1. Atrial Fibrillation: Clinical Relevance, Pathogenesis and Therapy
Atrial fibrillation (AF) is a condition in which the electrical signals in the
upper heart chambers (atria) are rapid and disorganized, producing an irregular
and chaotical heartbeat. AF is the most common arrhythmia linked to noteworthy
mortality and morbidity [1]. The sinus rhythm should be between 60 to 100 bpm at
rest, while the heart rhythm in AF patients may be over 140 bpm [2]. The most
dramatic cardiovascular outcomes of AF are stroke and heart failure (HF) [3]. AF
affects over 33 million people worldwide with increasing prevalence because of
aging and obesity population [4]. Men are 1.5 times more likely to develop AF
compared with women. In addition to age, race and sex, risk factors for
developing AF include intrinsic cardiovascular diseases and modifiable noncardiac
risk factors, including smoking, alcohol or drug use, caffeine, lack of physical
activity, overweight, diabetes, high blood pressure or obstructive sleep apnea
(OSA) [5]. The symptoms of AF include rapid and irregular pulse, palpitations,
weakness, fatigue, chest pain, dizziness and shortness of breath, considerably
affecting the quality of life [6]. However, for many people AF may have no
symptoms. AF is classified based on aetiology or degree of persistence. In terms
of aetiology, AF can be classified as environmental factor induced-, congenital
or genetic [3]. A strong genetic component underlies the disease, since variants
in 160 genes associated with AF have been detected. Cardiac ion channel gene
variants are a underlying risk factor to AF development [7]. In terms of
persistence, AF can be paroxysmal, persistent (over 7 days), long-standing
persistent (over 1 year) or permanent.
The pathophysiology of AF is complex as described Fig. 1. Either structural and
electro-mechanical remodeling of the atrial tissue underlies the perpetuation and
evolution of AF from the paroxysmal to permanent forms [8]. AF genesis initiates
with well-established ectopic firings that initiate reentrant wave propagation
under a vulnerable atrial substrate [9]. Both trigger factors and a vulnerable
atrial substrate are critical for AF onset [9]. An important substrate for FA may
be conduction and refractoriness abnormalities, an inflammatory or fibrotic
background. Ectopic atrial foci are thought caused by an enhanced automaticity,
delayed afterdepolarizations (DADs) and/or early afterdepolarizations (EADs)
[10]. There is no doubt that the autonomic nervous system (ANS) also cooperates
in the triggers, substrate and perpetuators of AF [11].
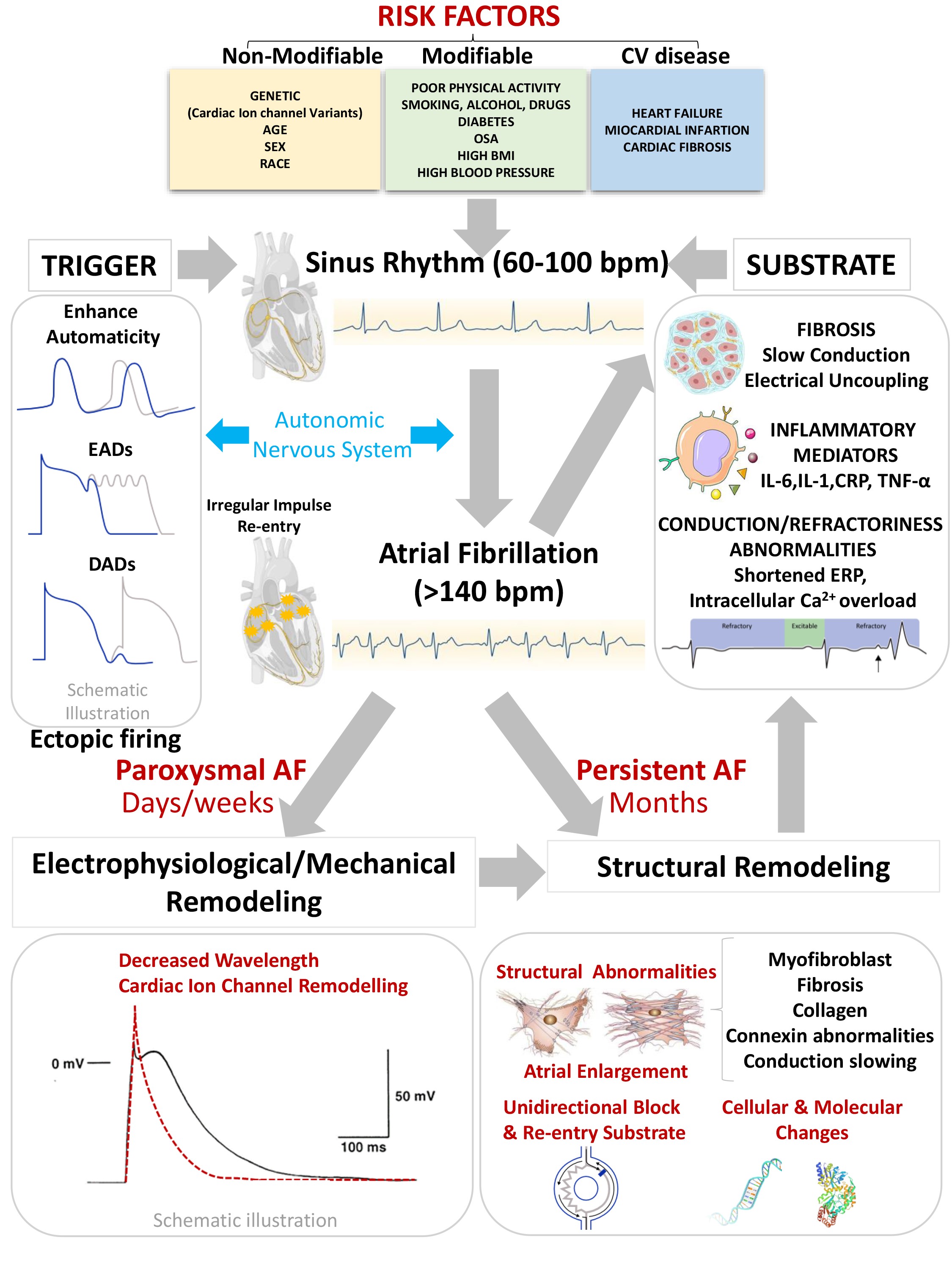 Fig. 1.
Fig. 1.
Pathophysiology of Atrial Fibrillation. AF, Atrial
Fibrillation; OSA, Obstructive Sleep Apnea; BMI, Body Mass Index; bpm, Beats per
minute; ERP, Effective Refractory Period; EAD, Early afterdepolarizations; DAD,
Delayed afterdepolarizations.
Electrical remodeling is caused by a dysfunction of the atrial ion channels that
essentially increasing outward K currents and/or decreasing inward L-type
Ca current, accelerating repolarization which accelerates atrial
repolarization lead to a short atrial action potential (AP) duration and
refractoriness, and thus favoring reentry [12]. All these changes have a strong
impact not only on the atria electrophysiology but also on atria structure [13].
The disruption of Ca handling, secondary to electrical remodeling, cause
the alteration of sarcomere proteins promoting the contractile remodeling which
induce atrium dilatation and blood clot formation perpetuating AF [9]. The
structural remodeling particularly atrial enlargement, fibrosis and
cellular/molecular changes causing localized conduction slowing and enhance
re-entry [14]. Electrophysiological, structural, and mechanical irregularities
also encourage AF perpetuation by stabilizing unidirectional block and re-entry
[12]. Brief refractoriness, slow conduction and conduction barriers favor the
induction and maintenance of reentry [15].
The main goal of AF treatment is relief of symptoms and prevention of stroke.
Pharmacological treatment mainly includes anticoagulation, heart rate or rhythm
control, and non-arrhythmic supportive pharmacological therapy. A variety of
medicines are available to convert and maintain the patient in a normal sinus
rhythm such as atenolol or bisoprolol (beta-blockers) or diltiazem or verapamil
(Ca channel blockers) could be prescribed. These medications can be
combined with digoxin, which helps controlling the heart rate and preventing the
rapid ventricular response [16]. Electrical cardioversion and interventional
ablation procedures can also help for correcting abnormal heart rhythms. These
pharmacological strategies often fail with the progression of AF to the
persistent or permanent forms; even ablation procedures have poor success [16].
Consequently, it is necessary to find new therapeutic targets for the relief of
persistent or chronic AF forms, as well as the development of new and more
effective pharmacological tools.
2. Atrial-Specific Ion Channels
The cardiac PA reflects the integrated conductance of numerous individual ionic
currents, largely dominated by the movement of Na, Ca and K
ions, which change the membrane potential (Vm) as a function of time. Cardiac ion
channel function is highly regulated and orchestrated, being influenced by
multiple factors, such as voltage, ligand binding, second messengers such as
cyclic adenosine monophosphate, and post-translational modification. The
summarized input and output of all ionic currents expressed in a specific cardiac
cell determines the duration and the cardiac AP shape [17]. Consequently
pacemaker, atrial, and ventricular cells possess heterogeneous morphology and AP
duration due to cardiac ion channel differences which confers distinctive
electrophysiological properties [17]. In atrial and ventricular cardiomyocytes,
most of the ion channels responsible for determining the AP are essentially the
same; however, the expression patterns, biophysical properties, and regulatory
pathways may differ. The atrial and ventricular AP are described in 5 main phases
(0–4), with different duration and morphology as described Fig. 2.
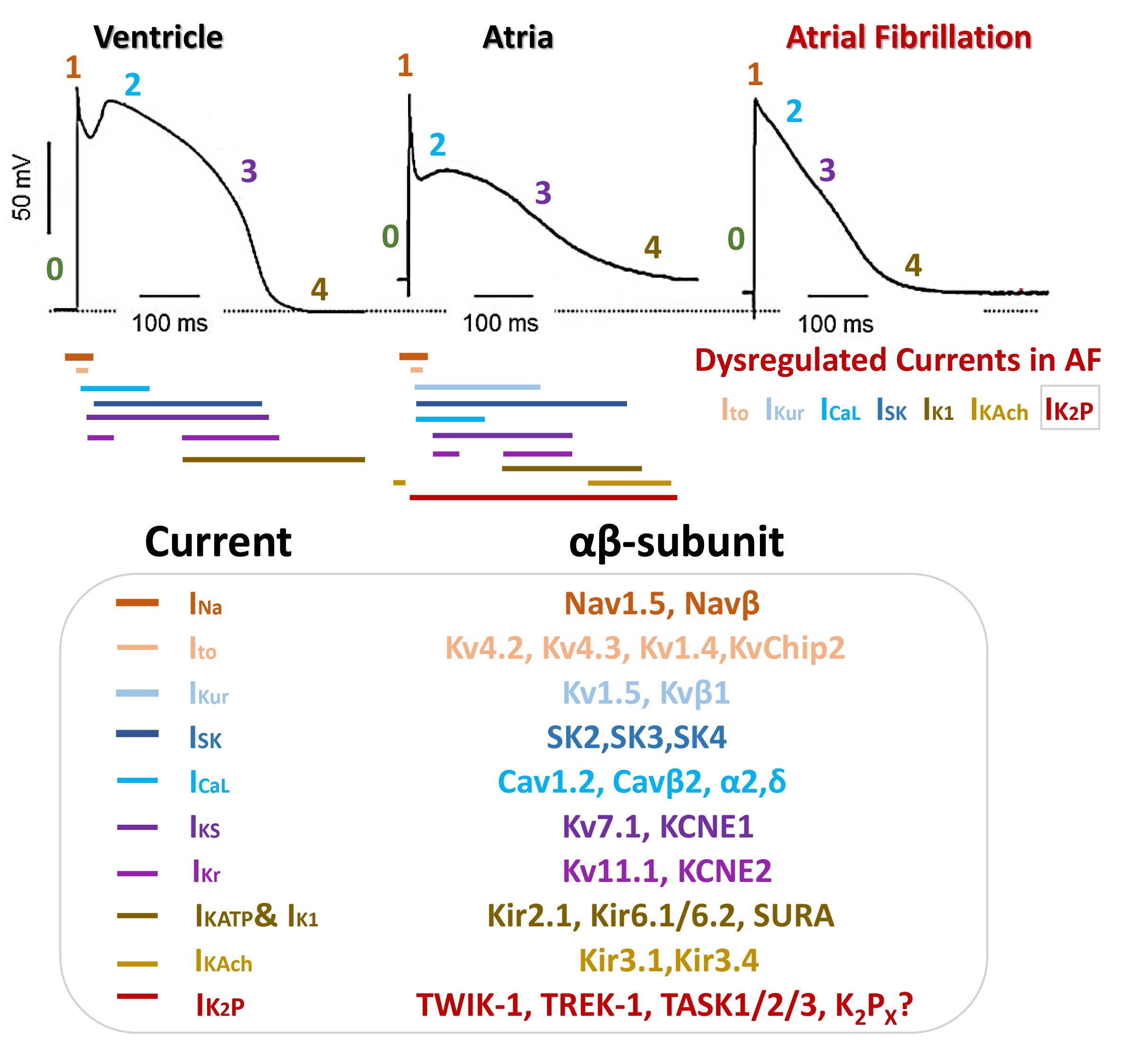 Fig. 2.
Fig. 2.
Schematic illustration of cardiac ion channels, currents and
proteins implicated in action potential (AP) morphology of ventricular, atria,
and AF-atrial cardiomyocytes.
The ventricular AP exhibits a peak-and-dome morphology with a prominent plateau
phase; conversely, the human atrial AP usually has a triangular morphology
compared to its ventricular homologue (Fig. 2). Moreover, electrophysiological
heterogeneity is included in AP in different zones of the atria. The atrial
resting membrane potential (Vrest) varies between -65 and -80 mV and is more
depolarized than that in the ventricle mainly due to the differences in the
expression density of the inward rectifier K current (Fig. 2) [17, 18]. The
human atrial AP duration at 90% repolarization (APD90) and the atrial maximum
upstroke velocities (Vmax) have been reported between 150–500 ms and 150
and 300 V/s, respectively; while for ventricular cells the APD90 and Vmax ranging
200–450 ms and 300–400 V/s [17, 18]. Atrial APs lose acquire a more triangular
shape in AF, the APD is significantly shortened and shows a decreased APD rate
with poor adaptation and abrupt changes (Fig. 2) [19]. The atrial AP depends on
three prime time- and voltage-dependent currents: I, I, and
I. Alterations in atrial ion currents due to AF pathogenesis have a strong
impact on the shape of atrial AP. I current density or biophysical
properties remain unaltered in patients with chronic AF. I current
density is decreased by 60–75% in chronic AF; whereas it appears to be
unaltered in AF. I and I currents are also downregulated in atrial
cardiomyocytes from AF patients. Conversely, the inwardly rectifying potassium
current (I) and acetylcholine-activated inward rectifier (I),
essential currents in the late phase repolarization, are increased in AF. The
excessive activity of I and I currents could accelerate
repolarization shortening the atrial AP and encouraging AF [17, 18]. Not much
information is available on the role of the rapid (I) and slow (I)
components in FA [12, 15, 18, 19].
Some K channels may be distinctively or predominantly expressed in atria
compared with ventricles or/and possess distinctive biophysical properties that
differentiate them from their counterparts in the ventricles, making them ideal
drug targets for AF. So far, the ultrafast activation delayed rectifier
voltage-dependent Kv1.5 current (I), the I current, and the
TWIK-related acid-sensitive K channel type 1 (TASK1) conducting I
current have been best validated atrial-specific ionic currents [20]. The atrial-
and ventricular-K-currents could be formed by the participation of several
-subunits as well as -regulatory subunits as is described in
Fig. 2.
Numerous variants in K channel encoding genes are associated with rare
forms of genetic AF, such as Kv1.5, Kv4.2, Kv4.3, Kir2.1, Kir3.4, and
KP (TASK-1) [11], these last two specific to atria cells. So far,
many previous studies have demonstrated that the dysregulation of the cardiac ion
channel conducting I, I, I, I, I,
I, and I currents is critical in the electrophysiological
remodeling of AF (Fig. 2). Hence, it has highlighted that targeting these
atrial-specific K channel as a promising therapeutic principle for AF.
KP channels are fascinating ion channel group since are responsible of the
leak or conductance voltage-independent K current and appear to have
functionality for most of the duration of the atrial AP [21, 22]. In addition,
knowledge of the regulation and physiological role of KP channels is not
yet fully understood. Therefore, throughout this review we will mainly emphasize
the role of KP channels in AF and their therapeutic potential.
3. Outlook KP Channels in Atrial Fibrillation: KP Channels
Druggability
KP channels are formed by a structure of two pore-forming loop domains in
each alpha subunit; two of these alpha subunits assemble into a dimer to form the
channel [23]. I currents are involved in background K conductance,
stabilizing the Vrest and the repolarization in atrial cells. KP channels
have been considered voltage-independent ion channels, since there is no
voltage-sensing domain (VSD) in their structure, where their strong outward
rectification arises from the asymmetric K gradient across the membrane
following the Goldman-Hodgkin-Katz equation. KP channels produce basically
instantaneous and non-inactivatable currents an extensive range of the membrane
potential (Vm) [24].
Due to these characteristic biophysical properties KP channels are also
known as background or “leak” K channels with important implications in
stabilizing Vrest and contributing to repolarization. Whereas the TASK1 currents
conform to the Goldman-Hodgkin-Katz equation, other KP channels may exhibit
variations: i.e., TREK1 have a slight outward; TWIK1/TWIK2 channels have inward
rectification; there is a slow inactivation component that represents for
approximately 50% of TWIK2 current; and TRESK shows asymmetric gating behavior
[24, 25, 26]. Interestingly a noteworthy voltage-dependent activation has been found
in some KP channels, nevertheless the exact mechanism remains uncertain due
to the lack of a canonical voltage-sensing domain in channel structure [27].
Then, the modulation of KP currents provides a mechanism for regulating
cellular excitability [27]. KP channels can be modulated by several
physiological and chemical factors such as temperature, pH, lipids, stretch,
kinases, neurotransmitters, unsaturated fatty acids, antidepressants and
anesthetics [28]. The KP channels is categorized into six groups: Two
pore-domain weakly inward rectifying K channel (TWIK), TWIK-related
alkaline-sensitive K channel (TALK), TWIK-related acid-sensitive K
channel (TASK), TWIK-related spinal cord K channel (TRESK), TWIK-related
K channel (TREK) and tandem pore domain halothane-inhibited K channel
(THIK) [25]. To date, only TWIK-1, TREK-1, TASK-1, TASK-2 and TASK-3 channel have
been identified as responsible for background I current in atrial cells
[21, 22]. The I current persists throughout all phases of the atrial AP,
stabilizing the membrane potential toward Vrest (Fig. 2), prevent EADs, and could
be involved in adjusting the availability of the Na channel for
depolarization (phase 0) [29]. Patch clamp recordings have indicated that
alteration in I current can alter the shape and the duration of action
potentials in human atrial cells [30]. Then, the alteration of I current
either by changes in channel expression, trafficking or activity, can contribute
to changes that can affects the processes involved in the generation of
pro-arrhythmic phenomena as development of EADs or DADs and enhancing the
automaticity, constituting a trigger for the development and maintenance of AF.
However, it is not excluded that other KP subunits or subfamilies
have physiological roles in atria. New research in this area may improve our
knowledge about KP channel function. KP channel family is really
fascinating since several studies have demonstrated that the expression of
TWIK-1, TREK-1, TASK-1, TASK-2 and TASK-3 are dysregulated in AF
[6, 11, 23, 24, 31, 32, 33]. Moreover, the loss of function (LoF) mutations on
TASK1-enconding gene have been reported in patients with familiar AF [34].
KP channel modulators could have enormous therapeutic potential in AF and,
luckily, they are known to be very good “druggable” targets [28]. As most studies
have focused on elucidating the physiological role of these channels in
cardiomyocytes, the pharmacological profiles developed so far have not been very
satisfactory. Many studies have shown that several of the medications used to
control heart rate and heart rhythm in AF exert their mechanism of action in part
by modulation of KP channels. To date, many marketed and non-marketed
compounds capable of targeting KP channels direct or indirectly have been
identified. However, many of these drugs are not selective for a unique K
channel subfamily or subtype, but rather target several ion channels. Although
the exact pharmacology of TWIK channels is not yet known, many potent
activators/blockers for TASK and TREK have been identified. Our recent advances
in the physiology and pharmacology of KP channels have helped us to better
understand many of the intricate mechanisms that can modulate the KP
activity or expression.
Here we will review the information on KP channels expressed in atria and
review available drugs to modulate potentially the activity of specific KP
channels and their possible use for AF treatment.
3.1 TWIK-1
TWIK-1 or KP channels are encoded by KCNK1 gene and
expressed robustly in atria; however, is still a matter of ongoing debate its
physiological significance. The TWIK-1 channel appears to have a well-conserved
role in cardiac function. A study in zebrafish embryos demonstrated TWIK-1
channel is needed for a normal atrial morphology and heart rate. The TWIK-1
knockdown results in bradycardia and atrial dilatation [28]. However, despite its
functional importance in atria, genetic variation in KCNK1 has not been
revealed to be a common direct cause of AF. Gaborit N and colleagues [35] also
showed that TWIK-1 is upregulated in AF associated to valvular heart disease.
Therefore, TWIK-1 inhibitors may be useful for AF treatment. Another study also
demonstrated that the TWIK-1 channel could change its permeability to Na by
promoting membrane depolarization under conditions of hypokalemia and acidosis.
The changes in TWIK-1 ion selectivity could increase excitability of atrial
cardiomyocytes resulting in enhanced automaticity under these conditions
predisposing to tachycardia and AF development [24]. In this case, where the
selectivity of K is replaced by Na will be also effective TWIK-1
blockers to preventing AF.
As stated above, the TWIK channels-related pharmacology is still very lacking.
To date, it has not been identified any TWIK-1 channels activators or enhancers
and any TWIK-1 inhibitor/blockers reported are not useful in the submicromolar
range. Bupivacaine, a local anesthetic, showed a low potency block effect on TWIK
subfamily [36]. Antiarrhythmic such as quinidine, useful for controlling heart
rhythm in AF, and Ibutilide, useful for the cardioversion of recent AF or
flutter, have a blocker effect on TWIK-1 channels [37, 38, 39]. Quinine, a
antimalaria treatment, also have been demonstrated that can inhibit these
channels [37] (Fig. 3). It should be noted that TWIK-1 is also expressed in
brain, kidney, and pancreatic cells; therefore, drugs targeting these channels
for the treatment of AF could have side effects on these tissues. In brain,
TWIK-1 channels contribute to the regulation of AP firing and excitability in
dentate gyrus granule cells. TWIK-1 channel also participates in the ion and
water transport in kidney and in the regulation of Vrest on pancreatic beta cells
[25, 28, 40].
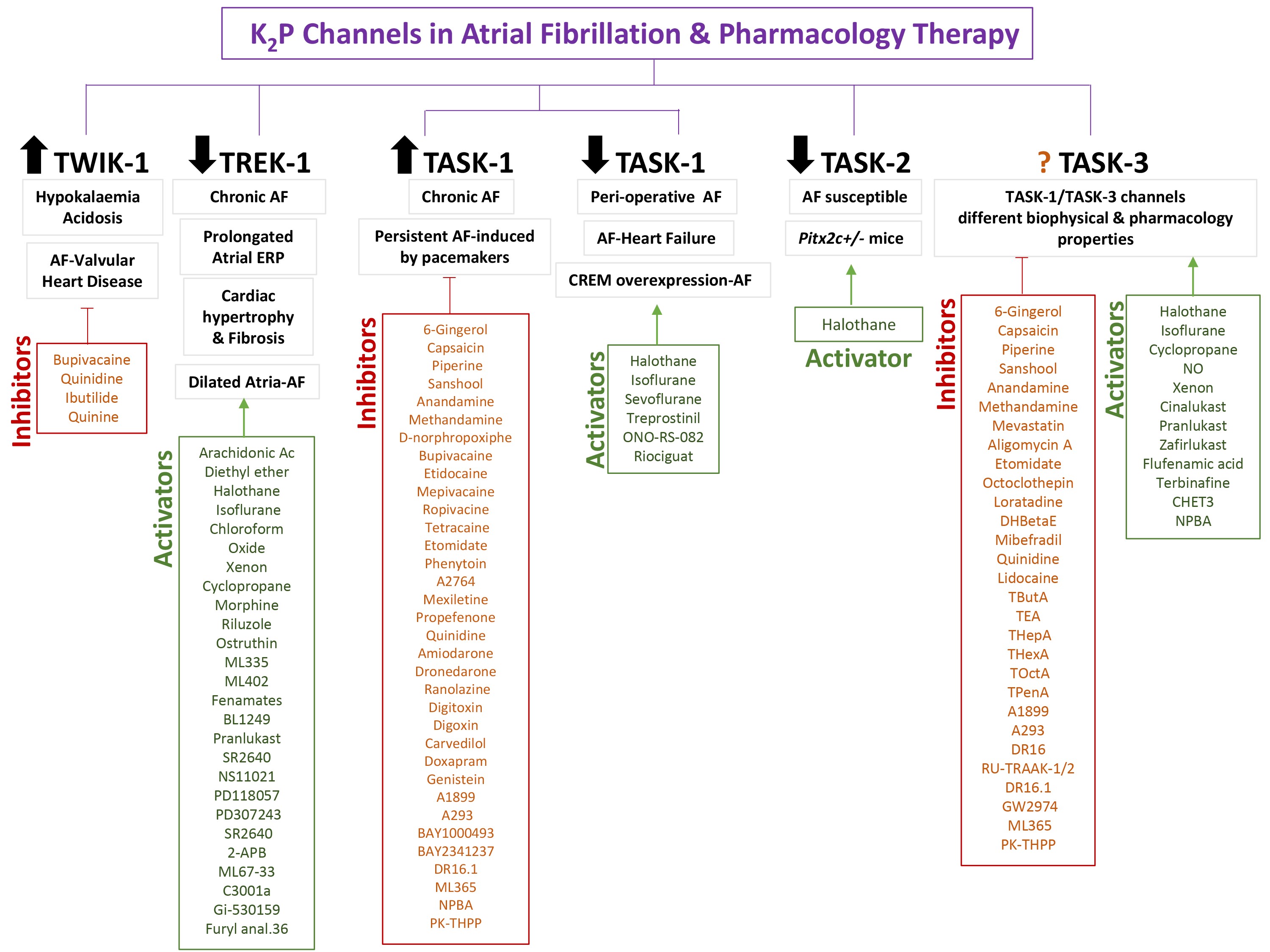 Fig. 3.
Fig. 3.
KP channels in the pathogenesis of atrial fibrillation and
available drug therapy.
3.2 TREK-1
TREK-1 or KP channels, encoded by the KCNK2 gene, are
stretch-sensitive contributing to mechanoelectrical feedback, AP regulation and
atrial electrophysiology. It was identified with a time-dependent reduction of
TREK-1 protein expression in the right atrium by 70% at 7 days and 80% at 21
days of induced-AF in a porcine model; however, TREK-1 channel expression in the
left atrium, AV node, and ventricles was not affected [26]. Lugenbiel P
et al. [41], also demonstrated that atrial TREK-1 mRNA levels were
reduced by 82% (left atrium) and 81% (right atrium) in patients with chronic AF
and HF which was associated with prolongation of atrial effective refractory
periods (ERP). The authors propose that functional correction of the TREK-1
channel by gene therapy could represent a new paradigm for the AF treatment [41].
Schmidt C et al. [42], study also recapitulated in CREM transgenic mice
the development of AF-associated with TREK-1 mRNA/protein downregulation
suggesting a mechanistic contribution of this channel to cardiac
arrhythmogenesis. The dysregulation of TREK-1 channel has also been implicated in
cardiac fibrosis and hypertrophy as well as heart failure due to their
mechano-active properties [24].
Only downregulation of the TREK-1 channel has been reported in the pathogenesis
of AF, therefore specific activators or enhancers of this channel could be a
pharmacological therapy for the disease (Fig. 3). A distinctive feature of TREK
channels with respect to other KP channels is the activation through the
C-terminal region by polyunsaturated fatty acids (arachidonic acid) or
lysophospholipids [43]. As other KP channels, TREK channels can be
activated at clinically concentration by many general anesthetics (cyclopropane,
nitrous trichloroethanol, xenon and oxide) and volatile compounds (chloroform,
halothane, isoflurane and diethyl ether) [44, 45]. Opioids such as morphine can
induce an opioid receptor-independent TREK-1 channel activation by binding
directly to TREK-1 structure [46]. Many fenamates (diclofenac, flufenamic acid,
mefenamic acid and niflumic acid), fenamate-like compound (BL1249, Pranlukast)
and other negatively charged activators [NCAs] (SR2640 NS11021, PD118057,
PD307243, and SR2640) are activate TREK-1 through direct unlocking and
stabilizing the selectivity filter gate [47, 48, 49].
2-aminoethoxydiphenyl borate (2-APB) also activates TREK-1 channels through
C-terminal region [50]. Riluzole, a drug used for amyotrophic lateral
sclerosis treatment, exhibits a dual effect on TREK-1 channels. Riluzole can
transiently activate TREK-1 followed by a longer lasting inhibitory effect
attributed to increased intracellular cAMP and protein kinase A (PKA)-dependent
inhibition [51]. Then, Tolbutamide, cAMP inhibitor, has the contrary to
riluzole’s long term-effect of on TREK-1 channels. A plant extract named
aristolochic acid used in pain treatment, and caffeic acid esters such as CAPE
and CDC also are TREK-1 channels enhancers [52, 53]. Joseph A et al.
[54], showed that ostruthin may exert its anxiolytic and antidepressive effects
in part through TREK-1 channel activation. Recently, two small molecular
activators (ML335 and ML402) capable of binding near the selectivity filter of
the TREK-1 channel have been identified [55]. Other small molecules as ML67-33,
C3001a and gi-530159 have been also identified as activators of this channels
[56, 57, 58]. However, many of these TREK-1 activator compounds described here are no
selective for TREK1 channel presenting other side effects on other potassium
subunits, which does not make them good candidates [59]. One study identified a
furyl analogue 36 as one of the first promising selective TREK-1 agonist (Fig. 3)
[60]. Several studies have demonstrated the functionality of this channel in
neurons, endothelial, vascular smooth muscle cells, and in gastrointestinal tract
cells. TREK-1 is targeted for general anesthesia by volatile and gaseous agents
playing a role in ischemic and epileptic neuroprotection, pain sensing and
depression [61].
3.3 TASK-1
TASK1 or KP channels, encoded by KCNK3 gene, exhibited
predominantly atrial expression which is conserved in different species including
human, pigs, dogs, chicken, mice, rats, and zebrafish [23]. Importantly, TASK-1
current is considered the major component of background conductance in human
atrial cardiomyocytes which is inhibited by 1A-adrenergic receptor
stimulation and extracellular acidosis [30]. These specific properties are shared
by the sustained cardiac outward K current, indicating that TASK1 channels
largely conduct the cardiac plateau K current known as I [23]. Its
functional significance in atria has been strongly supported by several studies
suggesting TASK-1 channel dysregulation has a huge impact on atrial electrical
activity and morphology supposing an arrhythmogenic substrate for AF [30].
It has been evidenced that atrial TASK1 mRNA/protein levels are upregulated in a
human chronic AF cohort (5-11) compared with people in sinus rhythm; while, TASK1
expression was not affected in paroxysmal AF patients [23]. TASK1 channel
upregulation causes a shortened AP duration in the right atrium which is restored
using pharmacological TASK1 inhibitors [62]. Animals studies in pigs, have
also demonstrated that TASK1 channel expression is upregulated in persistent
AF-induced by atrial burst stimulation via implanted pacemakers. The TASK1
current upregulation as well as the shortening of the APD is prevented by the
intravenously treatment of a TASK1 specific blocker, A293, administered one per
day during 14 days [63]. Constanze Schmidt, Felix Wiedmann and colleagues
[64] also demonstrated that TASK1 genetic ablation using Anti-TASK-1
adeno-associated virus suppresses AF and corrects the electrical remodeling in a
pig AF animal model. For instance, TASK channel inhibition seems to be a
promising therapeutic approach for the treatment of AF (Fig. 3). Many plant
extracts (6-Gingerol, capsaicin, piperine, sanshool) [50, 65], cannabinoids
(anandamine and methandamine) [66], and the opioid D-norphropoxiphe [67] can
inhibit TASK1 channels. A large group of anesthetics such as bupivacaine,
etidocaine, mepivacaine, ropivacine, tetracaine, etomidate has been demonstrated
to block TASK1 channels [38, 68]. The anticonvulsant phenytoin, the antipsychotics
fluoxetine and a cloxiquin analogs (A2764) also showed to exert a TASK1 blocker
effect on several studies [67, 68, 69]. The therapeutic effect of several drugs used
and commercialized for targeting different cardiac diseases among them AF could
be mediated by the TASK1 inhibition such as antiarrhythmics (mexiletine,
propefenone, quinidine, amiodarone, dronedarone, ranolazine), cardiac glycosides
(digitoxin and digoxin), and the blocker carvedilol
[32, 69, 70, 71, 72, 73, 74]. Doxapram, a ventilatory stimulant, and genistein, TK inhibitor, also
inhibit TASK1 channel; however, these two drugs also act indiscriminately on
TASK3 channels [75]. A new potent TASK-1 inhibitor, doxapram, is being
investigated under DOCTOS clinical trial. This study will reveal in the near
future whether doxapram is a good option for the acute conversion of
paroxysmal/persistent AF to sinus rhythm [76]. Selective Kv1.5 channel blockers
(A1899 and A293) designed as antiarrhythmic drugs for AF treatment, have showed
interestingly to be much about 70-fold more potent on TASK-1 channels than Kv1.5
channels, making them TASK selective at low doses [77]. Furthermore, A293
treatment significantly reduced AF burden in a persistent AF animal model. A
limitation of this study was an increase in pulmonary arterial pressure after
acute TASK-1 inhibition. However, no adverse effects on the central nervous
system were observed [63]. Other small molecules have been developed to exert a
blocker effect on TASK1 currents such as BAY1000493, BAY2341237, DR16.1, ML365,
NPBA and PK-THPP [68, 78, 79, 80, 81, 82].
On the other hand, many other studies had supported the downregulation of the
TASK1 channel or the absence of TASK1 current in peri-operative AF (peri-op AF),
which is common complication after thoracic surgery; and in AF associated to
heart failure (AF-HF) which is increasingly encountered in patients with HF (Fig. 3). In 2013, Harleton and colleagues [83] investigated that canine perioperative
AF was associated with loss of TASK-1 current function due to an increased
phosphorylation at threonine 383 in the C-terminus of TASK-1 channel. In
2015, the same group found TASK-1 current was present in human and canine atrial
myocytes with regular sinus rhythm, but was absent in humans with AF undergoing
cardiac surgery and in canine atrial myocytes after induction of AF by chronic
tachypacing [84]. In this study, phosphatase treatment rescued TASK-1 current in
atrial myocytes with AF, indicating that inhibition in TASK-1 current is
phosphorylation-dependent; however, the specific phosphorylation site in the
channel remains unidentified [84]. Other study in pigs also showed TASK1 channel
down-regulation in pacing-induced AF with HF [31]. Wiedmann F et al. [85], also found atrial TASK-1 channel expression was pointedly
reduced in HF murine model where the cardiac dysfunction was induced by
transverse aortic constriction. The same group also assessed the TASK1 channel
modulation in (CREM)-IbC-X transgenic mice since human AF
susceptibility has been associated with CREB/CREM transcription factors target
genes downregulation [85]. Myocardial overexpression of the transcriptional
repressor CREM-IbC-X in these transgenic mice (CREM-AF) results in
downregulation of target genes, among them TASK-1 channels, showing a phenotype
of atrial ectopy and AF [85]. Then in peri-op AF, AF-HF and
CREM-AF cases, TASK1 channels activators would be also of therapeutic interest in
AF. Nevertheless, few TASK-1 or TASK-3 channel activators are known so far.
Volatile anesthetics (halothane, isoflurane and sevoflurane) have been identified
as TASK1 activators probably through the anesthetic binding pocket, between M4
and M3 segment [59, 68]. A prostacyclin analog (potent pulmonary vasodilator)
named treprostinil activates the TASK-1 channel at clinically relevant
concentrations. Activation is mediated via cyclic AMP (cAMP)–dependent
phosphorylation of the channel induced by protein kinase A (PKA) [86]. The loss
of TASK-1 current function can be reversed by application of the phospholipase
inhibitor ONO-RS-08288 [87]. A recent drug licensed for the pulmonary arterial
hypertension treatment, riociguat, a soluble guanylate cyclase (sGC) stimulator,
activates protein kinase G (PKG) and stimulates the production of cGMP which can
enhance TASK1 current [88]. Drugs targeting TASK1 channel could have side effect
in neurons, vascular smooth muscle, and endocrine cells. Loss-of-function
mutations at multiple sites in the TASK1 encoding gene are one of the causes of
pulmonary arterial hypertension [89].
Taking together all these findings, TASK1 ion channel is a strong regulator of
the atrial repolarizing phase of the atrial AP and drugs targeting this
atrial-specific ion channel could provide a treatment for AF patients. The full
understanding of its role in atria will help to develop better therapeutic
approaches.
3.4 TASK-2
TASK-2 or KP channel, encoded by KCNK5 gene, is expressed
uniquely in left atria and its function is still uncertain [90]. The
transcription factor homeodomain-2 (PITX2) may regulate gene expression
and electrical function in the adult left atrium. Mice with low levels of atrial
Pitx2 expression have a shortened atrial AP and are more susceptible to AF.
Pitx2c+/- mice also showed atria Vrest more depolarized related to a
TASK-2 gene and protein expression downregulation (Fig. 3) [91]. In this case,
TASK-2 activators could prevent this phenotype; however, there are no selective
activators or inhibitors for this channel currently. Volatile anesthetics appear
to activate TASK-2, especially halothane [92]. To consider possible side effects
of treatment with TASK-2 activators, it is important to know TASK-2 is also
involved in breathing regulation by brainstem retrotrapezoid nucleus chemosensory
neurons and pH homeostasis by kidney proximal tubule cells [93].
3.5 TASK-3
TASK-3 or KP channel, encoded by KCNK9 gene, is
also expressed consistently in right human auricles. TASK-3 channel is the
closest relative of TASK-1 sharing many similarities. It has been demonstrated
that TASK-3 forms heteromeric TASK-1/TASK-3 channels at the surface membrane of
atria cardiomyocytes with a lower affinity for TASK-1 blockers [94]. Therefore,
the design of drugs against AF should consider the possible expression of
heteromers at the atrial level and antagonism could be useful for AF treatment
(Fig. 3). To date, it is uncertain whether TASK-3 channel is up- or
down-regulated in AF, but since it can form heteromers with TASK-1 subunits it
should be considered as a possible pharmacological target. In addition to
activating TASK-1 channels, halothane and isoflurane also activate TASK-3
channels [94]. Other gaseous anesthetics such as cyclopropane, NO and xenon
showed TASK3-enhancer properties [45].
Leukotriene receptor antagonists (LTRA) drugs used for asthma treatment such as
cinalukast, pranlukast and zafirlukast could activate TASK-3 current among other
potassium channel [48]. TASK-3 is also activated by flufenamic acid [47], the
antifungal terbinafine [48], the biguanide derivate CHET3 [95] or the small
molecule NPBA [82].
Many plant extracts (6-Gingerol, capsaicin, piperine, sanshool) [50, 65],
cannabinoids (anandamine and methandamine) [66], antibiotics (mevastatin,
aligomycin A) [96] and the general anesthtetic etomidate [68] have been
identified as TASK-3 channel inhibitors. Some neurotransmitters antagonists as
octoclothepin (D2), loratadine (H1) and DHBetaE (nAChR) also can inhibit TASK-3
channels [96]. We can use Ca channel blockers such as mibefradil and the
antiarrhythmics quinidine and lidocaine to inhibit these channels [96, 97].
Different quaternary ammonium (QA) ions (TButA, TEA, THepA, THexA, TOctA, and
TPenA) are binded to the interior of the TASK-3 pore with high-affinity leading
to inhibition of the I current [98].
Other small molecules such as A1899, A293, DR16, DR16.1, GW2974, ML365, PK-THPP,
RU-TRAAK-1/2 designed and modified to strongly inhibit TASK channels has a
blocker effect on TASK-3 channels, but share affinity for TASK-1 channels as well
[77].
TASK-3 gene is also imprinted in the brain and can participates in cognition,
sleep/wake control, and epilepsy [82]. On the other hand, several types of cancer
cells overexpress TASK-3 channel at the level mRNA and protein, suggesting that
upregulation of the TASK-3 channel may play a role in oncogenesis [99]. In
addition, mutations in KCNK9 gene are associated with Birk-Barel
dymosphyrm syndrome which is an inherited disease characterized by intellectual
disability, hyperactivity, hypotonia and unusual facial features [100].
4. Conclusion, Limitations and New Challenges
Remarkable efforts over several years have been made to define and improve the
molecular, structural and electrophysiological processes underlying the induction
and perpetuation of AF. Our mechanistic and biological understanding of AF is
incomplete and current therapeutic options have limited efficacy and are often
fraught with risk and side effects. There is a primordial need to explore and
propose new therapeutic approaches. Understanding the pathological mechanisms
involved in atrial tissue remodeling and arrhythmogenesis in AF is essential for
developing targeted approaches. Understanding the pathological pathways linked to
atrial arrhythmogenesis and remodeling in AF is crucial for developing new
targeted approaches. Atrial-specific KP channels constitute the background
current in atrial cardiomyocytes and modulate cell excitability emerging as novel
targets in this disease. This family was one of the last K channels to be
cloned, which makes them relatively unknown compared to other channels. By
advancing crystal structures, in silico experiments with molecular
dynamics simulations, mathematical models, docking studies and
electrophysiological studies have greatly helped to reveal the physiological role
of these channels and improve the drug design [101, 102, 103, 104]. Molecular dynamics
simulations provide information at molecular level that can be contrasted with
functional studies [101, 102, 103, 104]. These simulations are used for the study of
conductance, ion selectivity and ion channel opening where the channel is usually
embedded in a membrane patch model that allows following ion movements with a
high spatial-temporal resolution [30, 105]. Knowing in more detail the ion channel
structure allows to perform docking studies between the channel and candidate
drugs, to investigate the points of highest affinity for modulation and to
propose new modulatory structures. For instance, Xin Tan and colleges
investigated how Quercetin alleviate AF explored by network pharmacology combined
with molecular docking and experimental validation [106]. Limberg SH
et al. [30], demonstrated that TASK1 channel modulate atrial AP shape and
duration and shape of human atrial cells using mathematical modeling and
patch-clamp technique.
Given the recent advances related to their physiological importance in different
cellular, k2p channels have emerged as relevant pharmacological targets against a
wide variety of diseases, including AF. So far, TWIK-1, TREK-1, TASK-1, TASK-2
and TASK-3 channel have been identified as responsible for background currents
I current in atrial cells [21, 22]. However, it is not excluded that other
groups or subunits have physiological roles. To date, a great diversity openers,
activators and blockers of KP channel have been identified, particularly
those targeting TASK and TREK channels. However, a major limitation is that many
of these TASK modulators are not selective for TASK-1, TASK-2, or TASK-3 members,
but have affinity for all subunits due to their high homology. The main goal of
this review is to outline the pathways of KP channel modulation in AF and
how correct their dysfunction using different pharmacological KP
modulators. Interestingly, many of heart rate and rhythm controlling medications
used as current therapy in AF patients have a multichannel blocking profile,
among them KP channels. Blockade of KP channels in the heart causes
AP prolongation and may provide antiarrhythmic action in AF [72]. Here, it has
been summarized as several antiarrhythmic drugs exert their mechanism of action
in part by modulation of KP channels. Despite the multitude of known
KP channel modulators, more selective and potent compounds with adequate
pharmacokinetics are needed to avoid side effects on other tissues. There are two
TASK-1 channel blockers under clinical investigation against and AF (DOCTOS
Clinical Trial) and OSA (SANDMAN Clinical Trial) [77, 107]. The future therapeutic
applications of these two compounds for AF are a compelling incentive for further
study of the pharmacology of KP channels. However, a serious complication
of the use of TASK1 blockers for the treatment of AF may be their effect on the
pulmonary vasculature. For example, Wiedmann and colleagues [63] found that
in vivo TASK-1 channel inhibition in pigs with persistent AF was
associated with an increase in pulmonary arterial pressure (PAP), confirming that
TASK-1 plays a role in the homeostasis of the pulmonary vasculature. One way to
overcome this could be to encourage a more cardiac-targeted drug delivery
procedure, cardiac cell therapy or regulation of the expression of this ion
channel by an upstream component, i.e., using oligonucleotide therapeutics. For
example, one study showed that microRNA-34a might regulate the expression of
atrial TASK-1 channels and modulation of this miR-34a can help alleviating AF
[108]. Up to now, many studies have highlighted the pathological role and
pharmacology of TWIK-1, TREK-1, TASK-1, TASK-1, TASK-2 and TASK-3 channels in AF
and, undoubtedly, future research study in this field will provide more detailed
mechanistic knowledge about KP channels allowing the development of new
drugs modulation of these channels to alleviate AF.




