
† These authors contributed equally.
Background: Aberrant gene expression, including protein-coding and
non-coding genes (like long non-coding RNA, lncRNA), is associated with cervical
cancer development. To reveal the possible molecular mechanisms of cervical
carcinogenesis, this study conducted high throughput sequencing along with a
bioinformatics analysis. Methods: The differentially expressed lncRNAs
and mRNAs were assessed using a microarray technique in three pairs of cervical
cancer and paracancerous tissues and analyzed using the Gene Ontology (GO) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotations. Their
co-expression profiles, containing 653 nodes and 400 edges, were constructed and
analyzed using Pearson’s correlation and lncRNA-mRNA co-expression network
analyses. Results: There were 242 lncRNAs and 169 mRNAs upregulated and
1204 lncRNAs and 1131 mRNAs downregulated in cervical cancer (fold change
Cervical cancer is the second leading cause of morbidity and mortality among all gynecological malignancies [1]. Approximately 95% of cervical cancer is caused by infection with a high-risk persistent human papillomavirus (HR-HPV) [2], including 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 68, 69, and 82 HR-HPV subtypes [3]. Molecularly, cervical carcinogenesis, like most other human cancers, is mainly related to the activation of oncogenes or deactivation of tumor suppressor genes, which may also involve the integration of viral DNA with genomic DNA of human chromosome [4, 5]. Over the past 60 years, the simple and economic Papanicolaou (Pap) smear and cervical cytology have been widely used in the early detection of cervical cancer [5]. Traditional chemotherapy and radiotherapy are by far the most common treatments for advanced cervical cancer, but present serious side effects [6, 7]. Thus, research on the pathogenesis and biological functions are urgently needed for the early detection, treatment, and monitoring of cervical cancer.
Genetically, more than 70% of the human genome sequences has been transcribed into non-coding RNAs (ncRNAs) [8]. Long non-coding RNAs (lncRNA), a class of RNA molecules with transcripts longer than 200 nucleotides (nt), have shown to play an important role in human cancer development and progression via the epigenetic, transcriptional, and post-transcriptional regulation of protein-coding genes [9]. In general, the molecular mechanism of lncRNA can be categorized in four aspects, i.e., signals, decoys, guides, and scaffolds [10]. LncRNA interacts with DNA, RNA, and proteins to affect the formation and function of the secondary and tertiary structures of these molecules. When localized in the nucleus, lncRNAs can guide chromatin-modifying complexes or transcription factors, while in the cytoplasm, it can control the stability of mRNA or compete with endogenous mRNA for protein expression mechanisms [11]. miRNAs can also downregulate expression of target gene mRNA in cytoplasm by binding to the seed region of the three prime untranslated region (3’-UTR). LncRNAs can compete for large numbers of miRNAs in the cells, acting like a sponge and reducing or interfering with the ability of the target gene mRNA to encode proteins. Recently, there is growing evidence that shows that lncRNAs and miRNAs possess particular expression in cervical cancer and different pathophysiological stages of tumor growth, confirming the involvement of certain lncRNAs in cervical carcinoma [12, 13, 14, 15, 16]. Despite these findings, the expression patterns, targets, and functions of lncRNAs involved in the pathogenesis of cervical cancer remain largely unknown. Therefore, further research is of great importance.
MTMR3, a member of the myotubularin family, is an inositol lipid 3-phosphatase that can hydrolyze PtdIns3P (PI3P) [17]. PI3P is required for the process of autophagy [18]. Some studies have reported that the suppressive effect of MTMR3 on PI3P can inhibit autophagosome formation [19]. Nevertheless, whether MTMR3 is involved in the regulation of cervical cancer has not been elucidated. Recently, both lncRNA and mRNA have been identified as a new class of gene expression regulators [20], although the functions of most lncRNAs remain to be elucidated. In contrast, the alteration and function of some differentially expressed mRNAs have also been recognized in cervical cancer. In this study, we assessed the differentially expressed lncRNA in cervical cancer tissue samples using microarray analysis and analyzed the lncRNA-mRNA network to explore the possible molecular mechanisms of cervical carcinogenesis.
In this study, we collected three pairs of invasive cervical cancer tissues (S102_TN_5_NS, S369_TN_3_NS, S372_TN_2_NS) and paracancerous tissues (Ctl_1_NS, Ctl_2_NS, and Ctl_3_NS) from the Obstetrics and Gynecology Hospital of Fudan University (Shanghai, China). These patients were histologically diagnosed with squamous cervical carcinoma (SCC) or adenocarcinoma and classified according to the National Comprehensive Cancer Network (NCCN) criteria [21]. Immunohistochemical results showed that these tumor tissues were CK7(+), P16(+), and Ki-67(+). The clinical characteristics of these cases are shown in Table 1. Tissue specimens were collected from the surgery room and snap-frozen for RNA isolation (see next subsection). This study was approved by the Ethics Committee of the Obstetrics and Gynecology Hospital of Fudan University (2018-11).
| Sample ID | Histologic diagnosis | Group |
| Ctl_1_NS | Normal | N_1 |
| Ctl_2_NS | Normal | N_2 |
| Ctl_3_NS | Normal | N_3 |
| S369_TN_3_NS | SCC | SCC-1 |
| S372_TN_2_NS | SCC | SCC-2 |
| S102_TN_5_NS | Adenocarcinoma | Adenocarcinoma-1 |
Total RNA was isolated using a RNAiso Plus kit (cat. #9109; Takara Bio, Kusatsu, Shiga Prefecture, Japan) according to the manufacturer’s instructions, quantified using a spectrophotometer, and then analyzed with an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) for RNA integrity. The resulting RNA samples were further purified with the NucleoSpin RNA Clean-up XS kit (Macherey Nagel, Strabe, Germany) and RNase-free DNase set (Qiagen, GmBH, Germany) according to the manufacturers’ protocols.
Six human ceRNA arrays (4
The microarray data of differentially expressed lncRNAs and mRNAs in cervical cancer tissues vs. normal ones were first analyzed using the GO database (http://www.geneontology.org/) for their functional categories as the specific biological GO terms [22]. Then, we performed the pathway analyses of the differentially expressed lncRNAs and mRNAs using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database, assisted by the Shanghai Biotechnology Corporation, to examine their potentially related gene pathways in cervical cancer tissues vs. normal ones.
We further predicted and evaluated the targeting mRNAs of the differentially
expressed lncRNAs for their cis/trans-regulatory effects on
mRNAs. Specifically, cis-targeted mRNAs were considered as targets of
lncRNAs when the mRNA genes were localized within 10 kb of the paired lncRNAs,
while trans-targeting occurred when lncRNAs contained the specific gene
sequences. After selecting the complementary or similar gene sequences paired
with lncRNAs using the Basic Local Alignment Search Tool (BLAST) online software,
we calculated the RNA duplex energy level between the two sequences using the RNA
duplex energy parameter of
To assess the relationship between lncRNAs and mRNAs, we conducted the
lncRNA-mRNA co-expression network analysis of differently expressed lncRNAs
(total of 1446, including 242 upregulated and 1204 downregulated) and mRNAs
(total of 1300, including 169 upregulated and 1131 downregulated) using Pearson’s
correlation coefficients (PCCs). The lncRNA-mRNA co-expression network was
constructed by Cytoscape software (Version 3.7.1; The Cytoscape Consortium, San
Diego, CA, USA) and analyzed statistically. Correlation with a value of –0.99
To verify the microarray data, we selected 15 mRNAs and 10 lncRNAs randomly and
assessed their expression levels in three cases of cervical cancer and normal
tissue specimens using RT-qPCR. Total RNA was isolated from tissue samples and
reversely transcribed into cDNA using an RNAiso Plus kit (Takara) and HiScript II
Q RT SuperMix (Vazyme Biotech, Nanjing, China), respectively, according to the
kits’ instructions. These cDNA samples were subjected to qPCR amplification using
the SYBR Green RT RCP master mix (Genetech, Shanghai, China) with specific
primers in a 7900 HT Sequence Detection System (Applied Biosystems, Foster City,
CA, USA). The primers of mRNAs and lncRNAs were designed and synthesized by
Sinotech Genomics (Shanghai, China) and are listed in Table 1. The relative
fold-change of each mRNA and lncRNA was calculated using the
2
The data were statistically analyzed using SPSS (Version 17.0; SPSS Inc.,
Chicago, IL, USA) and are presented as the mean
To assess the RNA expression profiles in cervical cancer, we first conducted a
whole transcriptome microarray analysis in three pairs of cervical tumor and
normal tissues using the ceRNA chips (Shanghai Biotechnology, China), which
contain 88,371 circRNA probes, 68,423 lncRNA probes, and 18,853 mRNA probes. A
total of 1300 DE-mRNA and 1446 DE-lncRNA transcripts were identified in cervical
cancer development with a threshold of fold-change
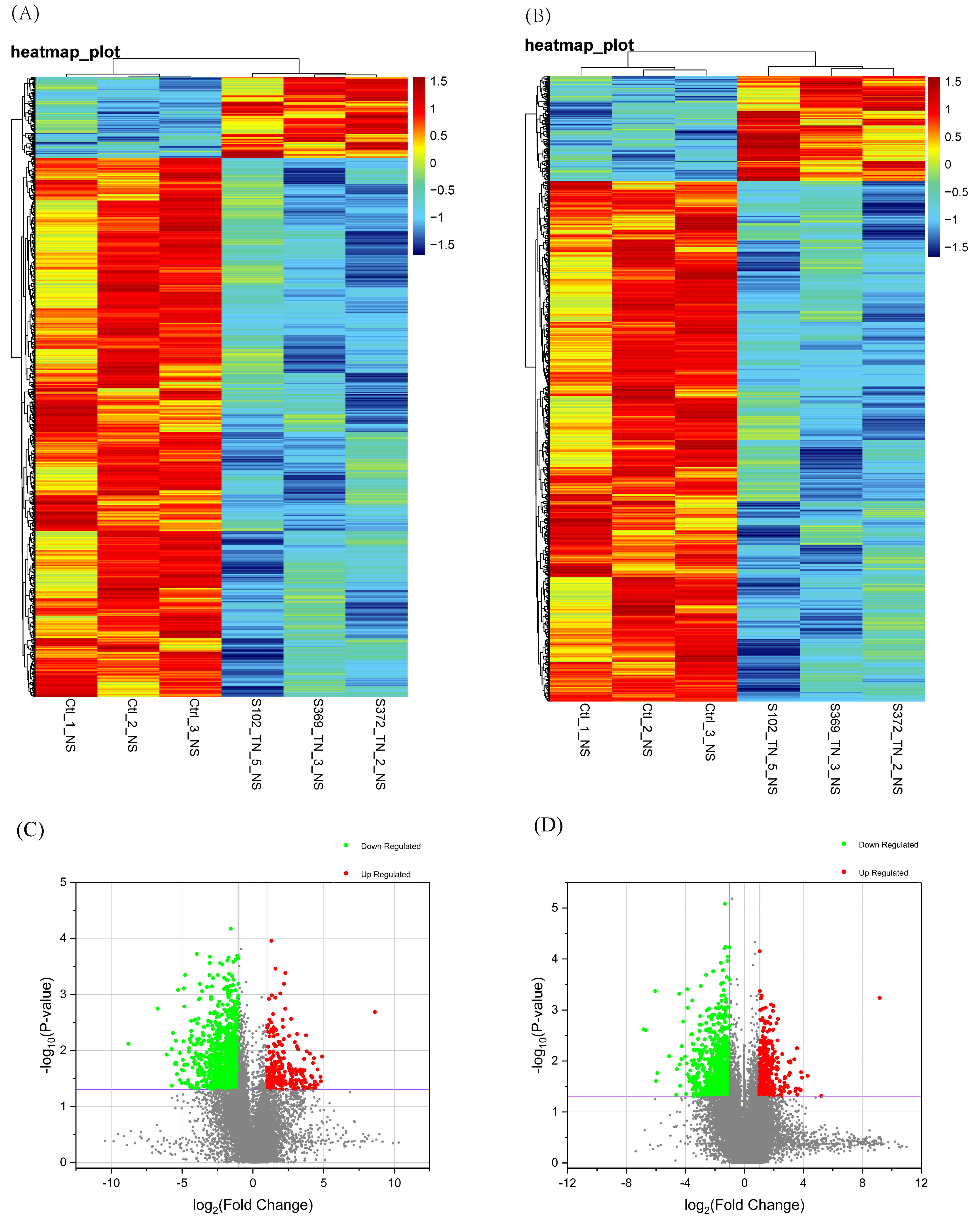 Fig. 1.
Fig. 1.Identification of differentially expressed mRNAs and lncRNA in
cervical cancer. (A) Heat maps of differentially expressed mRNAs. (B)
Heat maps of differentially expressed lncRNAs, which are based on the expression
values of all lncRNAs and mRNAs detected through the microarray analysis. (C)
Volcano plots of differentially expressed mRNAs. (D) Volcano plots of
differentially expressed lncRNAs, and cutoff criterion was fold-change of
To explore the possible functions of lncRNA, we predicted its potential targets
in cis-regulatory relationships by searching for protein-coding genes
10-kb upstream and downstream of all the identified lncRNAs. To gain further
insight into the potential biological and pathophysiological processes that may
be moderated by these DE-RNAs in cervical cancer, we performed the GO term and
KEGG pathway analyses. Concerning the biological process, we found that DE-lncRNA
was mostly enriched in extrinsic apoptotic-signaling pathway via death domain
receptors and was involved in nuclear-transcribed mRNA poly(A) tail shortening
and metalloaminopeptidase activity. The top 30 GO terms are shown in Fig. 2A.
These results suggest that one of the primary roles of lncRNA may be
post-transcriptional regulation of gene expression. Undistinguishably, the GO
terms of DE-mRNA were mainly enriched in protein K11-linked ubiquitination,
mitochondrial fusion, and the anaphase-promoting complex. The KEGG pathway
analysis indicates that these up- and downregulated mRNAs function in
ubiquitin-mediated proteolysis and molecular pathways involved in cancer,
respectively (Fig. 2A). In particular, the lncRNAs and cis-targeted
mRNAs showed to play in a role in the TGF-
 Fig. 2.
Fig. 2.Data obtained from the Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of differentially expressed mRNAs and lncRNAs in cervical cancer. (A) GO term annotations and KEGG pathway enrichment analysis of mRNAs with top 30 enrichment scores. (B) KEGG pathway enrichment analysis of cis-targeted lncRNAs and trans-targeted lncRNAs with the top 30 enrichment scores.
The functions of lncRNA primarily depend on interaction with mRNAs, which may be
an important target of the former. It has been reported that the functions of
most lncRNAs include the targeting of transcription, translation, and
post-translation of their targeting mRNAs. To gain a better understanding of
these DE-RNAs in cervical cancer, we determined the potential targets of lncRNAs
in cis-regulatory relationships and performed a co-expression analysis
using Pearson’s correlation analysis with Psych packages (Corr. Test method,
parameter CI = F, adjust = BH) in the R program. The screening correlation
coefficient was determined to be r
 Fig. 3.
Fig. 3.Data on the differentially expressed lncRNA-mRNA co-expression
network. The lncRNA-mRNA co-expression network was constructed using the
Cytoscape software (https://cytoscape.org/,
version 3.7.0) and analyzed statistically. Correlation values of –0.7
| mRNA | Gene symbol | Connecting RNAs |
| NM_033318 | SMDT1 | NR_029480, NR_029514, lnc-SIPA1L3-3:7, lnc-RSPO3-2:3, ENST00000362160, lnc-ACTR8-1:2 |
| NM_021090 | MTMR3 | lnc-C5orf30-6:1, lnc-AC016745.1-2:1, lnc-LPL-2:1, ENST00000391313 |
| NM_021964 | ZNF148 | lnc-RSPO2-2:1, lnc-CNTNAP1-2:1, lnc-PAX5-7:2 |
| NM_003026 | SH3GL2 | lnc-UQCRFS1-9:1, ENST00000562082, lnc-ZNF570-2:1 |
| NM_018127 | ELAC2 | lnc-CNTNAP1-2:1, lnc-PAX5-7:2, lnc-FAM83F-4:1 |
| NM_017934 | PHIP | lnc-ZNF101-6:1, lnc-TM4SF1-4:1, ENST00000601239 |
 Fig. 4.
Fig. 4.Data on GO terms and KEGG analysis of genes in the differentially expressed lncRNA-mRNA co-expression network. (A) GO analysis genes in co-expression network. (B) KEGG analysis of genes in co-expression network.
To assess the prognostic value of these above hub genes, we searched and downloaded data from the online database GEPIA2 (http://gepia2.cancer-pku.cn/#index), which contains gene expression and overall survival data. Importantly, GEPIA2 is an enhanced web server for large-scale gene expression profiling and interactive analysis that includes 198,619 isoforms and 84 cancer subtypes from TCGA and GTEx samples [23]. Our results demonstrate that MTMR3 has a lower expression in cervical squamous cell carcinoma and endocervical adenocarcinoma. Based on the Kaplan-Meier data analysis data, patients with higher MTMR3 expressed tumor had poor overall survival (OS), although the p-value was not statistically significant (HR = 0.94, p-value = 0.78; Fig. 5A). Similarly, the low expression of SMDT1 was also correlated with poor overall survival (OS) (HR = 0.65, p-value = 0.077; Fig. 5B). As for the other hub genes, association of their expression with survival of patients was not statistically significant (Supplementary Fig. 1).
 Fig. 5.
Fig. 5.Different expressions of the hub genes in cervical cancer vs. normal tissues and association with overall survival. (A and C) Expression levels of MTMR3 or SMDT1. (B and D) Overall survival analysis using Kaplan-Meier curves and the log-rank test.
To verify the reliability of our microarray analysis results, we performed RT-qPCR validation in 15 randomly selected mRNAs and 10 randomly selected lncRNAs from cervical cancer and normal tissue samples (Primers used were shown in Table 3). We found that two lncRNAs (lnc-CLRN3-3:1 and lnc-TMEM238-2:1) were upregulated in cervical cancer tissues, whereas ENST00000369391, ENST00000547898, ENST00000561507, lnc-SEMA3A-2:1, lnc-STOM-1:1, NR_110418, and NR_026867 were verified to be downregulated in cervical cancer tissues vs. those of the control tissues (Fig. 6A). Moreover, according to the validation of differentially expressed mRNAs, we discovered that CCNE2, FN1, INHBA, and UBE2S were upregulated, while APPL1, GSN, KAT2B, MITF, PIK3R1, PRICKLE1, SAV1, SMAD4, TCF7L2, TGFBR2, and UBE2D3 were downregulated in cervical cancer tissues vs. control tissues (Fig. 6B). The qPCR results confirm that the expression patterns of these RNAs are consistent with their expression levels calculated from the RNA-seq data (The comparative results were shown in Supplementary Fig. 2), which indicates the reliability of high-throughput sequencing.
 Fig. 6.
Fig. 6.RT-qPCR validation of microarray data in cervical cancer vs. normal control tissue specimens. (A) Relative expression levels of lncRNAs in cervical cancer tissues. (B) Relative expression levels of mRNAs in cervical cancer tissues.
| Gene name | DNA sequences | |
| PIK3R1 | 5’-TCTTTTGACGAGAGGGAGGA-3’ | 5’-GTCAAGGAGGCACACTACCC-3’ |
| TCF7L2 | 5’-ACCATCTTCGTTTCCCCTTT-3’ | 5’-GGTCAGTCCGGGTCCTAAG-3’ |
| PRICKLE1 | 5’-CGTCCCACACAAGGGTTT-3’ | 5’-GCACCAGACCAGGGAATGT-3’ |
| GSN | 5’-ACGGCTGAAGGACAAGAAGA-3’ | 5’-TTCGATCACAAAACGTCCAA-3’ |
| CCNE2 | 5’-CATGGTGTTCAACCTGTGCT-3’ | 5’-CATGTCAAGAAAGCCCCAGT-3’ |
| KAT2B | 5’-CATAACCCCCTAAAATCCATCA-3’ | 5’-CAGAGTTGGGAATGGCAGAT-3’ |
| MITF | 5’-TGGATTCATTTCAGGGGCTA-3’ | 5’-GGGAGTGGTGTGGATCATTT-3’ |
| UBE2D3 | 5’-GTTTGTTGAAATGGCTTAGTTGA-3’ | 5’-CAAAACATGGCTGAGTTACCC-3’ |
| UBE2S | 5’-GCATGAGGGAAGCAAACAGTC-3’ | 5’-TTTCAGTGGGTTCCGAGGC-3’ |
| TGFBR2 | 5’-GGGTGGGCTGAGAGTTAAAG-3’ | 5’-AGGTCAATGGGCAACAGCTA-3’ |
| FN1 | 5’-AGCATCACCCTGGGAGTTT-3’ | 5’-CGAAGCAGAACAGGCAATG-3’ |
| INHBA | 5’-TGCACCTTTTGGCAACTTCT-3’ | 5’-TACAATGTCCACCCCCAAAC-3’ |
| SMAD4 | 5’-ATGCCAGAAGCCAGAGAAGA-3’ | 5’-CCTGGGACTTTCAACTGACC-3’ |
| APPL1 | 5’-TGTAAAACCTGCCACCTGAA-3’ | 5’-CCAAGATCACACCACTGCAC-3’ |
| SAV1 | 5’-GACGAGGCAGTGGAGAGTTC-3’ | 5’-CCCTGGTTGTCATTTTTGGT-3’ |
| ENST00000369391 | 5’-CCAAGCCCAGGAAACAGATA-3’ | 5’-CCAAGCCCAGGAAACAGATA-3’ |
| ENST00000547898 | 5’-TGACACCCTCCTTCTCACAG-3’ | 5’-TCCTCTCCATGCTTTCTTCC-3’ |
| lnc-STOM-1:1 | 5’-TGCCCAGTGGTGATGAGGTT-3’ | 5’-GGCCTGAAGGGTTCTTGGA-3’ |
| NR_110418 | 5’-CCAGCAATAGACCCTCCTTCAA-3’ | 5’-ACCGGTGGATCCTGCTTTC-3’ |
| ENST00000561507 | 5’-CCATCATTTTTCCAGCAGTT-3’ | 5’-AGGGGGTGGCATTAAAAGAG-3’ |
| lnc-SEMA3A-2:1 | 5’-TCGTGGATGACCTCTCCTATTGT-3’ | 5’-CCCCCAAATTAGTGCCGTAT-3’ |
| lnc-TMEM238-2:1 | 5’-AGACCCAGGACACCCAAGTA-3’ | 5’-ACAAACTCACCACTCGTTGC-3’ |
| lnc-CLRN3-3:1 | 5’-GATCTTTCAGGTAAAAACGTAGTCT-3’ | 5’-TTCACAGCAGATAAACAAAGTTAGC-3’ |
| lnc-IGSF11-5:1 | 5’-AAGGTGCTGGCAGATCTAGTTGT-3’ | 5’-TTGCCCTGTGAAGACATAGTAAGAA-3’ |
| NR_026867 | 5’-AAGCCTTCAGGGTCAGTTCA-3’ | 5’-AGTCCCCAAAATCATCAAATCCA-3’ |
The risk in developing cervical cancer is higher in developing countries [24], where there is an increase in prevalence of HVP infection. In addition, the underlying molecular mechanisms of cervical carcinogenesis is still poorly understood, lncRNAs have been reported to be altered in many cases of cervical cancer [1, 25, 26, 27]. In the current study, we conducted a microarray analysis of differentially expressed lncRNAs and mRNAs and then performed the integrated bioinformatical analysis of these differentially expressed lncRNAs and mRNAs. In the present study, we identified a total of 2746 differentially expressed lncRNAs and mRNAs using the ceRNA microarray chips. Of these, 242 lncRNAs and 169 mRNAs were found to be significantly upregulated, and 1204 lncRNAs and 1131 mRNAs were downregulated in cervical cancer tissues compared with the control tissues. In addition, various dysregulated lncRNAs and mRNAs were randomly chosen for qRT-PCR validation, and the results confirm the microarray analysis findings to some extent.
To predict the potential functions of the differentially expressed lncRNAs
identified in this study, GO and KEGG pathway analyses were performed using the
coding genes associated with the significantly differentially expressed lncRNAs.
Our GO term and KEGG pathway analyses revealed that different biological events
and signaling pathways may be associated with cervical carcinogenesis. The GO
term analyses divided these differentially expressed mRNAs in cervical cancer
tissue specimens into functional modules of cell process, like protein
ubiquitination, cell migration, cell proliferation, and immunity. Our KEGG
pathway analysis revealed that these differentially expressed mRNAs are involved
in ubiquitin-mediated proteolysis and diverse cancer pathways. Protein
ubiquitination was also reported to regulate the functions and signaling of a
profusion of proteins in various cell pathways [28, 29], while the chains of
K11-linked polyubiquitination participate in cell mitosis [30, 31]. Furthermore,
our pathway analysis of these differentially expressed lncRNAs revealed their
association with CAMs, TGF-
Furthermore, we also employed a lncRNA-mRNA network analysis to identify
interactions between differentially expressed mRNAs and differentially expressed
lncRNAs, as previously described [32, 33]. Our results showed that a total of 328
lncRNAs and 315 mRNAs were included in the co-expression network, which consisted
of 653 network nodes and 400 connections. We also found that SMDT1, an mRNA, was
correlated with up to 6 lncRNAs; MTMR3 was correlated with 4 lncRNAs; and ZNF148,
SH3GL2, ELAC2 and PHIP were associated with 3 lncRNAs. Particularly, as a member
of the myotubularin family, MTMR3 has demonstrated to participate in tumor
development in oral [34], gastric [35], and breast cancers [36]. According to the
functional analysis, the integrated mRNA-lncRNA co-expression network was linked
to autophagy, regulation of vascular genesis, transcriptional repressor complex,
and regulation of gene expression. Based on previous studies, vascular genesis
and angiogenesis are under the tight regulation of growth factors, like
transforming growth factor-
Overall, the presented study confirms the usefulness of the cDNA microarray to identify the differentially expressed genes in human cancer. Further research is warranted to verify and evaluate these genes as biomarkers for the early detection and prognosis of cervical cancer.
Despite our promising findings, this study is preliminary, and deeper research is needed to confirm the importance of these differentially expressed mRNAs and lncRNAs in cervical cancer development. Therefore, the limitations of this study include the small sample size and limited data for analysis. In conclusion, this study effectively profiled differentially expressed mRNAs and lncRNAs in cancerous tissues and confirmed their association with the development of cervical cancer. Future study of some of these differentially expressed mRNAs and lncRNAs using a larger sized cervical cancer tissue sample is highly recommended.
TJ contributed to the conception of the study, acquisition of the financial support, oversight, and leadership responsibility for the research activity planning and execution. LYY conducted the research, performed the data analyses, and wrote the manuscript. ZJF contributed significantly to the provision of the study’s patients and laboratory materials. PJQ helped perform the analysis with constructive discussions.
Patients have given their written informed consent before enrolled into our study. The ethics committee of the Obstetrics and Gynecology Hospital of Fudan University approved our study protocol with a reference number of 2018-11.
We are grateful to Shanghai Biotechnology Corporation (http://www.shbio.com/, Shanghai, China) for providing technical support.
This study was supported in part by grants from the Shanghai Key Specialty Project of Clinical Pharmacy (#AB83110002017005) and Science and Technology Support Planning of Shanghai Science and Technology Commission (#19401900900).
The authors declare no conflict of interest.