




1 Department of Medical Genetics and Fundamental Medicine, Bashkir State Medical University, 450008 Ufa, Russia
Abstract
Frontotemporal dementia (FTD) develops in proteinopathies involving TDP-43 (transactive response DNA-binding protein 43 kDa), tau, and FUS (fused in sarcoma) proteins, which possess antiviral properties and exert inhibitory effects on human transposable elements. Viruses and aging have been suggested to trigger FTD by activating specific retroelements. FTD is associated with multiple single nucleotide polymorphisms (SNPs), most located in intergenic and regulatory regions where many transposable element genes are found. Therefore, genetic predisposition to FTD may influence the interaction between retroelements and the TDP-43, tau, and FUS proteins, causing pathological conformation changes and aggregate formation. Subsequently, these aggregates lose their ability to inhibit retroelements, leading to the activation of transposable elements. This creates a harmful negative feedback loop in which TDP-43, tau, and FUS protein expressions are further enhanced by retroelement transcripts and proteins, resulting in protein aggregate accumulation and pathological disease progression. Hence, epigenetic inhibition of pathologically activated retroelements using micro-ribonucleic acids (microRNAs) derived from transposable elements has been proposed as a potential treatment for FTD. Finally, a review of the current scientific literature identified 13 appropriate microRNAs (miR-1246, -181c, -330, -345-5p, -361, -548a-3p, -548b-5p, -548c-5p, -571, -588, -659-3p, -708-3p, -887).
Keywords
- antiviral proteins
- viruses
- frontotemporal dementia (FTD)
- microRNA
- retroelements
- aging
Frontotemporal dementia (FTD) presents as behavioral or language disorders, characterized by significant changes in social and personal behavior, blunted emotions, apathy, deficits in both receptive and expressive language, and atrophy of the frontal and temporal lobes of the brain [1]. FTD is the second most common presenile dementia after Alzheimer’s disease [2]. The incidence of FTD in Europe averages 2.36 per 100,000, with a marked increase with age, peaking at age 71 (13.09 per 100,000 for men and 7.88 per 100,000 for women) [3]. In 60% of cases, FTD is a multifactorial disease, while in 40%, it is autosomal dominant due to mutations in the MAPT (microtubule associated protein tau), GRN (progranulin), C9orf72 (chromosome 9 open reading frame) genes [4].
The pathogenesis of FTD, in both familial and sporadic forms, involves the accumulation and misfolding of three primary proteins: TDP-43 (transactive response DNA binding Protein 43 kDa), FUS (fused in sarcoma protein) and microtubule-associated protein Tau, leading to the formation of pathological intracellular aggregates [1]. In 50% of FTD patients, TDP-43 pathology is detected, while 40% show frontotemporal lobar degeneration (FTLD) with tau pathology, and the remaining 10% display FUS and UPS (ubiguitin/p62) proteinopathies. Sporadic cases of FTD have been linked, through numerous genetic studies, to single nucleotide polymorphisms (SNPs) in various gene loci, such as BTNL2, HLA-DRA, HLA-DRB5, RAB38, TMEM106B [5], MOBP, SORT1, PGRN [6], DPP6, HLA-DQA2 [7], UNC13A, TBK1, VIPR1, RBPJL, L3MBTL1, and others [8], including genes associated with Alzheimer’s and Parkinson’s diseases (APOE, HLA, MAPT). Most FTD-associated polymorphisms are found in introns, untranslated regions (UTRs), or intergenic regions [9], which is typical for multifactorial diseases [10].
Thus, FTD is one of the common neurodegenerative diseases [2] associated with aging [3] and in most cases is a multifactorial disease [4]. According to the latest research results in the current area of research, many polymorphisms have been identified in the human genome [5, 6, 7, 8, 9], the identified association of which with frontotemporal dementia cannot be explained, since there are very many of these SNPs and since most of these SNPs are located in intronic and intergenic regions. Therefore, this article aims to explain the mechanisms of influence of multiple SNPs on the development of frontotemporal dementia in terms of changes in the activity of retroelements (REs) located in intergenic and intronic regions (where FTD-associated polymorphisms are located). Moreover, the potential academic value of the mechanisms of REs influence on the development of FTD described in this article is due to the fact that the “vicious circle” of interactions between REs and antiviral proteins under the influence of aging, viruses and SNPs located inside retroelements, identified in the pathogenesis of FTD, may be a universal mechanism for the development of other neurodegenerative diseases. Therefore, currently used treatment methods for other neurodegenerative diseases aimed at suppressing pathological activity of REs can be proposed as promising methods for FTD therapy. The mechanisms of influence of retroelements on the development of FTD described in the article explain the mechanism of influence of disease-associated polymorphisms on the progressive accumulation of tau, TDP-43 and FUS proteins in the brain of patients.
Approximately 15% of FTD patients also exhibit symptoms of amyotrophic lateral sclerosis (ALS), and a subset of ALS patients develop FTD symptoms [11]. This overlap may be explained by shared pathogenetic mechanisms involving proteinopathies, particularly of the TDP-43 protein [2, 12]. Familial forms of ALS and FTD can be caused by mutations in the same genes: C9ORF72, TARDBP, FUS, TIA1, and SQSTM1/p62 [13]. It is plausible that similar epigenetic mechanisms underlie both FTD and ALS. One potential driver of epigenetic regulation is REs - genomic regions capable of relocating to new loci through a copy-and-paste mechanism. REs include human endogenous retroviruses (HERVs) with long terminal repeats (LTR) and non-LTR REs such as long interspersed nuclear elements (LINEs), short interspersed nuclear elements (SINEs), SVAs (SINE-VNTR-Alu) [14]. Abnormal activation of REs has been proposed as a cause of epigenetic abnormalities in FTD. Elevated levels of HERV-K have been detected in the serum and affected brain tissues of FTD patients [15]. Additionally, ERV activation has been observed in CHMP2BIntron5 models of FTD in Drosophila, where the gypsy endogenous retroviruses (ERVs) had a toxic effect on the central nervous system. Genetic blocking of gypsy and inhibition of reverse transcriptase in REs were found to prevent neurodegeneration [2].
Pathological activation of REs in FTD could be linked to individual polymorphisms within RE-associated genes, as REs are primarily located in intergenic, regulatory, and intronic regions of the human genome [16] - regions where most FTD-associated polymorphisms are found [5, 6, 7, 8, 9]. The influence of FTD-associated SNPs localized in REs regions can be illustrated by the ORF1p (open reading frame) translation product of LINE1, which forms cytoplasmic aggregates and shares similarity with RNA-binding proteins implicated in neurodegeneration. Changes in specific amino acids of ORF1p affect retrotransposition efficiency and protein aggregation dynamics. Key FTD proteins co-localize with ORFp-LINE1 RNP particles in cytoplasmic RNA granules, suggesting that FTD-associated polymorphisms in RE regions similarly enhance the ability of RE expression products to form TDP-43 aggregates [17]. An additional factor is the age-related activation of REs [18], as the incidence of FTD rises significantly with age [3]. This activation leads to aseptic inflammation in the brain, with the interferon response and other antiviral systems being triggered by RE expression products [19]. This can explain the neurodegenerative processes observed during physiological aging, resembling those at FTD, but not leading to such serious consequences as in this disease. Indeed, the high frequency of TDP-43 proteinopathy was determined in the elderly with normal intellect [20], as well as the accumulation of tau in the brain during normal aging of people [21]. In this case, mutual potentiation of REs and proteins involved in the pathogenesis of the disease can be observed. Accumulated TDP-43 [22], FUS [23] and tau aggregates contribute to the derepression of REs [24, 25, 26]. It is possible that the proteins TDP-43, FUS, and tau, whose pathological conformations form aggregates, are components of an antiviral system that responds to transcripts and proteins from REs. To better understand the role of REs in FTD pathogenesis, it is necessary to examine more closely the interactions between REs and the proteins TDP-43, FUS and tau.
Despite the pathogenic role of the REs in the development of the FTD, due to the influence of the aging [18] that cause inflammation [19], it should be noted that in the normal brain REs play an important regulatory role. This role is primarily associated with the regulation of genes expression during differentiation of neuronal stem cells, since REs are drivers of epigenetic regulation [14]. The results of programmed activation of retroelements in specific neurons that cause activation of genes involved in these processes [27, 28] are somatic mosaicism detected in nerve cells by REs integrations into their genomes [29]. Compared with the liver and heart, a significantly higher number of retrotranspositions of LINE1 [30], SVA and Alu (Arthrobacter luteus) retroelements are found in the brain of healthy people [31]. As a result of new integrations, unique transcriptomes of individual neurons are created in various areas of the brain, which affects their functioning features [32]. The largest number of REs insertions is determined in the neurogenesis zone – in the dentate gyrus of the hippocampus [33]. At the same time, programmed retrotranspositions affect the expression of precisely those genes that are involved in the differentiation of neurons and their specific functioning [34]. A study of the features of L1 retrotranspositions in more than 30 brain regions revealed many cell lines specific for insertions of various L1 [35]. In experiments on mice, specific LINE1 expression was also shown depending on the central nervous system region and the age of the animal [36]. Although changes in gene activity in these processes may occur not only due to direct integrations into specific regions of the genome (Fig. 1), but also in connection with the role of REs in epigenetic regulation, since REs transcripts can function as long non-coding ribonucleic acids (ncRNA) molecules [37, 38], and also as competitive endogenous RNAs, binding as “sponges” with complementary micro-ribonucleic acids (microRNAs) [39]. This is due to the origin of many microRNAs in evolution from REs, which exert epigenetic control over the functioning of various genes, including in the brain [40]. Therefore, it can be assumed that in the evolution of the brain, a control system was formed for strictly defined REs in the brain, the expression of which does not cause an immune response and the production of antiviral proteins. However, with the pathological activation of unplanned REs, transcripts and proteins of such retroelements can cause immune reactions, inflammation [19], as well as interact with TDP-43 [12], FUS [23] and tau proteins [24, 25, 26].
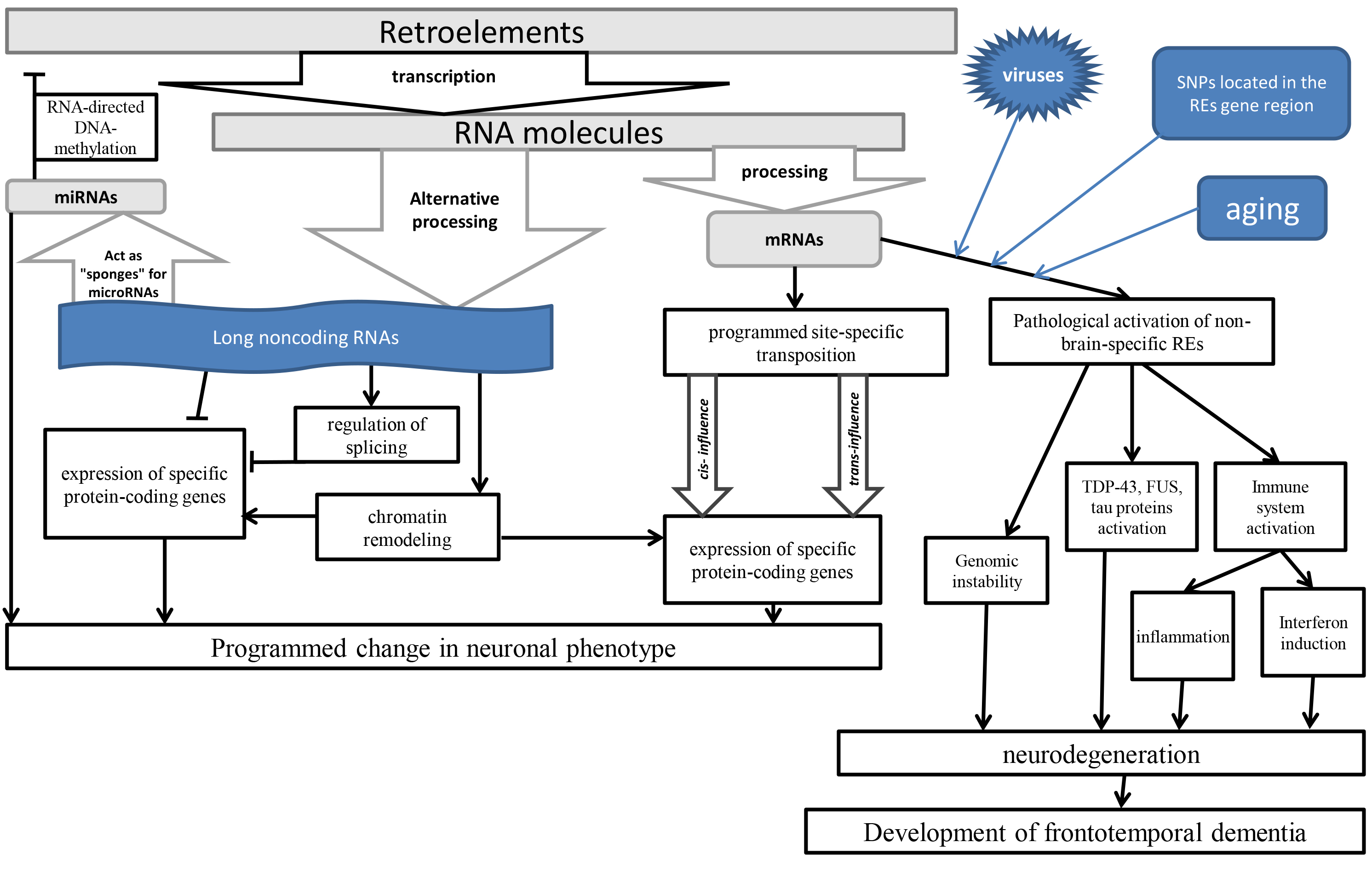 Fig. 1.
Fig. 1.
The scheme of the influence of REs on brain functioning. TDP-43, transactive response DNA binding protein 43 kDa; FUS, fused in sarcoma protein; REs, retroelements; SNPs, single nucleotide polymorphisms; miRNA/microRNA, micro-ribonucleic acids; mRNA, messenger RNA.
Aggregated forms of TDP-43 specific to FTD significantly increase the accumulation of HERV-K viral proteins [41]. In contrast, normal TDP-43 protein plays a crucial role in limiting LINE1 retrotranspositions [17]. TDP-43 protein is involved in the epigenetic regulation of RE expression, as its loss in FTD is associated with chromatin decondensation at LINE loci, leading to their activation [42]. The dysfunction of aggregated TDP-43 may contribute to the observed reduction in methylation of retrotransposition-capable LINE1 in the motor cortex of ALS patients [43]. The study of cerebral cortex of patients who died from ALS has demonstrated that the loss of TDP-43 leads to the overexpression of LINE1 and other REs, as TDP-43 directly binds retrotransposon mRNA [44]. The role of TDP-43 in inhibiting LINE1 has been confirmed in mouse embryonic stem cells and preimplantation embryos, where functional analysis shows that TDP-43 interacts with the ORF1p protein of LINE1, protecting the genome from insertions [45]. In brain samples from FTD patients, TDP-43 aggregates were found to colocalize with the HERV-K pol protein, indicating a pathological interaction in the development of FTD [15].
The role of tau in the epigenetic regulation of REs has been determined - chromatin tags associated with tau have been found at the loci of the HERV-Fc1 location. The conducted profiling of REs in drosophila throughout the brain showed heterogeneous response profiles, including those depending on the age and genotype of REs activation under the influence of tau [24]. In the brain of mice, taupathy leads to activation of endogenous retroviruses (ERVs) [25]. In experiments on SH-SY5Y cells, overexpression of various tau isoforms and their interaction with beta-amyloid led to locus-specific patterns of REs dysregulation (which indicates the participation of strictly defined REs in the pathogenesis of the disease, rather than random or total changes) [26]. The accumulation of tau protein in human postmitotic neurons [46], as well as the derepression of REs [47] was caused by knockout of the same BMI1 (B cell-specific Moloney murine leukemia virus integration site 1) gene [47]. Interferon I activated by retroelements [19] also contributes to the accumulation of FUS by increasing the stability of its mRNA. FUS-expressing cells become hypersensitive to the toxicity of double-stranded RNAs [23].
REs also influence the aggregation of TDP-43, a critical factor in FTD pathology. HERV activation has been shown to dramatically increase prion-like protein propagation in ALS-model cell cultures [48]. Research [22] of RNA-protein interaction and gene expression profiling have revealed extensive binding of RE transcripts to TDP-43. In mouse models of TDP-43 pathology, most REs were derepressed, highlighting the importance of TDP-43 as a protective protein against RE activity [22]. This is further supported by the observation that TDP-43 regulates the accumulation of immunostimulatory double-stranded RNAs formed from REs, preventing them from triggering a lethal RIG-I (retinoic acid-inducible gene)-dependent interferon response [49]. Experiments in Drosophila have shown that TDP-43 proteinopathy and ERV expression mutually reinforce each other [12]. In Drosophila with a null mutation in the TBPH gene (the homolog of TDP-43), increased RE expression was detected in the brain. This increase was attributed to TBPH (TAR DNA-binding protein-43 homolog) proteins normal interaction with Dicer-2 mRNA, promoting the suppression of REs through small non-coding RNAs [50].
A common genetic cause of familial FTD is the expansion of hexanucleotide repeats in the C9orf72 gene, which results in global derepression of transposable elements [51]. This suggests that, like TDP-43, C9orf72 functions as an antiviral protein and an inhibitor of RE expression in the brain [52]. Toxic forms of tau protein also cause heterochromatin decondensation leading to RE activation and subsequent neuroinflammation [53]. This suggests that tau, under normal conditions, inhibits RE expression, and its loss contributes to RE activation in FTD pathogenesis. Additionally, the antiviral response can trigger FUS proteinopathy. FUS becomes incorporated into stress granules, and the resulting aggregates sequester the autophagy receptor optineurin and nucleocytoplasmic transport factors. Virus-activated interferon I further promotes FUS accumulation by stabilizing its mRNA, making FUS-expressing cells hypersensitive to double-stranded RNAs [23]. Overall, the pathogenesis of FTD is associated with the antiviral properties of TDP-43, tau, and FUS, as viruses share an evolutionary relationship with REs [54].
In addition to the relationship with the proteins TDP-43, tau and FUS, REs
influence the pathogenesis of FTD in other ways associated with inflammation and
degeneration in the brain. For example, the RNA of the HERV-K envelope gene has
been shown to bind to human Toll-like receptor 8 (TLR8) and activate it in
neurons and microglia, promoting neurodegeneration [55]. Since ALS and FTD are
characterized by identical developmental mechanisms [11, 12, 13, 14], studies [56] of the
mechanisms of REs influence on ALS development indicate the same mechanisms in
FTD. Thus, the role of REs as inflammatory stimulators in the brain has been
determined in the etiopathogenesis of ALS [56]. The stimulators of the interferon
response and subsequent inflammation in the brain in neurodegenerative diseases
are the most common in the genome LINE1 [57] and Alu [58]. At the same time,
signal inflammation molecules NF-
Since neuronal death or degeneration is the fundamental cause of FTD, it is important to characterize the evidence of recent scientific research on the role of REs in these processes. In an ALS-modeled fruit fly, it was shown that activated REs cause neurodegeneration and activation of DNA damage-mediated programmed cell death [60]. In drosophila, the initiation of toxic expression of human TDP-43 in small groups of glial cells contributed to the death of neighboring neurons caused by endogenous retroviruses in glia. Activated HERVs caused DNA damage and neuron death [12]. HERV-W env has been shown to amplify the expression and activity of human inducible nitric oxide synthase, which contributes to demyelination, oligodendrocyte damage, and disruption of the blood–brain barrier, promoting neuronal apoptosis, axon damage, and neurodegeneration [61].
Past viral infections can act as triggers in the development of FTD. For instance, COVID-19 infection has been implicated in the onset of FTD [1]. Similarly, the role of SARS-CoV-2 [62] and enteroviruses [63] has been reported for ALS, a disease with overlapping etiopathogenesis to FTD. Enterovirus D68, in particular, has been shown to promote TDP-43 aggregation and neurotoxicity [64]. The antiviral function of TDP-43 has been demonstrated through its interactions with Drosha [65] and Dicer [50], as well as through its direct modulation by viral protein during infections caused by enteroviruses [66], HIV (human immunodeficiency virus) [67], and influenza A virus [68]. In FTD patients, aberrant TDP-43 accumulation in hippocampal astrocytes contributes to progressive memory loss and localized changes in antiviral gene expression, correlating with impaired astrocytic defense against viral infections [69].
In experiments on primary hippocampal neurons infected with HSV-1 (herpes simlex virus 1), it was revealed that tau protein acts as an acute antiviral response [70]. Herpesviruses interacting with tau are also REs activators [71], contributing to the development of autoimmune processes in the brain [72]. The FUS protein limits the lytic reactivation of herpes viruses, and depletion of FUS significantly enhances the expression of viral mRNAs and proteins, which leads to the formation of infectious virions [73]. EBV (Epstein-Barr virus), HHV6 (human herpesvirus 6), HSV-1, VZV (varicella zoster virus) viruses transactivate HERV-W promoters, thus acting as inducers of neurodegeneration progression due to immune-inflammatory processes in the brain [74]. The correlation between the autoantites against Peptide EBV (EBNA1386-405) and HERV-W486-504 [75], and infections caused by the Caposhi sarcoma and EBV can cause HRV transactional surfacing [76]. Neuroinflammation and tau hyperphosphorylation have also been observed in HSV-1-infected brains [77, 78], and antiviral treatments such as acyclovir and penciclovir have been shown to reduce tau accumulation [79]. Additionally, the FUS protein, normally involved in the antiviral response, directly inhibits viral replication by physically interacting with viral genomes and its removal leads to increased viral RNA transcription [80].
The hypothesis that viral infections act as FTD triggers via their effects on REs is supported by evidence that the same viruses inhibited by TDP-43 also activate REs. For example, a comparative analysis of colon biopsies and peripheral mononuclear cells from HIV-infected patients revealed the activation of 47 out of 59 REs compared to healthy controls [81]. Increased HERV expression has also been documented in the presence of influenza A [82] and enterovirus infections [83]. Mechanisms involving SARS-CoV-2 in RE activation have been proposed to explain the neurological complications of COVID-19 [84]. HSV-1, which promotes tau proteinopathy [77, 78], is also known to activate RE expression [71, 85]. Given this, it can be suggested that FTD develops through a combination of genetic predisposition, such as SNPs in RE loci, and environmental factors like aging and viral infections. These factors likely promote RE activation, leading to the formation of aggregates as REs interact with TDP-43, tau, and FUS. Since these proteins normally suppress the expression of viruses and REs, their dysfunction - triggered by pathologically activated REs - creates a “vicious circle”. This dysfunction eliminates the inhibitory effects of TDP-43, tau, and FUS on RE expression, further accelerating the progression of FTD.
REs play a key role in the epigenetic regulation of the human genome [14], and their hyperactivation in FTD pathogenesis can influence the expression of specific microRNAs, many of which evolved from transposable elements [86]. These RE-derived microRNAs, like the proteins that arise from REs, are implicated in disease processes. For example, the REs-derived proteins CXX1B (RTL8A retrotransposon Gag like 8A) and PEG10 (paternally expressed 10) are implicated in the pathogenesis of familial FTD linked to UBQLN2 gene mutations. The UBQLN2 gene product prevents the degradation of CXX1B and PEG10 through ubiquitination [87]. This suggests that RE-derived microRNAs may also contribute to both sporadic or familial forms of FTD, as evidenced by changes in their expression during the disease. An analysis of scientific literature identified 13 microRNAs linked to FTD (Table 1, Ref. [88, 89, 90, 91, 92, 93, 94, 95, 96]), five of which originated from LINE elements, two from SINE, one from LTR-RE, and five from DNA transposons. The INT (integrase) domain of LTR-REs belongs to the DDE family of transposases, which are involved in the transposition of DNA transposons [97]. It has been proposed that LTR-REs may have evolved through recombination between non-LTR-REs and DNA transposons [98], suggesting that altered expression of DNA transposon-derived microRNAs may also be a consequence of REs involvement in FTD pathogenesis. The involvement of miRNAs derived from retroelements in the mechanisms of FTD development suggests their influence on other epigenetic factors, including DNA methylation, histone modifications and changes in lncRNAs expression. This is due to the fact that REs are drivers of epigenetic regulation [14], and are also key sources of lncRNAs nucleotide sequences [86]. This is reflected in changes in the methylation of promoter regions of genes involved in FTD pathogenesis, such as the GRN gene [99], C9ofr72 [100]. A significant increase in the expression of lncRNAs NEAT1 and NORAD was determined in patients with FTD compared with healthy controls [101].
| Transposon-source | microRNA | Changes in microRNA expression [Author] (function) |
| ERVL-MaLR | miR-1246 | decreased [88] (inhibits FAM53C (family with sequence similarity 53 member C), C12orf71 (chromosome 12 open reading frame 71), LENEP (lens epithelial protein), GLRB (glycine receptor beta) genes) |
| LINE-RTE-BovB | miR-181c | decreased [89] (inhibits NHEJ1 (non-homologous end joining factor 1), G3BP2 (G3BP stress granule assembly factor 2), IRS1 (insulin receptor substrate 1), STK24 (serine/threonine kinase 24) genes) |
| SINE/MIR | miR-330 | increased [90] (directly interacts with 3′UTR of mRNA of TARDBP I (encodes TDP-43 protein) gene) |
| SINE/MIR | miR-345-5p | increased [91] (inhibits EPN3 (epsin 3), CTTNBP2NL (CTTNBP2 N-terminal like), YEATS2 (YEATS domain containing 2), RFC1 (replication factor C subunit 1) genes) |
| DNA-hAT-Charlie | miR-361 | increased [92] (inhibits FBXL19 (F-box and leucine rich repeat protein 19), CAMK2B (calcium/calmodulin dependent protein kinase II beta), ALDH3B2 (aldehyde dehydrogenase 3 family member B2), EFNA5 (ephrin A5), TRAF3 (TNF receptor associated factor 3) genes) |
| DNA-TcMar-Mariner | miR-548a-3p | decreased [93] (inhibits ZFHX3 (zinc finger homeobox 3), CDC14A (cell division cycle 14A), ATRNL1 (attractin like 1), SKIL (SKI like proto-oncogene), CTTNBP2 (cortactin binding protein 2), CDK13 (cyclin dependent kinase 13) genes) |
| DNA-TcMar-Mariner | miR-548b-5p | increased [94] (inhibits BAI3 (brain-specific angiogenesis inhibitor 3), GK (glycerol kinase), SLC23A2 9 (solute carrier family 23 (nucleobase transporters), member 2), CNR1 (cannabinoid receptor), FAM134B (family with sequence similarity 134, member B), HS2ST1 (heparin sulfate 2-O-sulfotransferase 1), MYT1L (myelin transcription factor 1-like), NCAM1 (neural cell adhesion molecule 1) genes) |
| DNA-TcMar-Mariner | miR-548c-5p | increased [94] (directly interacts with 3′UTR of mRNA of BAI3 (brain-specific angiogenesis inhibitor 3), GK (glycerol kinase), SLC23A29 (solute carrier family 23 (nucleobase transporters), member 2), CNR1 (cannabinoid receptor), FAM134B (family with sequence similarity 134, member B), HS2ST1 (heparin sulfate 2-O-sulfotransferase 1), MYT1L (myelin transcription factor 1-like), NCAM1 (neural cell adhesion molecule 1) genes) |
| LINE1 | miR-571 | increased [94] (directly interacts with 3′UTR of mRNA of GK (glycerol kinase) and GTDC1 (glycosyltransferase-like domain containing 1) genes) |
| LINE1 | miR-588 | decreased [95] (directly interacts with 3′UTR of mRNA of PGRN gene) |
| DNA-hAT-Tip100 | miR-659-3p | decreased [96] (directly interacts with 3′UTR of mRNA of PGRN gene) |
| LINE2 | miR-708-3p | increased [93] (inhibits SCAMP1 (secretory carrier membrane protein 1), JARID2 (jumonji and AT-rich interaction domain containing 2), TRAF3 (TNF receptor associated factor 3), VIM (vimentin), BAZ1B (bromodomain adjacent to zinc finger domain 1B) genes) |
| LINE2 | miR-887 | decreased [94] (inhibits CASK (calcium/calmodulin dependent serine protein kinase), CCPG1 (cell cycle progression 1), PLD2 (phospholipase D2), TCTN3 (tectonic family member 3) genes) |
FTD, frontotemporal dementia; ERVL-MaLR, Endogenous Retrovirus-Like Mammalian Apparent LTR Retrotransposon; LINE-RTE-BovB, long interspersed nuclear elements – retrotransposable element - Bovine-B; SINE, short interspersed nuclear elements; MIR, mammalian wide-interspersed repeat; DNA-hAT, DNA-transposon hobo-Ac-Tam3.
Numerous studies indicate that the epigenetic changes in FTD and other neurodegenerative diseases can be both similar and distinct [102, 103]. These differences are most clearly reflected in the altered expression of microRNAs, which serve as guides for DNA methylation (RdDM – RNA-dependent DNA methylation) at specific loci [104, 105, 106]. For example, miR-26a, miR-326, miR-484, miR-361 are associated with both FTD and Alzheimer’s disease [92]. MiR-320a and miR-328-3p are downregulated in FTD and Alzheimer’s disease compared to controls [107], while decreased expression of miR-127-3p has been specifically observed in FTD [108]. Other differences in microRNA expression have been noted between FTD and Alzheimer’s disease, such as miR-125b/29a, miR-125b/-874, miR-107/-335-5p; and between FTD and ALS, such as miR-129-3p/miR-206, miR-338-3p/let-7e [109]. Several microRNAs, including mir-124, miR-125, miR-132, miR-141, miR-143, miR-200a, miR-218, miR-659, are associated with both FTD and ALS [110]. These distinctions in RE activation between FTD and ALS may underlie epigenetic differences between the diseases, which are reflected in the expression changes of transposable element-derived microRNAs and the clinical manifestations of each disorder. Indeed, an analysis of scientific literature has shown that in ALS, which, like FTD, also involves TDP-43 proteinopathy, leading to neurodegeneration [2, 11, 12, 22, 25]. The microRNAs shown in the table are related to REs because they evolved from these REs. This feature allows to suggest the mutually regulatory effect of these REs and microRNAs: transcripts of these REs function as concreteness endogenous RNAs, binding like “sponges” to these microRNAs and leveling their effect on the expression of target genes [39]. In addition, the microRNAs themselves, due to the complete complementarity of the REs sequences, are able to influence the transcription of these REs due to the RNA-directed DNA methylation (RdDM) mechanism, acting as “guides” for DNA methyltransferases [104, 105, 106].
A literature review reveals that, similar to FTD, ALS – another disease involving TDP-43 proteinopathy and neurodegeneration – is associated with decreased expression of LTR-REs-derived miR-1246 [111] and DNA transposon-derived miR-548a [112]. However, most transposable element-derived miRNAs associated with ALS are not implicated in FTD. These include LINE-derived miRNAs such as miR-1249 [113], miR-130a-3p, miR-151a, miR-151b, miR-28-3p, miR-342-3p [114], miR-191c [115], miR-1825 [116], miR-374b-5p [117], miR-4455 [118], miR-582, miR-606 [112], as well as SINE-derived miRNAs such as miR-1268a [119], miR-378a-5p [120], and miR-6801-3p [121]. LTR-REs-derived miRNAs such as miR-3927-3p [121], miR-4286 [122], miR-4454 [123], miR-7977 [119], also show diseased expression in ALS. The distinct effects of these microRNAs on target genes may explain significant clinical differences between ALS and FTD. Nevertheless, the shared mechanisms of RE influence on TDP-43 in both diseases suggest that similar treatment approached could be effective. For instance, reverse transcriptase inhibitors have shown promise in treating ALS [124], and future therapies targeting the HERV envelope gene with antisense oligonucleotides, antibodies and non-coding RNAs [125], as well as chromatin remodeling drugs, are under investigation [126].
The neurotoxicity of the Env HERV-K protein in patients with ALS in the cerebrospinal fluid is eliminated with the help of antibodies against Env [127], which can be proposed for the treatment of FTD. The creation of monoclonal antibodies aimed at pathologically activated RES in FTD is promising. An example is the method of therapy for multiple sclerosis, which also leads to neurodegeneration due to an autoimmune response, in which the role of herpesviruses and REs activation has been proven [128]. In a phase II clinical trial, a randomized, placebo-controlled study demonstrated the efficacy of the monoclonal antibody GNbAC1 (Temelimab) against the HERV-W Env envelope protein in reducing central nervous system (CNS) damage and atrophy without significant side effects [129]. It can be assumed that similar methods can be used in the treatment of FTD. Currently, there are no REs-based drugs and therapeutic approaches for the treatment of FTD. However, the similarity of the pathogenesis of this disease with ALS suggests the possibility of using approaches used to treat ALS as a prospect for the development of FTD treatment methods. In addition, monoclonal antibodies successfully used against the HERV expression product in the treatment of multiple sclerosis, in the pathogenesis of which the role of REs and herpes viruses has been proven [128], similar to the mechanisms of FTD development. Taking into account the role of REs in the physiological processes of the brain, including neuron differentiation [130] and memory formation [131], a differentiated approach with targeted effects only on REs involved in the pathogenesis of the disease is most promising. For this purpose, it is possible to use specific microRNAs derived from REs in evolution and their completely complementary sequences [86]. Development in this direction can also become the basis for improving cognitive processes during physiological aging of the brain in connection with the reactive oxygen species (ROS) role as a key link of the progressive aggregation of antiviral proteins TDP-43 [20] and Tau [21]. The transposable element-derived miRNAs identified in this paper may serve as potential targets for epigenetic therapies aimed at inhibiting miRNAs involved in FTD pathogenesis.
FTD is the second most common presenile dementia after Alzheimer’s disease [2] and associated with aging [3], and with multiple polymorphisms located in intergenic regions, introns and UTRs [5, 6, 7, 8, 9]. To explain the association of FTD with multiple SNPs located in intergenic, intronic and regulatory regions of the human genome [5, 6, 7, 8, 9], a new hypothesis about the role of retroelements in the pathogenesis of the disease has been proposed. Indeed, most REs genes are located in intronic, intergenic and regulatory regions [16]. Therefore, FTD-associated polymorphisms in these regions may cause abnormal activation and dysfunction of REs expression products. As a result, activated REs cause increased expression and aggregation of tau, TDP-43, and FUS proteins, similar to viruses, since REs expression products activate the interferon response [19, 23, 41, 49, 57, 58]. These proteins are characterized by antiviral properties [64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80] and the ability to inhibit the expression of retroelements [17, 24, 25, 26, 41, 43, 45]. Therefore, transcription and aggregation of these antiviral proteins is activated by viruses [64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80] and retroelements (which is due to their evolutionary origin from viruses [54]) [19, 23, 41, 49, 57, 58]. The aggregates of tau, TDP-43, and FUS proteins formed under the influence of REs become unable to inhibit the expression of retroelements, resulting in derepression of REs [24, 25, 26, 41]. A “vicious circle” is created in which aggregates of antiviral proteins promote derepression of REs, which in turn enhance the expression and aggregation of tau, TDP-43, and FUS proteins. As a result, neurodegeneration progresses.
The following numerous facts support the described mechanisms of FTD pathogenesis. Elevated levels of HERV-K have been detected in the serum and affected brain tissues of FTD patients [15]. Additionally, ERV activation has been observed in CHMP2BIntron5 models of FTD in Drosophila, where the gypsy ERVs had a toxic effect on the central nervous system. Genetic blocking of gypsy and inhibition of reverse transcriptase in REs were found to prevent neurodegeneration [2]. The influence of FTD-associated SNPs localized in REs regions can be illustrated by the ORF1p translation product of LINE1, which forms cytoplasmic aggregates and shares similarity with RNA-binding proteins implicated in neurodegeneration [17]. An additional factor is the age-related activation of REs [18] as the incidence of FTD rises significantly with age [3]. This activation leads to aseptic inflammation in the brain, with the interferon response and other antiviral systems being triggered by RE expression products [19]. Direct interaction of retroelements with TDP-43 protein is described [44]. In brain samples from FTD patients, TDP-43 aggregates were found to colocalize with the HERV-K pol protein, indicating a pathological interaction in the development of FTD [15]. There is evidence of the role of tau protein in the epigenetic regulation of retroelements [24] and the influence of tauopathy on their derepression [25]. Toxic forms of tau protein also cause heterochromatin decondensation leading to RE activation and subsequent neuroinflammation [53]. Virus-activated interferon I further promotes FUS accumulation by stabilizing its mRNA, making FUS-expressing cells hypersensitive to double-stranded RNAs [23].
In addition to the described “vicious circle” in which REs enhance the
expression and aggregation of antiviral proteins, pathological activation of REs
influences the development of FTD in other ways. RNAs of the HERV-K envelope
genes has been shown to bind to human Toll-like receptor 8 (TLR8) and activate it
in neurons and microglia, promoting neurodegeneration [55]. Pathologically
activated REs stimulate inflammatory processes in the brain [56, 57, 58]. At the same
time, inflammatory processes are stimulators of REs expression [41]. The
transcript of the Env gene of HERVK encodes a conotoxin-like CTXLP protein that
affects the expression of innate immunity genes and the activity of p56 NF-
The mechanisms of involvement of activated REs in the pathogenesis of FTD described in the article allow us to propose new ways of treating this disease aimed at suppressing the activity of REs. The “vicious circle” of REs interactions with antiviral proteins described for FTD in this article may also be characteristic of other neurodegenerative diseases, since pathology of TDP-43 and FUS proteins is characteristic of ALS [11], and tau aggregation is determined in Alzheimer’s disease [9]. This is evidenced by the facts of detectable activation of REs in ALS [127], Alzheimer’s disease [132] and multiple sclerosis [128, 129]. Therefore, the methods used in the treatment of these diseases, directed against retroelements and their expression products, can be proposed in the therapy of FTD [127, 128, 129, 130, 131]. The most promising targeted therapy is using REs-derived microRNAs as tools, the association of which with FTD is described in this article [88, 89, 90, 91, 92, 93, 94, 95, 96].
A hypothesis has been proposed suggesting that retroelements (REs) play a central role in the pathogenesis of FTD, primarily through their interactions with antiviral proteins such as TDP-43, tau, and FUS. Throughout evolution, REs have been closely linked with viruses, and the expression products of pathologically activated REs in the brain stimulate the production of TDP-43, tau, and FUS, which in turn normally inhibit RE activity. However, in FTD patients, a “vicious circle” emerges, driven by both hereditary factors (such as polymorphisms in RE-related genes) and mutations in antiviral protein genes (in monogenic forms of FTD). In sporadic FTD, polymorphisms in retroelements cause conformational changes in TDP43, tau, and FUS, leading to their aggregation. The aggregates lose their ability to inhibit REs, resulting in further upregulation of RE expression. In response, increased levels of TDP43, tau, and FUS are produced to counteract the activated Res, but it only accelerates the accumulation of protein aggregates, thereby worsening the disease. In monogenic forms of FTD, mutations in tau and C9orf72 proteins directly impair their ability to inhibit REs, allowing excessive REs to interact with the defective proteins and promote their aggregation.
The primary triggers of these pathological mechanisms in FTD are aging, which enhances the activation of REs, and viral infections (Fig. 2). Both aging and viruses, particularly those that interact with TDP43, tau, and FUS, can stimulate RE activity, contributing to FTD development. The clinical differences between FTD and ALS, despite their shared pathogenic mechanisms, may be due to distinct patterns of RE activation, reflected in the differential expression of microRNAs derived from transposable elements. An analysis of the scientific literature reveals that FTD and ALS share only two such microRNAs, while FTD shows altered expression of 11 RE-derived not found in ALS. Conversely, ALS is associated with the decreased expression of 19 transposable element-derived microRNAs that are not altered in FTD. These microRNAs could potentially serve as targets for therapeutic strategies aimed at inhibiting pathologically activated REs in both diseases.
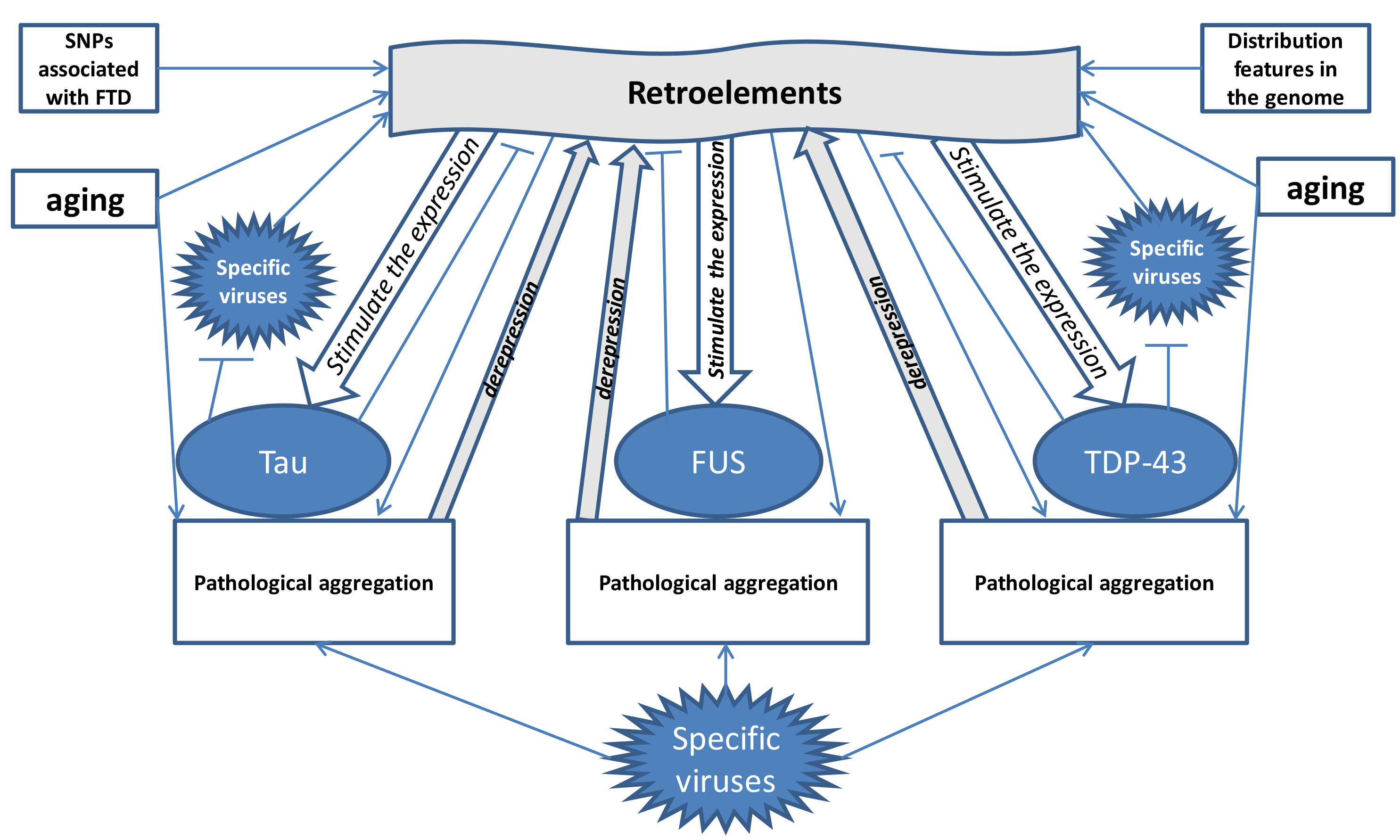 Fig. 2.
Fig. 2.
Mechanisms of influence of retroelements on the development of
FTD (formation of a “vicious circle”) with the participation of hereditary
predisposition, aging, viruses and antiviral proteins tau, TDP-43 and FUS. The
figure shows that under the influence of frontotemporal dementia (FTD)-associated
SNPs, aging, retroelement distribution patterns and viruses, retroelement
activation occurs. The arrows (
RNM made substantial contributions to conception and design, or acquisition of references, or analysis and interpretation of references; and been involved in drafting the manuscript or reviewing it critically for important intellectual content; and given final approval of the version to be published; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Not applicable.
Not applicable.
This research received no external funding.
The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.