in colorectal cancer agree upon the clinical validity of DPYD*2A DPYD*13, c.2846A$>$T and c.1236G$>$A-HapB3, a pre-emptive genotyping to avoid severe and unpredictable adverse drug reactions, translating such recommendations in the clinical practice remains a matter of open debate[21,22]. Prospective evidences of increased FL safety and efficacy when applying a DPYD*2A-guided dose reduction was reported, which strongly justify a drug label update limited to the well-known DPYD*2A variant[23,24]. $^{ }$However, a clinical validation of up-front genotyping considering the 4-SNP panel, including DPYD*2A, DPYD*13, c.2846A$>$T and c.1236G$>$A-HapB3 is still pending, warranting further investigation to support a genotype-guided dose adjustment for the entire set of variants.
Herein, we retrospectively evaluated the effect of DPYD variants on the risk to develop DLTs related to a FL-based chemotherapy, defined as grade $\geq $ 3 non-hematological or grade $\geq $ 4 hematological toxicity, in a large group of colorectal cancer patients from the clinical practice.
Traditionally, the pharmacogenetics studies published up to date on this gene-drug interaction, focused on the risk of developing NCI-CTC grade $\geq $ 3 hematological or non-hematological toxicity[25,26]. However, this approach could be poorly informative from a clinical point of view. To move a step forward from the laboratory diagnostics to the clinical setting, we decided to focus on the DLT. In the current clinical practice only the herein defined DLT usually guides the physician's treatment decisions including drug dose reduction or treatment interruption, with potential consequences on the anti-cancer efficacy. In fact, if a grade 3 hematological toxicity is hardly perceived by the patients, grade 3 non-hematological toxicity can hardly impact the patients' quality of life and usually requires medical intervention to manage the event that in some cases ($i. e$. cardiological grade 3 toxicity) can lead to patients' hospitalization.
Our findings showed that the presence of at least one decreased function DPYD variant in the 4-SNP panel, without any distinction among them, was significantly associated to a 2.7-fold increased risk of developing DLT, both acute and total.
A further step towards a better characterization of patients, who might benefit from a FL dose-adjustment according to DPYD genotype, was achieved by adopting the GAS model. According to the GAS, patients are stratified based on their catalytic enzyme activity[12]. According to the most updated CPIC guidelines, opting for this approach, patients bearing a GAS of 2.0 are classified as fully functional and can be treated safely with a standard FL dose. On the other hand, patients showing a GAS of 1.5 or 1.0 are classified as intermediate metabolizer and are recommended to be treated with a starting dose reduction of 25% or 50% of the standard dose, respectively. Whereas, patients with a GAS of 0.5 or 0 are classified as poor metabolizers and should avoid FL administration in order to prevent adverse drug reactions[8].
In the present study, the general trend observed for the 4-SNPs panel is maintained after patients' stratification according to the DPYD activity score. In fact, patients with a DPYD activity score of 1.0 showed an increased risk to develop DLT up to 10.1-fold when compared to full-metabolizers (GAS 2.0). On the other hand, consistent with a milder effect on the enzyme functionality, a weaker association was observed between 1.5 DPYD activity score carriers and DLT. These results are consistent with the clinical significance of the GAS model, highlighting the need to personalize the FL starting dose according to the residual DPYD activity.
Genotype-related toxicity tends to exacerbate earlier than a toxicity caused by a long period of drug exposure, putting the loss-of-function allele carriers at risk of early treatment interruption[5]. In support of this consideration, for 1.0 DPYD activity score carriers, the association between genotype and risk to develop DLT was stronger when considering the first three cycles of chemotherapy (OR $=$ 10.12, 95% CI $=$ 2.55--40.2) rather than the entire course of treatment (OR $=$ 7.09, 95% CI $=$ 1.69--29.65). On the other hand, patients with a DPYD activity score of 1.5 displayed an increased risk to experience DLT only when considering the entire course of treatment (OR $=$ 2.08, 95% CI $=$ 1.02--4.27), thus suggesting a minor involvement of DPYD genotype in FL related DLT and the need of a cumulative drug exposure for the phenotypic manifestations.
To better investigate the toxicity profile in relation to the GAS model, we evaluated the toxicity distribution among the three GAS groups by considering separately grade 3 and grade 4 toxicities, this time without distinguishing between hematological and non-hematological toxicity. Interestingly, a clear drop down of grade 3 toxicities in DPYD 1.0 activity score carriers in favor of a grade 4 enrichment was observed. The same trend concerning the grade 4 distribution was not maintained when considering the DPYD activity score 1.5 carriers, which showed an increased development of grade 3 toxicity. These results suggested that a lower DPYD activity score is strictly related to a more severe toxicity onset and, moreover, that the discrimination between mild toxicities and severe toxicities, intended as grade $\geq $ 3, could lead to loss of information concerning the safety profile of the drug.
Our results point out once again and in a wide cohort of more than 700 patients to the association between common DPYD genetic variants, for which the genetic test is recommended by the pharmacogenetics guidelines, and the risk of developing DLT, strictly related to drug discontinuation and the necessity of therapeutic management interventions. The implications of these results could be considered not only from a clinical but also from an economical point of view. The paucity of available data on the clinical utility of pre-emptive DPYD testing is among the major barriers for the test implementation. Our group previously demonstrated how patients genotype can discriminate between patients with differential toxicity management costs[27]. Concerning DPYD, current published data are limited to DPYD*2A[28]. In this regard, Deenen et al., demonstrated in a large prospective study that the genotype-guided dosing for the DPYD*2A variant carriers was not only safer but also cost-saving, hence paving the way to further economic evaluations in the pharmacogenetics field[29]. It is likely that a larger panel of DPYD variants, as that recommended by the pharmacogenetic guidelines, and adopted in the present study, could account for a greater fraction of toxic events, and consequently, of associated toxicity management costs.
Although the retrospective nature of this analysis actually limits its clinical value, the clear association between DPYD pharmacogenetic variants and the increasing risk to develop DLT was reported in a real-world setting, thus suggesting the necessity of prospective randomized clinical trials. It is important to stress that the pharmacogenetic guidelines do not represent a dogmatic statement. Instead, they aim at providing a scientific support to better tailor the starting drug dose, allowing the clinicians to increase the dose in well-responding patients, even in the presence of a risk-associated variant. With this in mind, it is highly sought that further prospective studies will substantiate the clinical validity of pharmacogenetic implementation in clinical practice and that pharmacoeconomic evaluations, which consider the DPYD 4-SNP panel, will provide insights in support of genetic test recommendation.
The authors declare no potential conflict of interest.
EC and GT conceived and designed the study; FS, ED, CDF, FE and RR performed the genotyping; JP performed statistical analysis of the data; CDF, JP, RR, and EC, critically reviewed the data; MG, LF, EP, AB, SN, EM, AF, MB, MD'A and ADP clinically addressed and recruited the patients to the study; CDF, MG, EDM, AB, ED and RR collected clinical data from clinical records; CDF and EC wrote the paper; GT contributed reagents, analysis tools, and administrative support; all authors read and approved the final manuscript.