



 , I Nyoman Bayu Mahendra 1,2, I Nyoman Gede Budiana 1,2, Kade Yudi Saspriyana 1,2, Ketut Suwiyoga 1,2
, I Nyoman Bayu Mahendra 1,2, I Nyoman Gede Budiana 1,2, Kade Yudi Saspriyana 1,2, Ketut Suwiyoga 1,21 Division of Gynecologic Oncology, Department of Obstetrics and Gynecology Prof. Dr. I G.N.G. Ngoerah Hospital, 80113 Denpasar, Bali, Indonesia
2 Faculty of Medicine, Udayana University, 80113 Denpasar, Bali, Indonesia
Abstract
Objective: To understand the basic mechanism and dynamic regulation
that underlies the epithelial-to-mesenchymal transition (EMT) in ovarian cancer
(OC) cells. Mechanism: A literature review using evidences from several
data bases (i.e., PubMed, EMBASE, Web of Science, Medline, Cochrane, Science
Direct, and Google Scholar) were conducted to describe the basic mechanism and
dynamic regulation of EMT in OC cells. Finding in Brief: EMT is a
complex epigenetic reprogramming orchestrated by specific transcription factors
(TFs) and multiple upstream activators and regulators, such as transforming
growth factor-
Keywords
- ovarian cancer
- epithelial-to-mesenchymal transition
- cellular reprogramming
In 2020, ovarian cancer (OC) became the third most common gynecologic malignancy
with a total of 313,959 new cases, and 207,252 new deaths recorded globally [1, 2]. Majority of OC are diagnosed in advance stage due to the ineffective
screening, and its silent progression at early stage [3]. OC imposes a
significant economic burden with annual average costs being significantly higher
in advance stages than early stage OC [4]. Epithelial OC is the commonest
histologic type, while only 10% belong to the non-epithelial type. The
epithelial subtype has five major histologic types, i.e., serous, mucinous,
endometroid, clear cell, and unspecified [5]. Surgical cytoreduction to attain no
gross residual disease (R0) followed by adjuvant chemotherapy is the current
standard treatment. Recently, maintenance therapies such as poly ADP-ribose
polymerase (PARP) inhibitors, bevacizumab, and drugs targeting homologous
recombination deficiency (HRD) are incorporated to prolong the survival [6].
However, despite advancement in the treatment of OC, recurrence rate remains high
(
Epithelial-to-mesenchymal transition (EMT) program has gained popularity among
researchers as the responsible cellular mechanism that confers OC cells with
increased metastatic potential and drug resistance, thus predisposing to
recurrence [8, 9, 10, 11]. EMT is a complex epigenetic reprogramming that results in
reversible phenotype transition where cancer cells lose their epithelial
phenotype and acquire mesenchymal phenotype [12]. The crucial orchestrator in EMT
promotion is a well-known group of transcription factors (TFs), which includes
SNAI1 (Snail1), SNAI2 (Snail2/Slug), Twist1/2, and zinc-finger E-box binding
homeobox 1/2 (ZEB1/2) [13]. These TFs induce epigenetic silencing of epithelial
markers (e.g., E-cadherin, Mucin 1 (MUC1), cytokeratin 18) while upregulating the
expression of mesenchymal markers (e.g., N-cadherin, vimentin, fibronectin, matrix metalloproteinases (MMPs))
[14]. The upstream regulators of these EMT-transctipion factors (EMT-TFs) are
multiple pathways which are mainly involved in embryonic development, such as
transforming growth factor-
This literature review will summarize the recent advances in the mechanisms underlying the complex regulatory network of EMT in OCs, with the focus on its key orchestrators such as EMT-TFs, signaling pathways, upstream activator, as well as the dynamics of its regulation.
EMT is a complex, reversible cellular process orchestrated by multiple activators and signal transduction pathways (Figs. 1,2). The downstream effector in this process is a series of activated TFs (EMT-TFs) that acts as either activator or repressor of the targets gene, with the end result of phenotypic transition from epithelial cells to more invasive mesenchymal cells. Thus, epigenetic reprogramming is at the heart of EMT regulation [15]. By undergoing EMT, OC cells gain characteristics crucial for distant metastasis, resistance to apoptosis, and thus, recurrence after therapy [16].
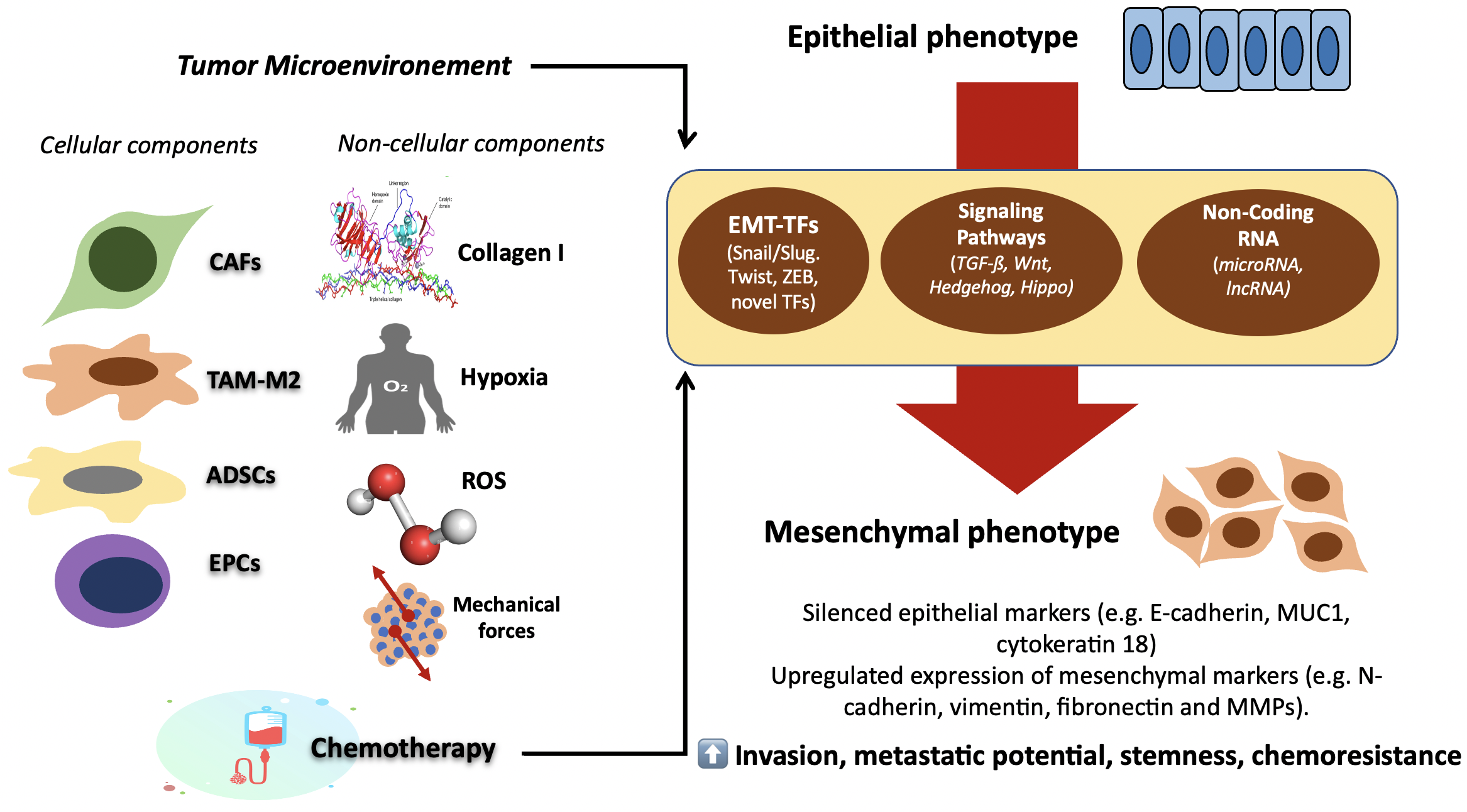 Fig. 1.
Fig. 1.Schematic illustration of complex regulatory interplays that
underlie EMT. Tumor microenvironment (TME) and exposure to chemotherapy are
believed to be the activators of EMT. The external factors then drive the
expression of specific TFs, and activate signaling transduction pathways
(TGF-
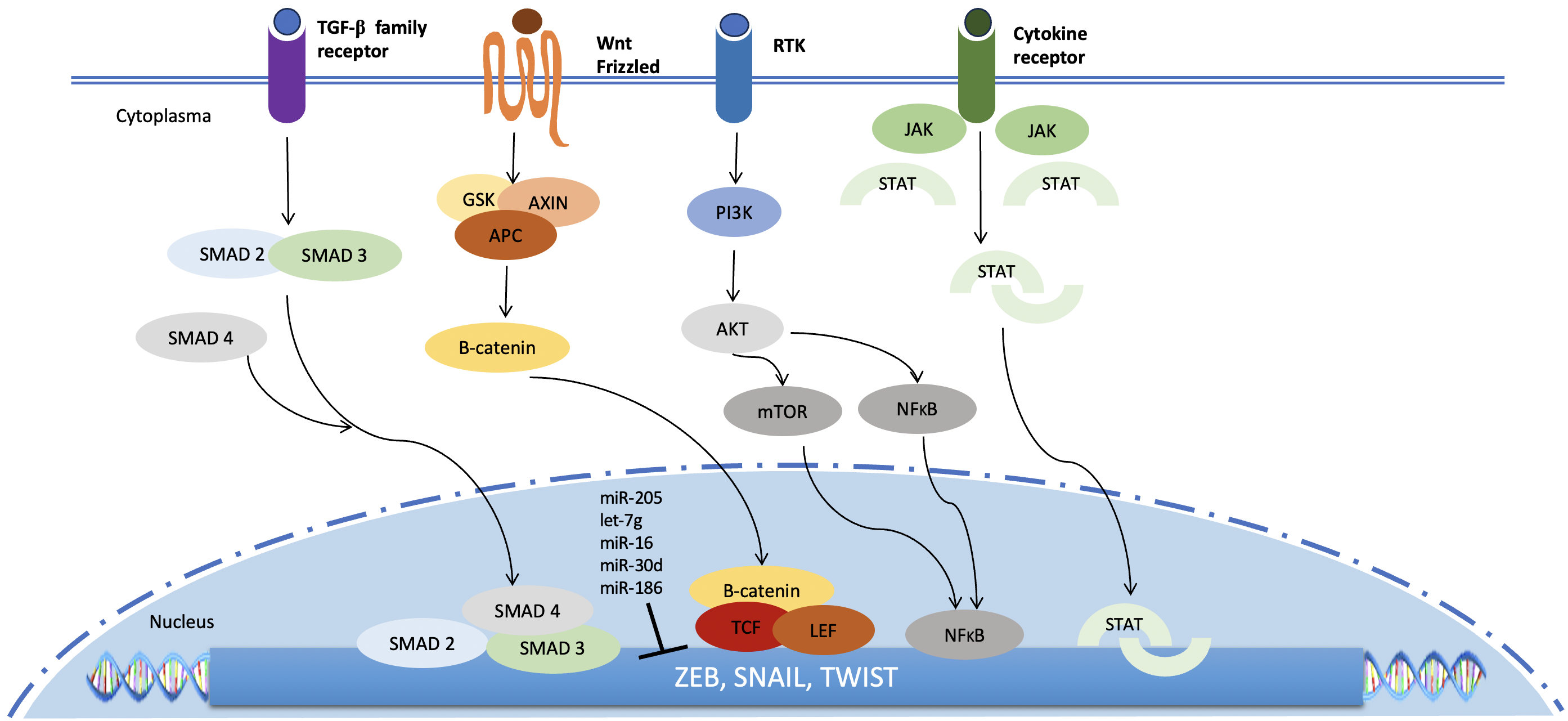 Fig. 2.
Fig. 2.Signaling pathways that regulate the induction of EMT. Several
signaling pathways play crucial role in EMT induction. All of these important
pathways culminate in the activation of genes encoding the key TFs zinc-finger E-box binding homeobox (ZEB), Snail and
Twist. Several miRNAs downregulate the expression of these TFs. SMAD 2/3/4, SMAD family
member 2/3/4; GSK, glycogen synthase kinase; APC, adenomatous polyposis coli;
AKT, AKT serine threonine kinase; JAK, janus kinase; STAT, signal transducer and
activator of transcription; TCF, T cell factor; LEF, lymphoid enhancer factor;
mTOR, mammalian target of rapamycin; NF
Snail and Slug, encoded by the snail family transcriptional repressor 1
(SNAI1) and 2 (SNAI2) gene, respectively, are transcriptional
repressors that play important roles in regulating EMT. Snail and Slug activation
results from several signaling pathways, such as receptor tyrosine kinases
(RTKs), TGF-
ZEB is the zinc finger E-box binding homeobox family of TFs with its two
members, ZEB1 and ZEB2. ZEB contain zinc-finger domain that allows binding at the
enhancer boxes within the promoter region of target genes [42]. ZEB can interact
with several TFs and cofactors, such as Smads (Suppressor of mothers against
decapentaplegic), protein 300 (p300)/P300/CBP-associated factor (pCAF), Brahma-related gene-1
(BRG1), Nucleosome remodeling and deacetylation (NuRD) complex,
and C-terminal binding protein (CtBP) [43, 44, 45]. The interaction determines ZEB
role either as transcriptional activator or repressor of the target genes. ZEBs
are crucial regulators of TGF-
Twist is a member of the basic helix-loop-helix (bHLH) family, and acts as
either transcription activator or inhibitor [52]. The two Twist genes, i.e.,
Twist1 (Twist) and Twist2 (Dermo-1), have 90% similarity [53]. Upregulated Twist
expression predicted shorter OS in OC patients [54, 55, 56]. Twist1 induce
upregulation of Akt upon cisplatin treatment in OC cell lines, which drives
resistance [57]. Twist2 also induces chemoresistance in OC cells through
induction of Akt serine threonine kinase (Akt)/Glycogen synthase kinase-
Heat shock transcription factor 1 (HSF1) is a proteotoxic stress-responsive
transcription factor that also contribute in EMT. Knockdown of HSF1 expression in
OC cell lines impaired TGF-
TGF-
Wingless/Integrated (Wnt) signaling pathway is one of the main orchestrator of
EMT in OC. In promoting EMT, Wnt signaling mainly depends on the activity of
Hedgehog (Hh) signaling cascade culminate in a balance between activator or repressor forms of transcription factor Glioma-associated oncogene TFs (Gli) with PTCH1, PTCH2, and GLI1 being the main target genes. Hedgehog (Hh) ligand, Patched (Ptch), and Smoothened (Smo) are proteins involved in the activation of Hh signaling cascade [102]. Hh signaling plays an important role in the regulation of invasiveness, chemoresistance, as well as maintenance of CSCs characteristics [103, 104, 105, 106, 107, 108]. A crosstalk between Sonic Hh-Gli1 signals and PI3K-Akt pathway regulates EMT induction in OC cells [109]. Inhibition of Hh signaling results in inhibition of EMT [110, 111]. Gli1 also regulates the expression of Snail1, Slug, and Twist. Gli1 and Gli2 repress the expression of E-Cadherin [112].
The Hippo pathway is a tumor suppressive pathway involved in regulating tissue
growth, and their component comprises a pair of related serine/threonine kinases,
macrophage stimulating 1 and 2 (MST1 and MST2), large tumor suppressor kinase 1
and 2 (LATS1 and LATS2), and lastly Salvador family WW domain containing protein
1 (SAV1), and Mps one binder 1 (MOB1A and MOB1B) [113]. This pathway
downregulates the activity of YAP/TAZ. Following Hippo inactivation, YAP and
transcriptional enhanced associate domain (TEAD) form complex within the nucleus
to direct transcription of target genes. EMT-TFs are capable of complex formation
with YAP/TEAD, which in turn upregulate the expression of YAP target genes in
inducing EMT in OC cells [114, 115, 116]. High YAP and TEAD expression is associated
with OC progression. Hippo pathway is also involved in regulating chemoresistance
in OC cells [117, 118, 119, 120, 121]. Hippo signaling interacts with other EMT-inducing
pathways, such as TGF-
MicroRNA (miRNA) is a single-stranded non-coding RNA that acts as antisense RNA
to downregulate expression of the target genes at the post-transcriptional level.
Several miRNAs are involved in the regulation of EMT in OC cells. miR-200 family
of miRNAs is a well-known EMT suppressor. Low level of miR-200 expression is
demonstrated in normal OSE, but the increased expression is present in OC [122].
Overexpression of miR-205 and/or miR-200 family result in downregulation of ZEB1
transcription factor and Wnt5a [123]. miR-200c upregulation induced
downregulation of ZEB1 and vimentin, and upregulation of E-cadherin [124]. In OC
cells line, miR-200c overexpression decrease Snail, increase E-cadherin, and
significantly reduce the invasiveness and tumorigenic potency of OC cell lines
[125]. miR-203 expression can attenuate TGF-
Long non-coding RNA (lncRNA) is non-protein coding RNA with length longer than
200 nucleotides that regulates target gene expression at both transcriptional and
post-transcriptional level [135]. lncRNA are capable of direct binding to DNA or
RNA to affect the transcription process. lncRNA H19 expression in the OC cells
promotes migration and EMT-related activity [136]. Silencing of lncRNA in colon
cancer associated transcript 2 (CCAT2) results in EMT inhibition. Knockdown of
lncRNA CCAT2 also inhibits the expression of
EMT activation is the result of dynamic and reciprocal interplay between OC cells and their tumor microenvironment (TME). Cellular or non-cellular component of TME can act either as activator or inhibitor to certain signaling transduction pathways associated with EMT. On the other hand, OC cells can induce differentiation of cellular component of TME into certain phenotypes that favor metastatic potential of drug resistance of OC cells. These complex regulations are also affected by external factors related to TME, such as hypoxia, oxidative stress, as well as mechanical forces.
TME is the niche or environment in which the cancer cells closely interact with
the host stroma, including cellular and non-cellular component of TME. The
cellular components include immune cells, endothelial cells and fibroblast, while
the non-cellular components include extracellular matrix and cellular
metabolites. The cellular components play a more dominant role in promoting EMT.
Cancer associated fibroblasts (CAF) are one of the key cellular components of TME
that play important role in EMT induction. CAF are more proliferative and have
higher metabolic states as compared to normal fibroblasts [141]. CAF-derived
exosomes are rich in TGF-
Tumor associated macrophage (TAM) plays a critical role in the interaction between TME and OC cells. TAMs are capable of differentiating into two disctinct phenotypes: M1, which has pro-inflammatory with anti-tumor activity, and M2, which has anti-inflammatory with pro-tumor activity. The path of TAMs is determined by the local TME [147]. In a mouse model of OC, TAM require ZEB1 expression to activate the tumor-promoting functions [148]. OC cells secretes macrophage colony-stimulating factor (M-CSF) to drive the differentiation of M2-TAM [149]. M2-TAMs are capable of inducing EMT of OC cells by releasing chemokine (C-C motif) ligand 18 (CCL18). On the other hand, CCL18 induces M-CSF transcription in OC cells through the activity ZEB1 transcription factor. Thus, a CCL18-ZEB1-M-CSF interacting loop exists between OC cells and TAMs that regulate the tumor progression and metastasis through EMT [149]. OC-derived exosomes containing miR-222-3p and miR‑940 are also capable in inducing TAM polarization into the M2 phenotype [150, 151].
Adipose-derived stem cells (ADSCs) are mesenchymal stem cells obtained from
adipose tissues. ADSCs have been shown to affect the proteomic profile of OC
cells via paracrine mechanism in favour of OC progression [152]. Secretion of
TGF-
Endothelial progenitor cells (EPCs), the bone marrow-derived stem cells, play a
significant role in tumor angiogenesis and growth. EPCs are recruited into the
neovascular bed of the tumor in response to certain signals or cytokines secreted
by tumor cells [158, 159, 160]. EPCs are able to invade into the OC cell clusters,
whereas normal human microvascular endothelial cells are not capable of invading
OC cell clusters [161]. Circulating levels of EPCs are significantly increased in
OC patients and correlate with tumor stage and residual tumor size [162]. OC
cells cultured in EPC-conditioned media (EPC-CM) demonstrate an increase in
TGF-
The non-cellular components of TME also participate in promoting EMT. Collagen I
enhance OC cells motility and invasiveness through the increased expression of
MMPs and
Hypoxia is one of the most important non-cellular factors in inducing EMT.
Hypoxia influences cellular processes such as angiogenesis, acquisition of stem
cell-like features, chemoresistance, as well as EMT [167, 168, 169, 170]. Hypoxia is the
characteristic of the peritoneal environment. Hypoxia induces the stabilization
of hypoxia-inducible factor-1
Reactive oxygen species (ROS), such as hydroxyl free radicals, superoxide, and
hydrogen peroxide can accumulate within the TME due to active metabolic patterns
of OC cells and tumor stromal cells. ROS accumulation can lead to lipid
peroxidation, antioxidants deprivation, and ultimately programmed cell death
which depend on iron, also known as ferroptosis. However, OC cells develop
several mechanisms that confer resistance to ferroptosis [178]. ROS are involved
in the regulation of EMT. ROS accumulation induced the increased expression of
HIF-1
OC cells endure several mechanical forces from their TME. The presence of ascites and interstitial fluid confer the shearing force to the OC cells, while tumor expansion against the extracellular matrix and TME exert tension on the tumor periphery. Furthermore, tumor expansion and the hydrostatic pressure of ascites also exert internal compression forces to the OC cells [181]. Those forces give impact into biochemical regulation and signaling pathways of OC cells, including the regulation of EMT. Oscillatory tension significantly decreases the expression of E-cadherin while increasing the expression level of Snail [182]. Tissue stiffness, recapitulated by substrate stiffness in vitro, promotes OC cells proliferation and nuclear translocation of the oncogene YAP. Substrate softening has been demonstrated to promote changes consistent with EMT [183, 184].
Results from in vitro studies have demonstrated that chemoresistant OC
cells express markers of EMT [8, 185, 186, 187, 188, 189, 190]. On the other hand, exposure to
chemotherapy has been demonstrated to induce EMT in OC cells. Cisplatin induced
EMT-associated morphological changes in OC cells [191, 192, 193]. Receptor cells
co-cultured with carboplatin- or etoposide-treated feeder cells in Transwell
co-culture system exhibited increased expression of EMT markers vimentin and
Snail. The altered microenvironment of either carboplatin- or
etoposide-16-treated feeder OC cells also significantly increased the migration
of the OC cells [194]. OC cells treated with carboplatin exhibited phenotypic
changes consistent with EMT [195]. Clinical studies in OC patients revealed that
treatment with platinum-based chemotherapy increased the proportion of EMT-like
circulating tumor cells (CTC), accompanied by the “de novo” emergence
of PI3K
The traditional concept viewed EMT as a binary process in which the phenotype of cancer cells can change completely into either epithelial or mesenchymal cells. However, a newer concept of “partial EMT” has been proposed, in which cancer cells can undergo phenotypic changes with both epithelial and mesenchymal phenotype [198, 199]. Tumours in a partial EMT state exhibit low expression of EMT-TFs, and co-express both epithelial and mesenchymal genes. A recent study in pancreatic ductal adenocarcinoma cells reported that partial EMT results from different mechanisms underlying complete EMT. Cancer cells lose their epithelial phenotype through an alternative post-translational process of protein relocalizations, which lead to “partial EMT”. In that study, E-cadherin protein was found to be confined to intracellular foci in delaminated cells exhibiting a mesenchymal morphology. Furthermore, cancer cells that exhibit partial EMT, migrate and form circulating tumor cell clusters, rather than disseminate as single cell as in “complete EMT” [200]. However, it remains unclear whether partial EMT represents an intermediate stage where cancer cells are in a paused transitional state within the mesenchymal differentiation continuum, or it is a final fate of the cancer cells. Interestingly, one study support the later hypothesis, in which they found no evidence that complete and partial EMT co-exist within the same tumor [50]. This finding implicate that the tendency of cancer cells to use either a complete or partial EMT program is a specific and stable feature of an individual tumor. Cells with partial EMT have been known to be more resistant to apoptosis and have greater tumor-initiating potential, as compared to those with complete EMT. EMT interconversions are also dynamically regulated during the development and progression of ovarian tumors [50, 201].
A review by Jolly et al. [198] stated that the core regulatory network for EMT or MET (Mesenchymal-to-Epithelial Transition) acts as a “three-way”, which give rise to three distinct phenotypes, i.e., epithelial (E), mesenchymal (M), and hybrid, or partial EMT (pEMT). The core regulatory network depends on two mutually inhibitory loops, i.e., miR-34/SNAIL and miR-200/ZEB. E phenotype is defined as high miR-200/miR-34, low ZEB/SNAIL; M phenotype is defined as low miR-200/miR-34, high ZEB/SNAIL; and partial EMT is defined as low miR-34/ZEB, high SNAIL/miR-200. Another theory proposed that miR-200/ZEB, with input from SNAIL, behaves as a three-way switch allowing for the existence of three phenotypes, i.e., E (high miR-200, low ZEB), M (low miR-200, high ZEB), and E/M or partial EMT (medium miR-200, medium ZEB). Therefore, ZEB activation is a necessary requirement for the acquisition of a complete EMT. However, results from experimental studies observing partial EMT appear to be more consistent with medium miR-200, medium ZEB theory. One study demonstrated that difference in expression and subcellular localization of transcription factor 21 (Tcf21) and Slug (Tcf21/Slug balance) is associated with phenotypic plasticity [202]. Downregulation of Slug by Tcf21 is known to maintain the epithelial properties of high grade serous carcinoma (HGSC). The study identified the association of Tcf21/Slug balance with additional intermediate phenotypic states (i.e., iE or iM), supporting the proposed hypothesis of a multistep EMT program [202]. Tcf21 high-Slug low expression was identified in E phenotype, Tcf21 low-Slug high expression was identified in M phenotype, and Tcf21 moderate-Slug moderate expression (E-M or pEMT) was identified as stable phenotypes. Intermediate E or M represents a metastable state associated with phenotypic switches. Intermediate E (iE) expressed epithelial markers, Snail, Twist1, but lacked Tcf21, while the tumor group with low EMT-TF expression was identified as intermediate M (iM). The dynamic regulation of EMT, which encompasses a range of phenotypic plasticity, is summarized in Fig. 3.
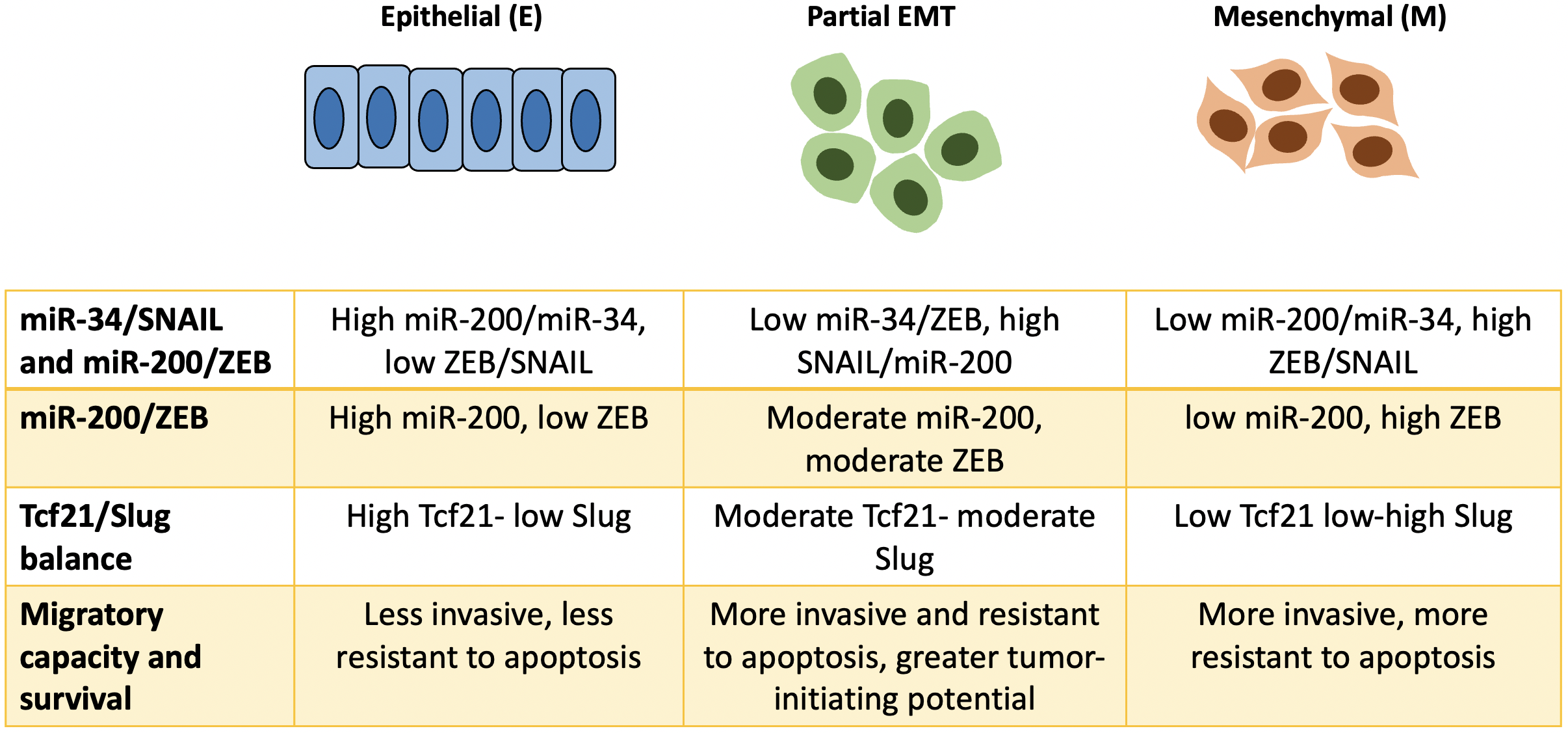 Fig. 3.
Fig. 3.Schematic illustration of dynamic regulation underlying phenotypic plasticity in EMT. A newer concept of EMT introduces the concept of partial EMT in which cancer cells exhibit both mesenchymal and epithelial characteristics. This phenotypic plasticity are determined by the balance of expression from certain TFs and their inhibitors (miRNA), such as the balance between miR-34/SNAIL and miR-200/ZEB. The expression and subcellular location of Tcf21 and Slug also influence the phenotype of cancer cells. Cancer cells undergoing partial or hybrid EMT tend to be more invasive, more resistant to apoptosis, and have greater tumor initiating potential.
Epithelial-to-mesenchymal transition or EMT is a form of epigenetic cellular reprogramming governed by complex regulatory networks that confers OC cells with increased invasiveness and drug resistance. EMT is orchestrated by multiple TFs, upstream activators, and regulators that result in the acquisition of mesenchymal phenotypes with increased metastatic potential, stemness properties, and chemoresistance. EMT activation is the result of dynamic and reciprocal interplay between OC cells and their tumor microenvironment. EMT is a dynamic process of phenotypic plasticity, which encompasses a range of hybrid states. Understanding this complex regulatory network is crucial in order to gain insight in developing novel and effective treatment strategies for OC.
ADSC, Adipocyte-derived stem cell; AKT, AKT serine threonine kinase; AMPK,
5
PKAP performed conception and design, acquisition of data, analysis and interpretation of data and drafting the manuscript. INBM and INGB performed conception and design, analysis and interpretation of data, and reviewing the manuscript. IGSW and KYS performed acquisition of data, analysis and interpretation of data and drafting the manuscript. KS performed conception and design, analysis and interpretation of data, reviewing and final approval of published version. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
We would like to express our gratitude to all those who helped us during the writing of this manuscript. Thanks to all the peer reviewers for their opinions and suggestions.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.