
Academic Editor: Andrea Tinelli
Background:
The vulvar lichen simplex chronicus (VLSC) is a common condition in gynecologic
clinics. Though VLSC is not life-threatening, it usually causes pruritus,
soreness and dispareunia, which cause general discomfort. The exact etiology of
VLSC is unclear. This study was performed to explore the vaginal microbiota of
VLSC and to identify the possible microbial
factors in attacks. Methods: Ninety women were recruited. 45 patients
with VLSC and 45 women without vulvar symptom were identified as lichen simplex
chronicus (LSC) and H groups respectively. The vaginal microbiota of the two arms
were compared by the V3-V4 region of 16S rRNA sequencing. Results: The
LSC group had less alpha diversity than H group (p
Lichen simplex chronicus (LSC) is a common condition and it is estimated to occur in 12% of the total population. It has been found that LSC affects female and adult population more frequently than the male and young population [1]. The incidence ratio of vulvar LSC is up to 1.7% in all gynecologic practices including prepubertal girls [2]. It is also found to comprise 10–35% of the patients in vulvar clinics [3]. Histopathologically, LSC lesions have hyperkeratosis, epidermal thickening, spongiosis and acanthosis [4]. Though it is not life-threatening disease compared with gynecological tumors, the vulvar LSC usually causes pruritus, soreness and dispareunia [3], which interrupt the work, sleep, sexual function, and cause general discomfort [5].
Vulvar LSC can occur de novo in healthy vulvar tissue named idiopathic vulvar lichen simplex chronicus (VLSC) or as a secondary complication of any pruritic vulvar condition such as psoriasis, lichen sclerosus, and contact dermatitis. The idiopathic VLSC is estimated to be found in up to 75% of affected women but the exact etiology is indistinct [6]. So far, many factors have been found to be associated with idiopathic VLSC such as immune factors, genetic factors, psychological factors, environmental factors and sex hormones [7]. Chen et al. [8] found that skin microbiota could affect immunity, epigenetics and epidermal barrier of the host, and it also played an important role in the pathogenesis of inflammatory skin diseases. Vaginal microbiota changes as response to the host biochemical and immunological change and vaginal secretion flows out and may make the vulvar skin in the similar microbiological environment with vagina, so we speculate that vaginal microbiota changes may play a role in the pathophysiologic conditions in VLSC. Along with the technology development, the high-throughput sequencing of the bacterial 16S rRNA can be used as a mature tool for assessing microbial communities with a high phylogenetic resolution. The goal of this study is to explore more accurate qualitative and quantitative information of the vaginal microbiota in females with VLSC, and to identify the potential microbiome pathogenesis which has not been studied before.
A total of 90 women were recruited at the Gynecology Outpatient Department of Sichuan University West China Second Hospital in Chengdu between January 2017 and May 2021. Forty-five patients diagnosed with VLSC confirmed pathologically were included as the LSC group. Forty-five women with no recorded vulvar complication and vaginitis were included as the control group (H group). The excluding criteria were (1) the vaginal smear indicated vaginitis such as bacterial vaginosis, vulvovaginal candidiasis or trichomoniasis, etc; (2) patients who had sexual intercourse within 48 hours before the visit; (3) patients received vaginal medications or douches within 48 hours before the visit; (4) pregnant women; (5) patients had taken any oral antibiotics,oral antimycotics, oral contraceptive medication or hormone replacement treatment in the past one month; (6) patients had received topical corticosteroids, topical immunomodulators, antihistamines, antidepressants or transcutaneous electrical nerve stimulation in the past one month. After obtaining informed consent, the mid-vaginal secretion samples were dipped using sterilized cotton swabs. Two sterilized swabs were collected for each sample and the swabs were put in tubes on ice until genomic DNA extractions within 3 hours. This study was approved by the Institutional Ethics Committee of Sichuan University West China Second University Hospital (NO.1356) and the work has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans.
The TIANamp Swab DNA kit (TIANGEN BIOTECH Beijing Co. Ltd, Beijing, China) was used to extract
the whole genomic DNA of vaginal bacteria species. The swab suspensions were
mixed with 20
The V3-V4 variable regions of the bacterial 16S rRNA gene were amplified with primers 338F (ACTCCTACGGGAGGCAGCA) and 806R (GGACTACNNGGGTWTCTAAT). The amplicon quality was visualized using gel electrophoresis. The PCR products were purified with AgencourtAMPure XP beads (Beckman Coulter Co., Bria, CA, USA) and quantified using Qubit dsDNA assay kit. The concentrations were then adjusted for sequencing. Sequencing was performed on an Illumina NovaSeq6000 with two paired-end read cycles of 250 bases each (Illumina Inc., San Diego, CA, USA; OE Biotech Company; Shanghai, China)
The
raw reads were deposited into the Genome Sequence Archive (GSA) database
(Accession Number: CRA005753. Paired-end reads were preprocessed using
Trimmomatic software (version 0.35, Usadel Lab, Aachen, Germany) to detect and cut off ambiguous bases (N). It
also cut off low quality sequences with average quality score below 20 using
sliding window trimming approach. After trimming, paired-end reads were assembled
using FLASH software (version 1.2.11, Center for Computational Biology, Baltimore, MD, USA). Parameters of assembly were 10 bps of
minimal overlapping, 200 bps of maximum overlapping and 20% of maximum mismatch
rate. Sequences were performed further denoising as follows: reads with
ambiguous, homologous sequences or below 200 bps were abandoned. Reads with 75%
of bases above Q20 were retained and reads with chimera were detected and removed
using QIIME software (version 1.8.0, Knight and Caporaso labs, Flagstaff, AZ, USA). Clean reads were subjected to primer
sequences removal and clustering to generate operational taxonomic units (OTUs)
using VSEARCH software (version 2.4.2, Institut Pasteur, Paris, France) with 97% similarity cutoff. The
representative read of each OTU was selected using QIIME package. All
representative reads were annotated and blasted against Silva database (Version
138) using RDP classifier (confidence threshold was 70%). The community
structure of LSC and H groups were compared. Venn diagrams were drawn to analyze
the amount of overlapped and unique OTUs in the two groups. In the
alpha-diversity analysis, Shannon and inverse Simpson indices were estimated. In
the beta-diversity analysis, principal co-ordinate analysis (PCoA) using
binary_jaccard was performed. Linear discriminant analysis effect size (LEfSE)
was performed to determine the dominant biomarkers of each group. Phylogenetic
investigation of communities by reconstruction of unobserved states (PICRUSt) was
performed to predict the function of different vaginal microbiota. Student’s
t-test was used to compare quantitative data and p
The average age of the LSC and H groups was 44.89
The data volume of valid tags ranges from 9359 to 74,949, and the length of the valid tags varied from 411.97 to 430.01 bps. 4475 OTUs were identified using a cutoff of 97% sequence similarity, including 31 phyla, 600 genera, and 905 species.
At the phylum level, the Firmicutes, Actinobacteria and Proteobacteria were the top three abundant bacterial phyla in H group. In the LSC group, the dominant three phyla were Firmicutes, Actinobacteria and Bacteroidota (Fig. 1A). At the genus level, the Lactobacillus and Gardnerella were the most abundant two bacterial genera in both of the two groups, with relative abundance of 64.32% and 13.64% respectively in the H group, and 64.82% and 9.36% respectively in the LSC group. Among the Lactobacillus, it was found that the Lactocillus iners. was the most abundant species. The relative abundance of Gardnerella decreased while Muribaculaceae and Streptococcus increased in the LSC group (Fig. 1B). The Venn diagram displayed that there were 2631 common OTUs of the two groups. H group had 402 unique OTUs, and LSC group had 1442 unique OTUs. The microbiota community of each group was different (Fig. 1C).
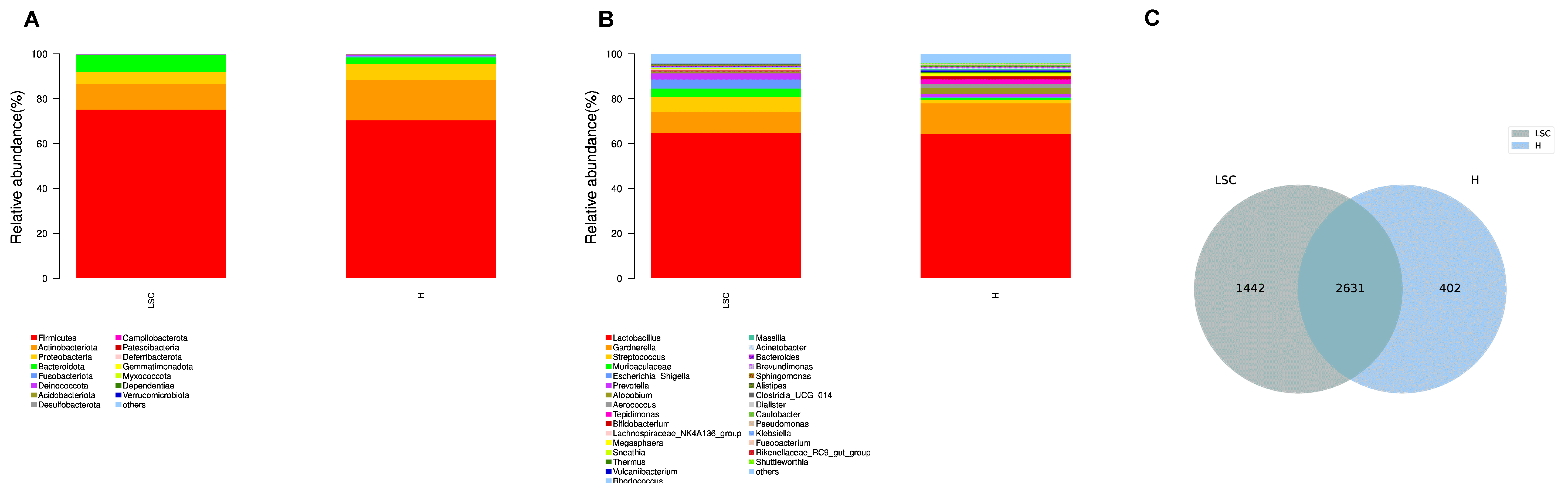 Fig. 1.
Fig. 1.Microbial community structure bar plots of the LSC and H groups. (A) The microbial community bar plots of the LSC and H groups at the phyla level. (B) The microbial community bar plots of the LSC and H groups at the genus level. (C) OTU Venn diagram. The number in the overlapped area indicates the amount of common OTUs of the two groups.
The median of Shannon index of LSC group was
0.79 which was significantly less than the index of H group (1.43, p
 Fig. 2.
Fig. 2.Alpha diversity comparison of LSC and H groups. Violin
plots of the Shannon (A) and Simpson (B) index for the two groups. ‘**’ indicates
p
 Fig. 3.
Fig. 3.PCoA analysis of the LSC and H groups. Some of the H group samples cluster separately from the LSC samples.
The analysis of similarity (anosim) of binary_jaccard revealed LSC group differed from H group significantly (p = 0.001). The top 5 discriminal genus with most relative abundance were Gardnerella, Atopobium, Tepidimonas, Sneathiaand and Massllia (Fig. 4). To identify the specific taxa, the LEfSE analysis was performed. Comparing with group H, family Leptotrichiaceae and genus Sneathia were discriminant taxa in group LSC (Fig. 5).
 Fig. 4.
Fig. 4.The box plot of the top 10 different genera in the LSC and H groups.
 Fig. 5.
Fig. 5.LEfSE analysis of the LSC and H groups. The rings represent the taxonomic levels in descending order, from phylum to genus. The circles in red or green represent more prevalent taxa of H or LSC groups. The diameters of the circles represent the relative abundances of the taxa.
To gain the function of vaginal microbiota in the etiology of VLSC, PICRUSt using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways was used to predicate the functional profile of bacterial communities. It found that in addition to the difference detected in bacterial composition and diversity, microbial genes related to the signal transduction, metabolism of terpenoids and polyketides, transporters, excretory system, nervous system, energy metabolism and many other functions were extremely enriched among the LSC group comparing with H group, while genes related to immune system, cardiovascular diseases and digestive system, etc were reduced in LSC group (Fig. 6).
 Fig. 6.
Fig. 6.Heatmap summarizing the predicted functional factors based on KEGG databases in the level 2.
In the recent dermatopathologic view, LSC is inflammatory dermatoses [9], but the mechanism of inflammation is not clear. So far, some gynecological diseases have been found associated with gut microbiota or vaginal microbiota dysbiosis. Chandni et al. [10] summarized that gut microbes might promote endometriosis lesion formation in the state of dysbiosis through disrupting gut barrier integrity and resulting macrophage activation. Pierluigi et al. [11] also found the lower genital tract microbiome would be affected by changes in polycystic ovary syndrome (PCOS). Both vaginal microbiota and vulvar skin microbiota can influence the status of labium skin. Compared with vulvar skin microbiota, vaginal microbiota is relatively more stable. Taking these into account, we tried to screen potential etiology by studying the composition of vaginal microbiota in women with VLSC through high phylogenetic resolution sequencing in the present research.
In our study, it was found that the LSC group had less alpha microbiota diversity than H group and the beta diversity of the two groups were different. Comparing with H group, the Sneathia was the discriminant genus and increased significantly in relative abundance in LSC group. Haggerty et al. [12] found that the presence of Sneathia in bacterial vaginosis was associated with increased risk of pelvic inflammatory disease. Muzny [13] found that as the secondary colonizers, Sneathia spp. and potentially other bacterial vaginosis-associated bacteria were more potent stimulators of the host-immune response to bacterial vaginosis and likely contribute to its signs and symptoms as well as its adverse outcomes. Another study [14] revealed that the presence of Prevotellaamnii and Sneathia sanguinegens was significantly associated with infection of Trichomonas vaginalis. All the findings indicated that VLSC was related to vaginal microbiota disturbance and the increased abundance of Sneathia in vagina might facilitate the secondary bacterial infection.
According to the PICRUSt analysis, it has been found that the function of the
vaginal microbiota differed in the LSC and H groups. For example, genes related
to folate, cysteine and methionine metabolism increased in LSC group in our
study. Fan et al. [15] compared the MTHFRC677T gene polymorphism between
the nonneopastic epithelial disorders of vulva and the normal control cases and
indicated that VLSC patients had significantly increased frequencies of MTHFR
677TT genotype and T allele which was associated with the folic acid and
homocysteine metabolism. Another study [16] investigated the changes of
transforming growth
factor-
So far, due to the lack of understanding of VLSC and its multifactorial nature, it is difficult to manage VLSC. Topical corticosteroids were accepted as the first line therapy and many other therapies such as topical immunomodulators, antihistamines, antidepressants, phototherapy, transcutaneous electrical nerve stimulation and focused ultrasound were also accepted as adjuvant therapy, but all the methods could only relieve the symptom and had a certain recurrent rate [17]. Now, the changes of microbiota in VLSC women may give hints for disease management that the vaginal probiotics might help regaining the vaginal microbial balance and assisting in therapy.
The richness and diversity of vaginal microbiota in VLSC group is different from H group. VLSC is associated with disturbed microbiota profiles.
The raw reads generated and analyzed during the current study are available in the Genome Sequence Archive (GSA) database. [https://ngdc.cncb.ac.cn/gsa/] (Accession Number: CRA005753).
LM took part in the data curation, formal analysis, investigation and drafting the article. YC, DW, TC, YZ and JM were involved in clinical resources. XZ, YL, QW and LD were involved in methodology. TW was responsible for methodology and project administration. XN made conceptualization, funding acquisition and final review of the manuscript. All authors read and approved the final manuscript.
This study was approved by the Institutional Ethics Committee of Sichuan University West China Second University Hospital (NO.1356) and the work has been carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans.
Not applicable.
This study was supported by the National Key Research and Development Program of China (2021YFC2009100) and the foundation of Science & Technology Department of Sichuan Province (2019YJ0044).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.