
Bogdan.Wlodarczyk@bcm.edu
Richard.Finnell@bcm.edu
Folic acid supplementation has been shown to significantly reduce both the occurrence and the recurrence of neural tube defects (NTDs) in human populations, yet the underlying mechanisms for reducing the risk of NTDs continue to be debated. This study examined genetic background and select folate metabolites as possible modifiers that may influence NTD risk. Specifically, several folate cycle and methylation metabolites were examined for their ability to reduce the occurrence of NTDs in two congenic mouse strains carrying targeted disruption of the folate receptor 1 (Folr1) gene. SWV-Folr1$^{tm1Fnn}$ and LM/Bc-Folr1$^{tm1Fnn}$ mice were provided with folate or several folate pathway metabolites, or combinations thereof, to determine the ability of these compounds to rescue nullizygous embryos from lethality and NTDs. Results demonstrated that SWV-Folr1$^{tm1Fnn}$ and LM/Bc-Folr1$^{tm1Fnn}$ mice exhibit different dose responses to folinic acid (5-formyl-tetrahydrofolate) supplementation; however, treating dams throughout gestation with downstream metabolites indicated that only folates rescued Folr1 nullizygous embryos from lethality and NTDs. Chemical inhibitors of folate metabolism were used to further elucidate essential enzymatic and biochemical metabolites. These data demonstrate that the inhibition of S-adenosyl-L-homocysteine hydrolase (AHCY) or selective inhibition of folate responsive isoprenylcysteine carboxylmethyltransferase (ICMT) results in embryo toxicity and fetuses with anterior NTDs. These data indicate that genetic background modifies NTD penetrance in folate-supplemented Folr1$^{tm1Fnn}$ mutants, while downstream metabolites of folate metabolism are not capable of rescuing Folr1$^{tm1Fnn}$ mutants. Moreover, these findings support the hypothesis that the methylation cycle and post-translational methylation of key signaling proteins such as Ras, by ICMT are essential to neural tube closure.
Clinical trials conducted on women of childbearing age have demonstrated that folic acid supplementation can decrease the risk of neural tube defects (NTDs) by up to 85%[1,2] Furthermore, to reduce the population risk of NTDs, the World Health Organization and United States Public Health Service now recommend that all women of childbearing potential, consume 400 micrograms of folic acid[3,4,5]. There is consistent evidence demonstrating the benefits of folic acid supplementation; however, the underlying mechanisms for reducing the risk of NTDs require additional elucidation. Similarly, the primary genetic risks underlying the etiology of NTDs have not been determined, despite the efforts of scientists worldwide[6,7,8].
The embryonic period, week three through eight in human development, is the timeframe during which organogenesis occurs [9]. It is also the most susceptible period for induction of structural malformations by teratogens [10]. The most common and severe malformations of the central nervous system that can occur during this time are NTDs. These malformations involve failure of the neural tube to properly develop and close during days 18 to 28 of human embryogenesis [11]. Developmental biologists, toxicologists, and clinicians study neurulation in an effort to understand early embryonic morphogenesis and identify the etiology of NTDs.
The pathogenesis of NTDs has been difficult to study for several reasons. First, neurulation begins early during gestation, so it is difficult to observe it in a clinical setting since it is normally completed prior to recognition of human pregnancies. Secondly, the isolation of human embryonic samples for scientific studies is presently subject to increasing political pressure and ethical debates. Due to these difficulties and concerns, animal models primarily shape our understanding of early embryonic development and the dynamic processes of neurulation. The cellular and morphogenetic events of neurulation are well described, but a molecular understanding of these events is only beginning to emerge[12,13,14]. NTDs are proposed to involve a failure in cellular proliferation, alterations in convergent-extension, altered differentiation in the developing neuroectoderm, or negative changes in vascular function and development supporting these cells[8,15]. The causes of these alterations are thought to be multifactorial, with contributions from both genetic and environmental factors [16]. The mechanisms that induce the formation, elevation, and fusion of the neural folds remain largely unknown, and no single gene has been individually implicated as a risk factor for a significant portion of the population burden for NTDs. Despite these gaps in knowledge, folic acid (FA) supplementation has emerged as a major nutritional modifier of NTD risk.
Folate receptor 1 (Folr1) mutant mice have folate responsive NTDs [17], and folate-responsive gene expression profiles have been reported in the anterior portion of the developing Folr1 mouse neural tube [18]. Genetic variations in humans and genetic background in mice is reported to influence the development of NTDs[19,20,21]. Multiple studies have likewise indicated that the background strain of mice can influence the occurrence of genetically induced NTDs. These include studies demonstrating background-dependent chromosomal linkage in Cecr2 mutants [22] and gene: gene interactions with curly tail (Grhl3) mutants [23]. To test the hypothesis that folate responsive defects are also largely influenced by gene-gene and gene-nutrient interactions, mutant Folr1$+$/$-$ mice were crossed onto SWV-Fnn and LM/Bc-Fnn background mouse strains following a speed congenic approach [24]. The resultant data from supplementation studies on these backcrossed inbred strains is presented to test the hypothesis that genetic background influences the penetrance of these folate-responsive NTDs.
SFA is a biologically inactive water-soluble B vitamin that is known to contribute to three metabolic pathways when it is transformed to its bioactive forms: activated methyl cycle, folate cycle, and trans-sulfuration [25]. These pathways are compartmentalized in the cell and donate methyl groups via the activated methyl cycle for the methylation of nucleic acids, lipids, and numerous other proteins[26,27]. These reactions are catalyzed by methyltransferases (MT), which utilize S-adenosylmethionine (SAM) as a methyl donor (Fig. 1). The metabolic process that generates SAM occurs primarily via the methylation pathway, where the conversion of homocysteine (Hcy) to methionine requires the transfer of a methyl group from 5-methyl-tetrahydrofolate (5-CH$_{3}$-THF). Methionine can then be converted to SAM by methionine adenosyltransferase and ATP. SAM, the universal methyl donor is then converted to S-adenosylhomocysteine (SAH) after donating the methyl group to acceptor molecules [28]. The enzyme S-adenosylhomocysteine hydrolase (AHCY) hydrolyzes SAH to adenosine and L-homocysteine (Hcy). This allows the cycle to continue as Hcy is converted back into methionine by 5-CH$_{3}$-THF and B12-dependent 5-methyltetrahydrofolate-homocysteine S-methyltransferase (MTR).
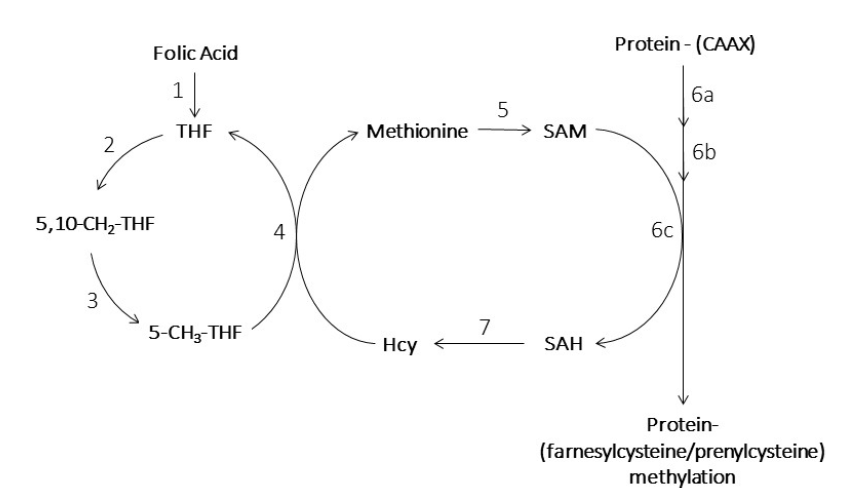 Fig. 1.
Fig. 1.Folate biochemistry and the methionine cycle.
Substrates: tetrahydrofolate (THF); 5, 10-methylene-tetrahydrofolic acid (5, 10-CH$_{2}$-THF); 5-methyl-tetrahydrofolate (5-CH$_{3}$-THF); S-adenosylmethionine (SAM); S-adenosylhomocysteine (SAH); homocysteine (Hcy)
Enzymes: 1. dihydrofolate reductase (DHFR); 2. serine hydroxymethyltransferases (SHMT1/SHMT2); 3. Methylene-tetrahydrofolate reductase (MTHFR); 4. methionine synthase (MS, MTR); 5. methionine adenosyltransferase (MAT); 6a. farnesyltransferase, 6b. CAAX prenyl protease 2 (Ras converting enzyme type 1, RCE1), 6c. isoprenylcysteine carboxyl methyltransferase (ICMT); 7. S-adenosylhomocysteine hydrolase (AHCY).
In the current study, small molecules were used to inhibit methytransferases globally (7, AHCY) or specifically (6c, ICMT). The methylation of CAAX proteins (e.g. Ras and Rho GTPases) utilizes 6a catalyzed lipidation (addition of prenylated lipids), 6b catalyzed endoproteolytic cleavage of the AAX amino acids, and 6c catalyzed carboxyl-methylation of the isoprenylcysteine.
Previous gene expression data in Folr1 mutant mice indicated folate-responsive differential expression of MT genes during neural tube closure, including isoprenylcysteine carboxylmethyltransferase (Icmt) [18]. ICMT is involved in post-translational methylation of isoprenylated C-terminal cysteines (consensus motif: CAAX, where: C $=$ cysteine, A $=$ aliphatic amino acid, and X $=$ undefined amino acid) [29]. Specifically, ICMT is localized in the endoplasmic reticulum and nuclear membrane, where it catalyzes the post-translational methylation of key proteins like Ras, Rho and other CAAX-containing proteins [30]. The CAAX pathway involves the linkage of an all-trans-farnesyl (C15) or an all-trans-geranylgeranyl (C20) moiety to the cysteine residue, followed by proteolysis that generates a carboxylated-terminal cysteine. Carboxylmethylation of the cysteine then completes this pathway. Genetic disruption of Icmt was reported to result in midgestational embryonic lethality [31]. Based on the link between SFA, MT, and folate responsive Icmt in the developing neural tube, the current study examined the effects of ICMT inhibition by CAAX analogs during murine neural tube development. This study also explored inhibitory adenosine analogs to test the hypothesis that AHCY inhibition during neurulation would result in NTDs.
Two highly inbred mouse strains, SWV/Fnn (SWV) and LM/Bc/Fnn (LM/Bc), were selected for Folr1 backcrossing. A panel of simple sequence length polymorphisms (SSLP) markers that spans the entire murine genome (except the X and Y chromosomes) was used to establish two congenic mouse strains carrying the Folr1$^{tm1Fnn}$. This panel consisted of 300 primer pairs (Invitrogen, Carlsbad, California, USA) designed against the Whitehead Institute Center for Genome Research markers. PCR was performed under the manufacture's recommended conditions, and amplicons were run on a 6% polyacrylamide gel. The gels were then stained with ethidium bromide, visualized under UV, and imaged. All polymorphisms were identified visually. Folr1$^{tm1Fnn}$ heterozygote females from a mixed background (10) were backcrossed to SWV or LM/Bc males, and subsequent male offspring were backcrossed for at least ten successive (N10) generations with females from SWV or LM/Bc strains. A minimum of 16 males from each generation were tested and those that carried Folr1$^{tm1Fnn}$ and the highest percentage of host-strain markers were selected for the next backcross, based on speed congenic polymorphic marker analysis [32].
The SWV-Folr1$^{tm1Fnn}$ and LM/Bc-Folr1$^{tm1Fnn}$ mice were used as model systems for this study. The established inbred mice were maintained in a closed breeding colony. Virgin females (2-3 months of age and 18-20 g of body weight) were mated overnight to males, and pregnancy was determined by the presence of a vaginal plug on the following morning. Based upon the likely time of ovulation, the beginning of gestation (day 0, hour 0) was set at 10 p.m. of the previous evening [33].
Starting on E0.5 and continuing to E18.5, dams received a daily oral gavage of an aqueous solution (10 $\mu $L/g body weight) of folinic acid (5-formyl-tetrahydrofolate, SFA; Acros, Geel, Belgium) dissolved in sterile water. To establish a dose response for SFA, supplementation regimens included eight females per group of $-$/$-$ and $+$/$-$ genotypes treated at 6.2, 12.4, 24.9, and 36 mg/Kg. On E18.5, the dams were killed by CO$_{2}$ asphyxiation and the offspring were collected by caesarian section for gross morphological examination.
To determine if folate metabolites could rescue the Folr1$^{tm1Fnn} $ induced lethality and NTDs, metabolites were tested individually, in biological groups, and collectively at 88 $\mu $mole/Kg via IP injections. Starting on E0.5 and continuing to E18.5, female mice received daily intraperitoneal (IP) injections of an aqueous supplementation solution. Controls included SFA administered via IP injection at 88 $\mu $mole/Kg in sterile water for injection. This SFA dose was selected based on preliminary testing. Individual metabolites were given at an equimolar concentration and included SAM, L-methionine, betaine, adenosine-5-monophosphate (A5MP), guanine-5-monophosphate (G5MP), thymidine-5-monophosphate (T5MP), in- osine-5-monophosphate (I5MP), deoxyadenosine-triphosphate (dATP), deoxyguanine-triphosphate (dGTP), and deoxythymidine-triphosphate (dTTP) (Sigma-Aldrich, St. Louis, MO, USA). The pooled-supplementation groups were divided into two folate-related biochemical pathways: methylation (SAM, L-methionine, and betaine) and nucleoside metabolism (A5MP, G5MP, T5MP, I5MP, dATP, dGTP, and dTTP). The compounds were also tested collectively, such that all compounds were pooled and the final concentration for each was 88 $\mu $mole/Kg. On E18.5, the dams were killed by CO$_{2}$ asphyxiation and the fetuses were collected by caesarian section for gross morphological examination.
Wild type SWV female mice were mated overnight to SWV males as described above. On gestational day E8.5, pregnant mice were treated by IP injection with the compounds listed below. On E18.5, the dams were killed by CO$_{2}$ asphyxiation and the fetuses were collected by caesarian section for gross morphological examination.
Adenosine dialdehyde (Sigma Aldrich), the periodate oxidation of adenosine (AdOx), was selected as a potent inhibitor of AHCY [34]. Three other AHCY inhibitors [35] were also selected for testing and the compounds were obtained from the National Cancer Institute (NCI): 3-deaza-adenosine (DZA), 3-deaza-neplanocin A (DZNep), and neplanocin A (NepA). Concentrations tested included were 5 mg/Kg, 7.5 mg/Kg, and 10 mg/Kg for DZNep (provided by Victor E. Marquez of NCI), 4 mg/Kg and 8 mg/Kg for NepA, 40 mg/Kg and 80 mg/Kg for DZA (provided by Robert J. Schultz of NCI), and 40 mg/Kg and 80 mg/Kg for AdOx (Sigma). All compounds were prepared in sterile water for injection, and injection volumes were 10 $\mu $L/g of body weight.
N-Acetyl-S-geranylgeranyl-L-cysteine (AGGC), or the structurally similar inactive control, N-Acetyl-S-geranyl-L-cysteine (AGC) (Enzo) were used for ICMT inhibition [36]. Both compounds were prepared in stripped corn oil (Sigma) at 4.8 mmoles/Kg, and injection volumes were 10 $\mu $L/g of body weight.
Initial backcrossing indicated that coat color was linked with Folr1 genotype in SWV backcrosses. Genome database query (NCBI) indicated the genetic loci for Folr1 and tyrosinase are both found on mouse chromosome 7 (Fig. 2). As males were selected for backcrossing using SSLP markers, coat-color linkage was separated from SWV-Folr1 genotypes and albinism restored. Supplementation with SFA indicated that both the SWV-Folr1$^{tm1Fnn}$ and LM/Bc-Folr1$^{tm1Fnn}$ mice exhibited congenital malformations at 24.9 mg/Kg (Fig. 3). The data also indicated that there was a differential response between the strains in penetrance of NTDs. Specifically, the LM/Bc-Folr1$^{tm1Fnn}$ strain was shown to have a higher percentage ($e.g. $ at 24.9 mg/Kg, 91% of Lm/Bc-Folr1$^{tm1Fnn}$ had NTDs versus 19% in the SWV-Folr1$^{tm1Fnn}$) of fetuses with NTDs (Fig. 3A, 3B). The maternal genotype also modified the SFA supplementation required to reduce the rates of NTDs and embryonic lethality. These data indicate that the SWV background has a higher rescue of lethality and lower incidence of NTDs, and maternal genotype is a modifier of lethality and NTD penetrance in nullizygote embryos.
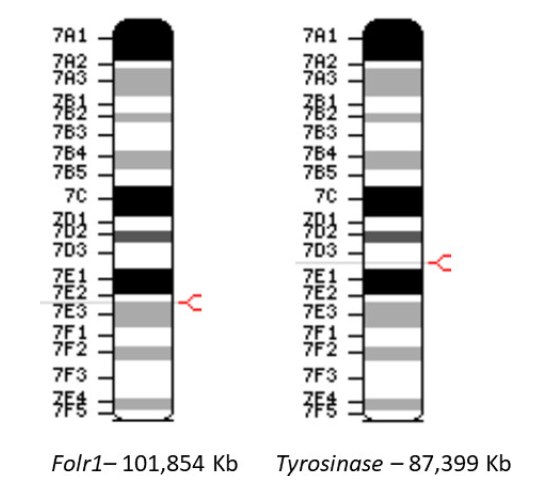 Fig. 2.
Fig. 2.Linkage of the Folr1 and Tyrosinase (SWV albinism) loci.
Due to the frequent occurrence of blonde coat color with Folr1 genotype during backcrossing onto the SWV strain, genomic analysis was performed. The 14.5 Mbase proximity is consistent with linked traits that can frequently segregate with mutant loci.
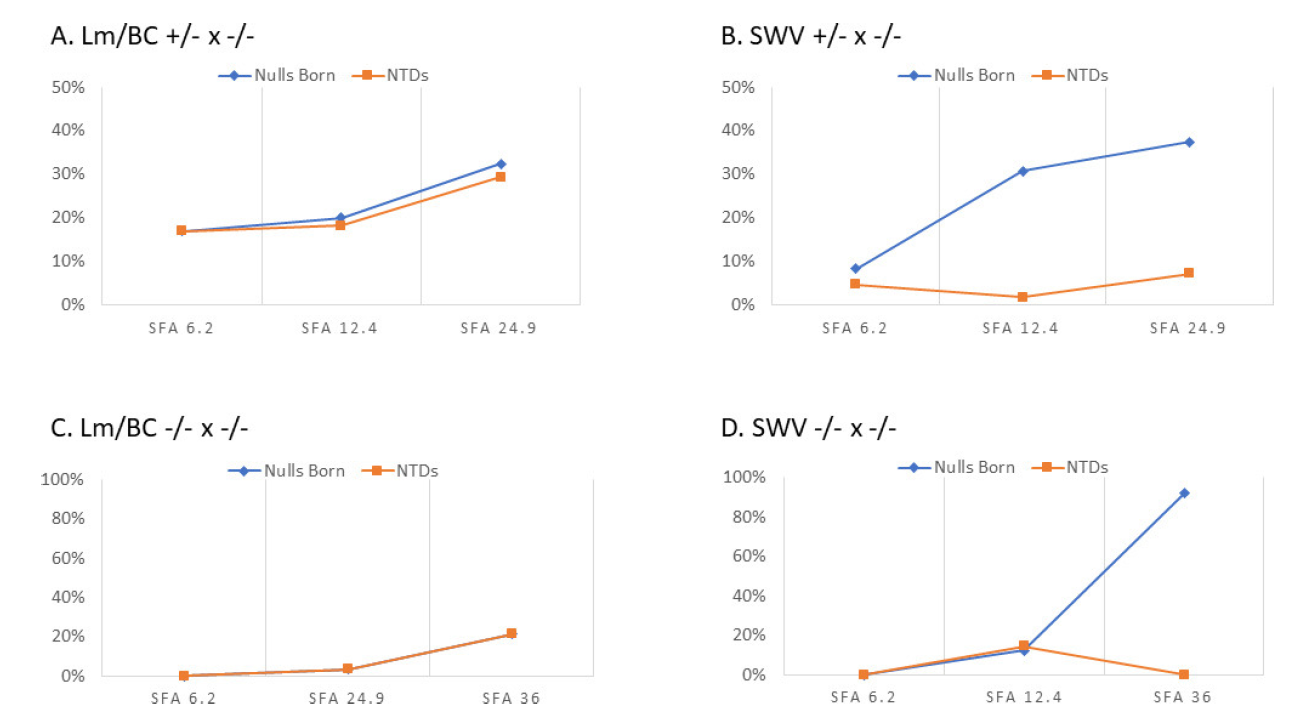 Fig. 3.
Fig. 3.Rescue response frequencies for Folr1.
Response frequencies are plotted for SFA and indicated that both the SWV-Folr1$^{tm1Fnn}$ and LM/Bc-Folr1$^{tm1Fnn}$ mice have folate-responsive NTDs. Genetic background strain is shown as a major modifier of NTD penetrance. The response to supplementation also differed by maternal genotype.
Testing of folate metabolites indicated that there were no nullizygous fetuses from females receiving either the negative control (water) or individual metabolites. Additionally, metabolites pooled by biological groups or collectively, did not exhibit the ability to rescue nullizygous embryos (Fig. 4). Follow up studies using 88 $\mu $mole/Kg/day hypoxanthine and thymidine (HT) or HT with methionine (HTM) did not result in rescue of any nullizygous Folr1 embryos from lethality. Folr1$^{-/-}$ fetuses from dams receiving equimolar treatment (88 $\mu $mole/Kg/day) of SFA were all rescued from lethality and exhibited no gross malformations.
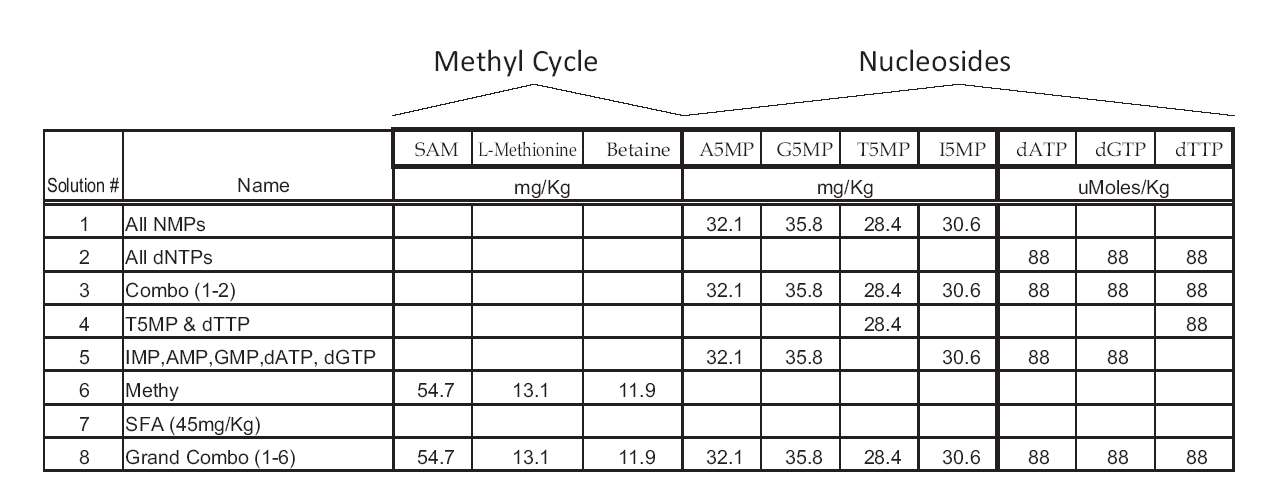 Fig. 4.
Fig. 4.Tested folate metabolites.
Testing strategy for methyl cycle metabolites and nucleosides are shown. All treatments were calculated as approximately 88 mmole/Kg equivalents of folinic acid (SFA). There were no nullizygous fetuses from dams receiving either the negative control (water) or individual folate metabolites. Only SFA resulted in any nullizygous fetuses surviving to term.
Chemical inhibition of MT or ICMT during neurulation significantly increased the response frequency of developmental toxicity and NTDs in SWV mice. Developmental toxicity data from AHCY inhibition are shown in Table 1 and Fig. fig5. NepA did not produce significant developmental toxicity at 4 or 8 mg/Kg. Initial treatment with NepA dosages of 12 or 20 mg/Kg resulted in maternal lethality and were discontinued. DZNep resulted in an increased incidence of embryo toxicity at 20 mg/Kg and NTDs at 5 mg/Kg. DZA resulted in embryonic lethality at 80 mg/Kg and NTDs in multiple litters at 40 mg/Kg. AdOx was maternally toxic at 40 mg/Kg; 4 of 18 dams died. Of the 14 surviving pregnant dams, one litter had a fetus with both exencephaly and midline facial cleft. Examination of fetuses after ICMT inhibition showed that the rate of malformations increased from 0% ($n =$ 8 litters) in the AGC control group to 9.1% ($n =$ 8, 3 litters with malformations) in the AGGC-treated group. The congenital malformations observed after AGGC treatment included exencephaly and gastroschisis ( Fig. 6).
| Treatment | Total | Total | % Unaffected | % Affected | |||||
|---|---|---|---|---|---|---|---|---|---|
| Litters | NTD Affected Litters | Implants | Normal | Dead | NTDs | Resorptions | |||
| DZA 40mg/Kg | 7 | 4 | 77 | 53 | 1 | 12 | 11 | 68.8% | 31.2% |
| DZA 80mg/Kg | 3 | 1 | 41 | 6 | 0 | 6 | 29 | 14.6% | 85.4% |
| DZNep 5mg/Kg | 14 | 1 | 171 | 148 | 7 | 3 | 13 | 86.5% | 13.5% |
| DZNep 7.5mg/Kg | 9 | 1 | 107 | 47 | 1 | 1 | 58 | 43.9% | 56.1% |
| DZNep 10mg/Kg | 6 | 0 | 69 | 13 | 0 | 0 | 56 | 18.8% | 81.2% |
| AdOx 40mg/Kg | 14 | 1 | 183 | 164 | 2 | 1 | 16 | 89.6% | 10.4% |
| AdOx 80mg/Kg | 3 | 0 | 38 | 36 | 0 | 0 | 2 | 94.7% | 5.3% |
| Nep A 4mg/Kg | 7 | 0 | 84 | 79 | 2 | 0 | 3 | 94.0% | 6.0% |
| Nep A 8mg/Kg | 6 | 0 | 78 | 74 | 2 | 0 | 2 | 94.9% | 5.1% |
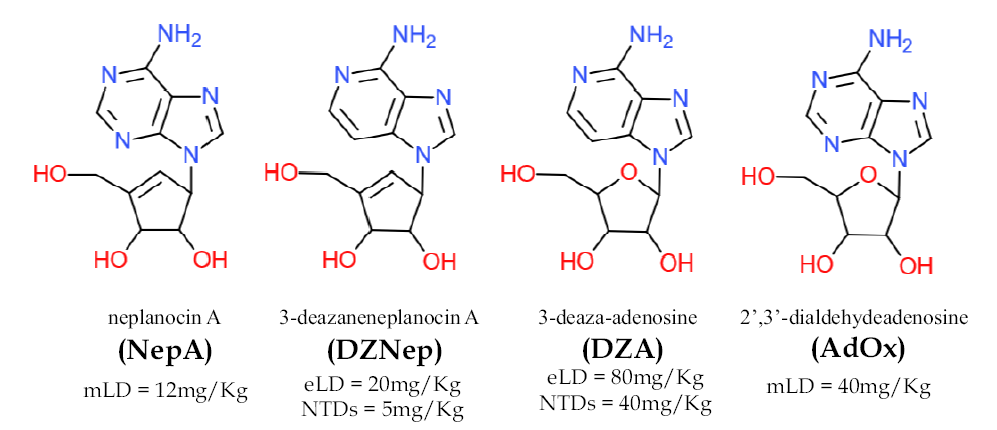 Fig. 5.
Fig. 5.Inhibitors of AHCY.
Summary of maternal and developmental toxicity of adenosine analogs and AHCY inhibitors. The order of teratogenicity was DZA $>$ DZNep $>$ AdOx* $>$ NepA. The maternally toxic dosage for NepA was reached without a significant increase in developmental toxicity. *There was one fetus with an NTD and midline facial cleft at maternally toxic dosages for AdOx, 4 of 18 moms died at 40 mg/Kg AdOx, $>$ 10% resorption. Both DZA and DZNep produced fetuses from multiple litters with NTDs.
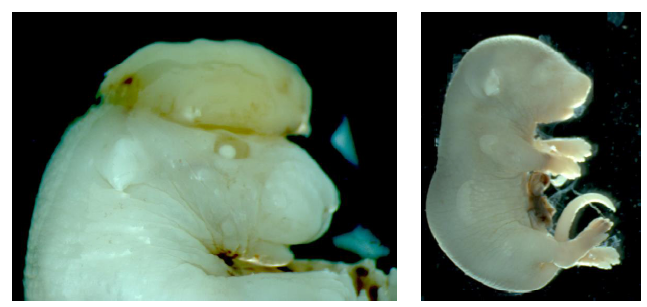 Fig. 6.
Fig. 6.Congenital Malformations due to ICMT inhibition.
The inhibition of ICMT via N-Acetyl-S-geranylgeranyl-L-cysteine (AGGC) administration at E8.5 resulted in three litters with congenital malformation including exencephaly and gastroschisis. There were no defects in animals exposed to N-acetyl-S-geranyl-L-cysteine (AGC).
The relative toxicities can be ordered by developmental toxicity as indicated by death and NTDs. The percent of affected fetuses were highest with DZA $>$ DZNep $>$ AdOx $>$ Nep A. DZA at 40 and 50 mg/Kg resulted in the highest number of NTDs. DZNep resulted in the highest percentage of resorptions compared to implants (81.2 %) at 10 mg/Kg.
In 1995, initial reports demonstrated that in the CF-1 strain, nullizygous epidermal growth-factor receptor embryos died in utero, but when the mutant allele was carried by the CD-1 strain, the nullizygous pups survived postnatally for up to three weeks [37]. This study demonstrated that although the time, cost, and energy to establish a mutant strain on multiple backgrounds can be daunting, genetic backgrounds can completely alter the phenotype observed in nullizygous embryos, making it of paramount importance that experiments conducted on knockout mice be conducted on multiple inbred strains. The reason for this includes the identification of differences in phenotype secondary to gene-gene interaction within the genomic background of the mice, which can then lead to investigations into the nature of these genetic variations. Genetic crosses and backcrosses using highly inbred mouse strains have previously revealed chromosomal loci contributing to differing sensitivity to NTDs due to in utero valproic acid exposure [38]. Ultimately, the variation in NTD penetrance from different strains provides a starting point from which to identify modifying genes [39]. Identification of modifier genes is performed by crossing the two inbred strains and tracking the modifier loci with marker genetic sequences such as the SSLPs used to initially establish the inbred strains within this study [40]. This technique has proven useful in identifying both naturally occurring and knockout strain variations[38,41,42].
In the current study, two congenic strains carrying Folr1$^{tm1Fnn}$ were established via speed congenics. Both strains demonstrated a dose-response to SFA supplementation, but the mice carrying the SWV-Folr1$^{tm1Fnn}$ were shown to have lower response frequencies of NTDs at equal concentrations of SFA when compared to LM/Bc-Folr1$^{tm1Fnn}$. We conclude that this is due to the presence of modifier genes associated with the different genetic backgrounds of the congenic strains. Future studies can cross these mouse strains and monitor the markers that are most highly associated with the Folr1$^{tm1Fnn}$ NTD phenotype using next generation DNA sequencing approaches. This is expected to allow genetic linkage with folate-responsive NTDs, which can then be mapped for identification of specific modifier genes. The identified gene(s) may also prove useful in the identification of genetic modifiers for human NTD risk and susceptibility.
Several metabolites of one carbon metabolism were tested for the ability to reduce the prevalence of NTDs and embryonic lethality in Folr1$^{-/-}$ embryos. Of the metabolites tested, none restored normal embryonic development, as would be evident by successful rescue of nullizygous Folr1$^{tm1Fnn} $ embryos from lethality or NTDs. On the other hand, SFA reduced the rates of NTDs and embryonic lethality in Folr1$^{-/-}$ embryos. These findings indicate that Folr1 mutants require folate for phenotypic rescue, and only folate provide benefit beyond that observed with equimolar dosage of any of the various metabolites. This benefit may be due to other metabolites that were not tested, such as the recently reported benefits of serine, glycine, or formate[43,44]. This benefit may also be dependent on compensatory FOLR2 signaling, such that Folr1 is functioning as a transcription factor or signaling molecule [45] and folates are required at a high enough concentration to allow the lower affinity FOLR2 to compensate for loss of the higher affinity FOLR1. Continued investigation into the nature of folate-mediated cascades within the developing embryo are needed to elucidate these interactions. Continuing this line of research may include the testing of additional supplements (e.g. hypoxanthine, thymidine, serine, glycine, or formate) that have proven useful in studying the changes induced by antifolates and other genetic models both in vivo and in vitro[46,47]. The identified gene(s) from the inbred mouse strains and the investigation into alternative strategies for rescue of Folr1$^{-/-}$ embryos may thereby also prove useful in the identification of NTD risk and susceptibility in humans.
Homozygous disruption of AHCY in mice was reported to result in embryonic lethality prior to organogenesis [48]. Compound heterozygosity for mutations in humans have also been reported; patients with AHCY deficiency have elevated methionine (hypermethioninemia), DNA hypermethylation, and delayed psychomotor development with demyelination and myopathy[49,50]. Small molecule inhibitors and shRNA knockout of AHCY are also being developed for anti-viral and suppression of cell proliferation [51]. Based on the data in this study, these compounds pose increased risk for NTDs and developmental toxicity, as AHCY is essential to early development and chemical or genetic disruption can both result in embryonic lethality.
The CAAX analog, AGGC, which is an inhibitor of ICMT [52] was shown to significantly increase the rate of NTDs after administration to pregnant dams. The teratogenicity of this compound indicates that ICMT is required for neural tube closure. Methotrexate, a chemotherapeutic folate analog and teratogen, can also inhibit ICMT activity[53,54]. In addition, it had been demonstrated in cell cultures that AGGC would produce improper localization of Ras and cytoskeletal proteins subject to post-translational modification by methylation[55,56]. These data indicate that AGGC is teratogenic and suggest that the mechanism of teratogenicity underlying AGGC and ICMT teratogenicity is impaired localization of GTPases and cytoskeletal proteins that undergo post-translational modification by methylation. In conclusion, these data support the conclusion that genetic background can readily modify folate responsive NTDs, and MTs and ICMT, specifically, are essential to neural tube closure.
This study was funded in part by the National Institutes of Health (NIH), Research Project Grants HD093758, HD096327 and HD081216.
The authors report no conflict of interest.