












 , Yoshiko Itoh 1
, Yoshiko Itoh 11 Department of pathology, School of Medicine, Tokai University, 259-1193 Isehara, Japan
Academic Editor: Angelo Facchiano
Abstract
Background: Pulsed ultraviolet (UV) irradiation can be used to generate a broad UV-C spectrum. The pulsing nature of such a spectrum helps increase the damage to cancer cells, leading to their injury and death. In contrast, non-tumor cells repair the damage and survive the same pulsed UV irradiation energy. Herein, we describe the development of a pulsed UV irradiation method for cancer cell dysfunction that irradiates cells with pulsed light by generating tremendous instantaneous UV energy—tens of thousands of times greater than that generated by UV lamps—to cause specific cell injury and dysfunction of cancer cells. Methods: A newly developed pulsed ultraviolet irradiation device was used. Features of the device used in this study. This device employs a quartz discharge xenon lamp. Cultured tumor cells and non-tumor cells were irradiated with pulsed light at different irradiation doses, and their reactions were observed using optical, electron, and laser microscopes. Results: Cancer cells have more FAS (CD95) receptor domains than non-cancer cells, and pulsed UV irradiation stimulates the production of reactive oxygen species (ROS) and OH, which exceeds the oxidative stress removal function, resulting in cell injury and death. That is, at low UV doses, only cancer cells underwent cell death, whereas non-cancer cells did not. The pulsed UV irradiation technique directly destroys cancer cells and minimizes the number of residual cancer cells while allowing minimum invasion into non-tumor cells, thereby improving their survival. This suggests the possibility of activating the host’s local immune response to eliminate residual cancer cells. Conclusions: A newly developed pulsed UV radiation system shows potential for use in the development of a drug-free cancer treatment system that selectively kills tumor cells by irradiating them with high-intensity pulsed UV rays over a broad UV-C range of 230–280 nm.
Keywords
- pulsed UV irradiation
- drug-free pulsed photon therapy
- targeted cancer therapy
- cancer cell injury
- cancer cell death
Traditionally, the three main cancer therapies include surgery (surgical resection, endoscopic resection, etc.), chemotherapy, and radiation therapy. However, these strategies are not specific to cancer cells and can cause off-target damage to the surrounding normal cells, tissues, and organs. For example, in anticancer drug therapy, cellular injury is observed in normal and cancer cells. While this therapy is based on the fact that normal cells recover more quickly than cancer cells, anticancer drug resistance often develops, rendering them ineffective. In addition, combinatorial therapies comprising multiple anticancer drugs are often administered, which may exhibit increased efficacy but also increases the burden on the patient’s body. Meanwhile, surgery requires marginal resection of the normal tissue cells surrounding the cancer, which may result in cancer cells remaining after the surgical intervention. Radiotherapy also damages normal cells and tissues within the irradiated area, which places a heavy burden on the patient. Therefore, developing a specific cancer therapy capable of specifically targeting cancer cells without damaging normal healthy cells and tissues is promising. To this end, near-infrared photoimmunotherapy (NIR-PIT, aka., photoimmunotherapy), using near-infrared light, has attracted attention as a new cancer treatment method [1, 2, 3]. Indeed Japan has led the way in NIR-PIT administration, having already demonstrated some success in cancer patients. In fact, the medical community and patients have predicted that photoimmunotherapy will represent the “fifth cancer treatment method”, following anticancer agents, surgery, radiation, and cancer immunotherapy.
Photoimmunotherapy is an innovative treatment that can be used to damage and kill cancer cells. As a pretreatment, a drug combining cancer cell-specific antibodies and light-reactive chemicals are administered intravenously, allowing the antibodies to reach the cancer cells in approximately one day [1, 2, 3]. When near-infrared light is incident on the affected area, a chemical reaction occurs, and the cancer cells are destroyed. In addition, Kobayashi and Choyke [4] reported further enhancement of NIR-PIT efficacy, designated as SUPR (super-enhanced permeability and retention effects). Under the SUPR effect, drug-based NIR-PIT only kills perivascular antigen-expressing tumor cells, leaving tumor vessels intact early after treatment while also allowing a significant increase in blood volume and nanodrug perfusion into the tumor bed within tumor vessels immediately after NIR-PIT. Cellular injury by drug-induced NIR-PIT reportedly occurs in the perivascular layer of tumor cells and is accompanied by marked dilation of tumor vessels in the dilated tumor stroma [4]. Notably, this regimen also requires the administration of a hybrid drug comprising cancer cell-specific antibodies and photoreactive chemicals as pretreatment. Although the mechanism of action is different, similar vasodilatation effects can be obtained with drug-free pulsed UV irradiation methods.
UV rays elicit bactericidal effects [5, 6, 7, 8]; low-pressure mercury vapor lamps (UV lamps) have long been used as sterilizing lamps. In recent years, “optical pulse sterilization” using xenon flash lamps has proved to be a novel technique [9, 10, 11, 12]. Xenon flash lamps can instantaneously emit light in the 200–300 nm wavelength range, which is considered highly effective for sterilization in the order of microseconds; each of which provides tens of thousands of times more energy than a UV lamp (~65 W). Light pulse sterilization with xenon flash lamps is a method that demonstrates high sterilizing power in an extremely short time (less than one second to several seconds) against bacteria, molds, and spore-forming fungi. Accordingly, this method is beginning to be put into practical use in food preparation, as well as in other fields [9, 13, 14]. However, its application in selective damage and death of tumor cells by pulsed light using xenon flashlamps or similar devices has not been reported. In this study, we developed a new cancer therapy using specific xenon lamp pulsed irradiation that selectively injures cancer cells and causes cell death without drug injection (drug-free).
A newly developed pulsed UV radiation system was used (S-BP60, Thunderlight
Corporation, Kanagawa, Japan). The luminescent characteristics of the UV field
are shown in Fig. 1A. This apparatus has a quartz discharged xenon lamp with a
pulse full-width half-maximum of 0.29
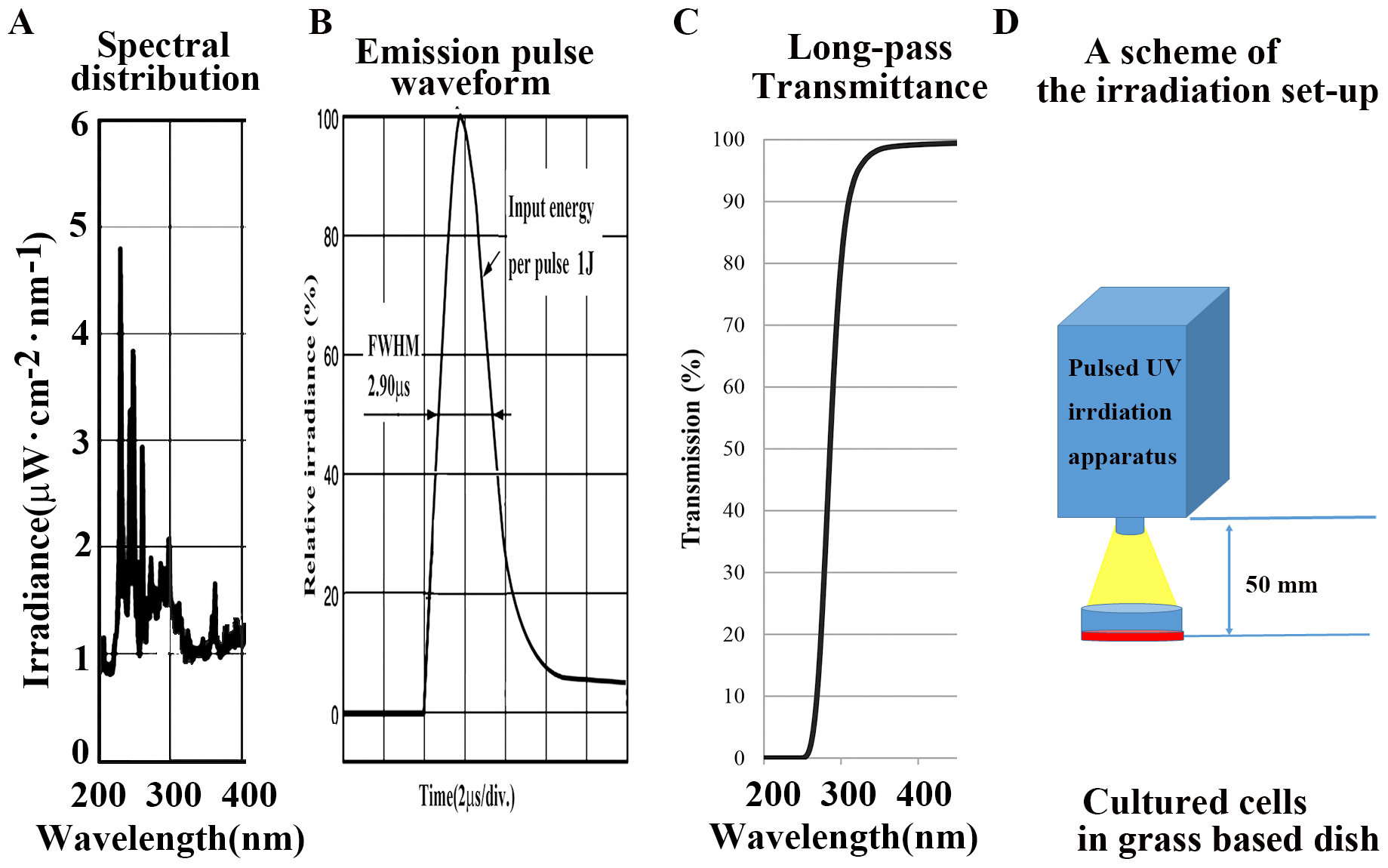 Fig. 1.
Fig. 1.Characterlizations of equipment used in this article. (A)
Spectra in the UV region (200–400 nm). (B) Emission pulse waveform. (C) Internal
transmittance curve in a long-pass filter (UV N-WG280; absorption 200–280 nm,
Edmund Optics Japan, Japan) in the UV region of a UV pulse flash to be indicated
from a xenon flash lamp (S-BP60, Thunderlight Corp. Japan) at a light source. (D)
Scheme of the irradiation setup. The total exposure energy was compared at 0, 25,
50, 100, 200, and 500 mJ/cm
An FUD-7010J system (FUJIFILM Corporation, Japan) was used for ultraviolet light intensity distribution analysis. This system enables digitization of UV, integrated light intensity values, as well as analysis and storage of the UV light intensity distribution by reading the color development of the UV scale.
The cellular effects of pulsed UV irradiation on tumors were compared with those
induced by pulsed UV irradiation at 0, 25, 50, and 100 mJ/cm
MCF-7 (human breast adenocarcinoma, EACC) and Cos7 (African green monkey cell
line producing the T antigen of SV40) were routinely cultured in DMEM (Dulbecco’s
modified Eagle’s medium) high glucose and 10% fetal bovine serum obtained from
Gibco (Thermo Fisher Scientific K.K. Japan) in 35 mm diameter glass-based dishes
(IWAKI (AGC), Japan) and maintained in a humidified incubator at 37 °C
with a 5% CO
Cultured MCF-7 and Cos7 cells were analyzed with and without fixation in 4%
paraformaldehyde (PFA), primarily using ZEISS LSM880 (ZEISS, Germany). Fixed
cells were covered with a 10% glycerol solution to prevent drying, while living
cells were observed under culture. Cells were observed using Plan-Apochromat
lenses (10
Pulsed UVC-irradiated, non-irradiated, and UV-C-irradiated cells were also compared. Cell viability (dead cells/total live and dead cells) was analyzed 4–40 h after irradiation using the IMRIS (Ver 9.0.2) image analysis software (Bitplane AG, Switzerland). Additionally, some 12- and 24-h time-lapse observations were performed.
The cells were fixed in glutaraldehyde solution for 1–2 h at 4 °C. Post-fixation, cells were incubated in 1% osmium tetroxide solution for 2 h at 4 °C. The cells were then embedded in an epoxy resin for 6 h. A plastic embedding plate (or gelatin capsule) with fresh epoxy resin was filled, and the tissue was placed in it and allowed to polymerize in an incubator at 60 °C for three days. Ultrathin sections were observed under an electron microscope JEM-1400 (JEOL, Japan).
Briefly, the cells were fixed in 4% paraformaldehyde dissolved in 0.01 M phosphate-buffered saline pH 7.4 (PBS). After fixation, cells were processed in a conventional manner using a JSM-6510LV electron microscope (JEOL, Japan).
To reveal the immunocytochemical localization of CD95, CD95 (FAS) antibody,
anti-human, PE, REAfinity™ (Miltenyi Biotec, Germany) were used. A
total of 2
For the cytochemical detection of Annexin V, a multi-parameter apoptosis assay kit (Cayman Chemical, MI, USA) was used. Using Annexin V-FITC reagent (maximum peak emission fluorescence wavelength: 517 nm) phosphatidylserine (PS) is redistributed to the external plasma membrane of apoptotic cells. Annexin V staining can result in morphological changes in the cells or even their release from the culture surface.
To determine mitochondrial membrane potential, the TMRE dye (maximal fluorescence wavelength: 595 nm) in the Multi-Parameter Apoptosis Assay Kit (Cayman Chemical, MI, USA) was used. TMRE is a cell-permeant, cationic, red-orange fluorescent dye that is readily sequestered by the active mitochondria. The membrane potential was thus preserved.
Briefly, Annexin V and TMRE staining was performed as follows. Cultured cells (2
Oxidative stress results from an imbalance between ROS production and the ability of cells to scavenge ROS. ROS plays an important role in the progression of several diseases, including inflammation, atherosclerosis, aging, and age-related degenerative disorders [16]. To detect oxidative stress in cells, CellROX™ Deep Red Reagent (Molecular Probes; Thermo Fisher Scientific K.K., Japan) was used. CellROX™ Deep Red Reagent is a novel fluorogenic probe for measuring cellular oxidative stress in both live and fixed cell imaging, with an emission maximum at 665 nm. The cell-permeant dye is non-fluorescent in a reduced state and exhibits bright fluorescence upon oxidation by reactive oxygen species (ROS).
To detect caspase-3/7 activity in the cells, CellEvent™ Caspase-3/7 Green Detection Reagent (Invitrogen, Thermo Fisher Scientific K.K. Japan) was used (maximum peak emission fluorescence wavelength: 530 nm). CellEvent™ Caspase-3/7 Green Detection Reagent is a novel fluorogenic substrate for activated caspases 3 and 7. The reagent consists of a four-amino acid peptide (DEVD) conjugated to a nucleic acid-binding dye. This cell-permeant substrate is intrinsically non-fluorescent because the DEVD peptide inhibits the ability of the dye to bind to DNA. Following activation of caspase-3 or caspase-7 in cytotoxic cells, the DEVD peptide is cleaved, enabling the dye to bind to DNA and produce a bright, fluorogenic response with an emission maximum of 530 nm.
To determine the ROS activity in the cells, 3,3-(p-hydroxyphenyl) fluorescein (HPF) was used as an indicator of ROS (Invitrogen, Thermo Fisher Scientific K.K. Japan) with a maximum peak emission fluorescence wavelength: 515 nm. HPF exhibits limited non-selective reactivity and relatively high resistance to light-induced oxidation. The new fluorescein derivatives are non-fluorescent until they react with the hydroxyl radical or peroxynitrite anion. ROS indicators can be used to selectively detect hypochlorite anions.
Briefly, cultured cells (2
To detect intracellular hydroxyl radicals (•OH), the Cell Meter™ Mitochondrial Hydroxyl Radical Detection Kit *Red Fluorescence* MitoROS™ OH580 (AAT Bioquest, California. USA) was used (maximum emission fluorescence wavelength: 598 nm). Detection of intracellular •OH is of utmost importance for understanding proper cellular redox regulation, and the impact of its dysregulation on various pathologies. •OH is an ROS that is highly reactive with other molecules to achieve stability. Hence, •OH is considered a harmful by-product of oxidative metabolism and can cause molecular damage in living systems. It shows an average lifetime of 10–9 ns and can react with nearly every biomolecule, such as nuclear DNA, mitochondrial DNA, proteins, and membrane lipids. This kit is optimized for detecting •OH in the mitochondria. MitoROS™ OH580 is a live cell-permeant probe that can rapidly and selectively target •OH in live cells after only 1 h of incubation. Red fluorescence is generated when it reacts with •OH.
In brief, the medium was removed, and 200
The integrated irradiance per unit area [J/cm
As shown in Fig. 2, when a xenon flash lamp (S-BP60, Thunderlight Corp.) was
used as the light source for pulsed UV irradiation and the distance between the
xenon flash lamp and the irradiated area was 5 cm, UV irradiation at 25
mJ/cm
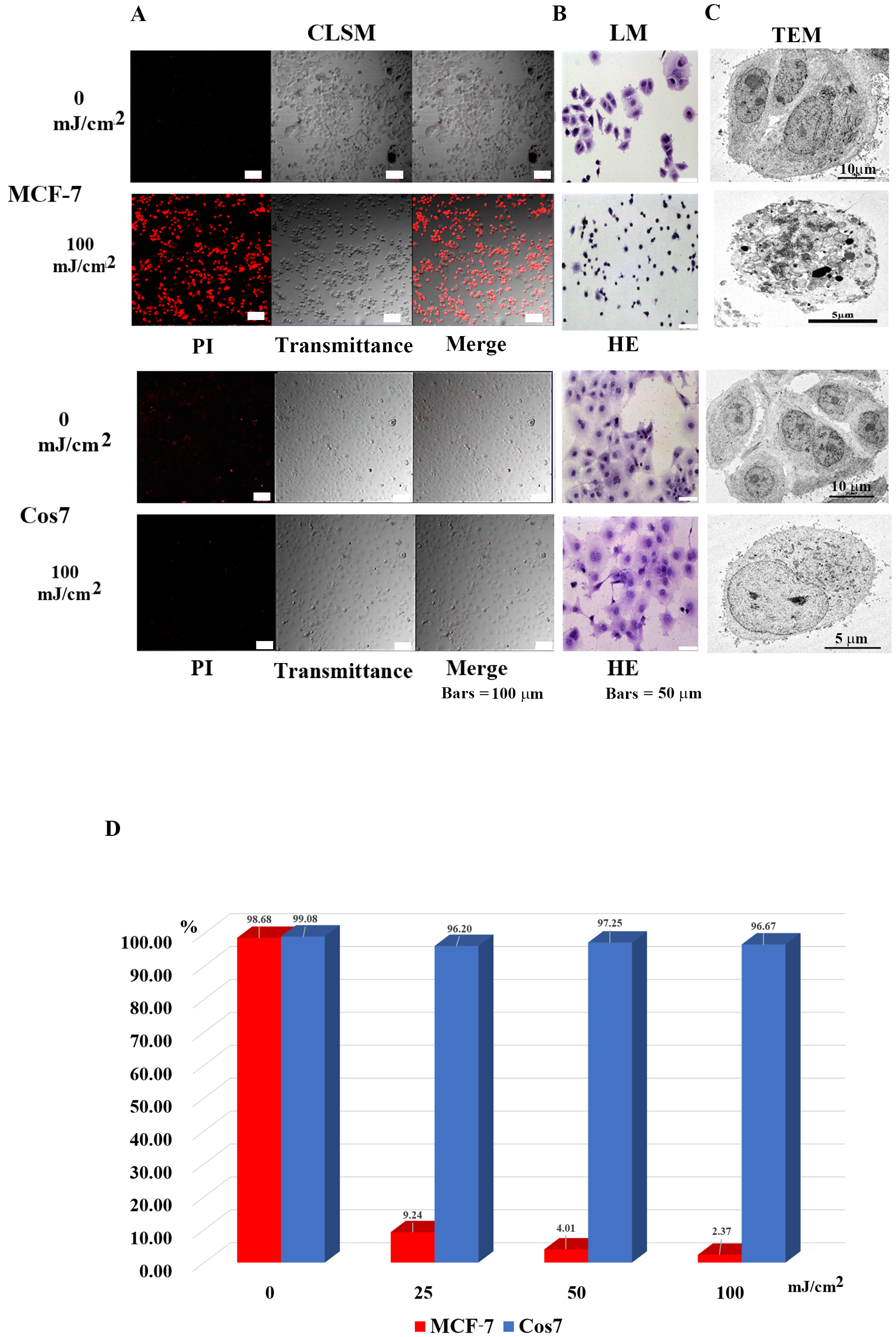 Fig. 2.
Fig. 2.Microscopic observation of tumor cells (MCF-7) and
non-tumor cells (Cos7) 24 h after irradiation. (A) Confocal laser scanning
microscopy images. In MCF7 tumor cells, most in the control group survived, while
most tumor cells in the 100 mJ/cm
As shown in Table 1, the tumor cells that showed efficacy in this method included cancer (malignant tumors of epithelial tissue origin), sarcoma (malignant tumors of non-epithelial tissue origin), leukemia, malignant lymphoma, benign tumor cells, and cell lines. In contrast, non-tumor cells are intact cells other than tumor cells. Hence, this method can be applied to sites where tumor cells and non-tumor cells are mixed.
| Tumor cells | Non-tumor cells | ||||
| Human | Type of cell | Cell name | Human | Cell type | Cell name |
| 1 | Human breast cancer cell line | MCF7 | 1 | Human mammary epithelial cell | HMEC |
| 2 | Human breast cancer cell line | BT474 | 2 | Leukocytes | Leukocytes |
| 3 | Human Cervical Cancer cell line | HeLa | 3 | plasma | plasma |
| 4 | Human leukemia cell line | MOLT/S | Monkey | ||
| 5 | Human leukemia cell line | MOLT/TMQ 200 (Trimetrexate (TMQ) anti cancer drag resistance 200 times) | 1 | African green monkey cell | Cos7 |
| 6 | Human leukemia cell line | K562/S1 | Dog | ||
| 7 | Human leukemia cell line | K562/S2 | 1 | Canis normal kidney | MDCK |
| 8 | Human leukemia cell line | K562/ARA-C (Cytarabine anti cancer drag resistance) | Mouse | ||
| 9 | Human fibrosarcoma cell line | HT-1080 | 1 | Mouse fetal fibroblast | NIH3T3 |
| 10 | Human prostate cancer cell lines | DU145 | |||
| 11 | Human prostate cancer cell lines | PC3 | |||
| 12 | Malignant cell lines of human choriocarcinoma | BeWo | |||
| 13 | Human renal cancer cell line | ACHN | |||
| 14 | Human renal carcinoma cell line | Caki1 | |||
| Mouse | |||||
| 1 | mouse lymphoma | EL-4 | |||
| 2 | mouse lymphoma | A20 | |||
| 3 | mouse lymphoma | RL male 1 | |||
| 4 | mouse lymphoma | B16 | |||
Additionally, transmittance observations and spectral analysis of PI
fluorescence was performed using LSM880 (Figs. 2,3). Atrophy was observed in
all cells used in the experiments; however, irradiation at 25 mJ/cm
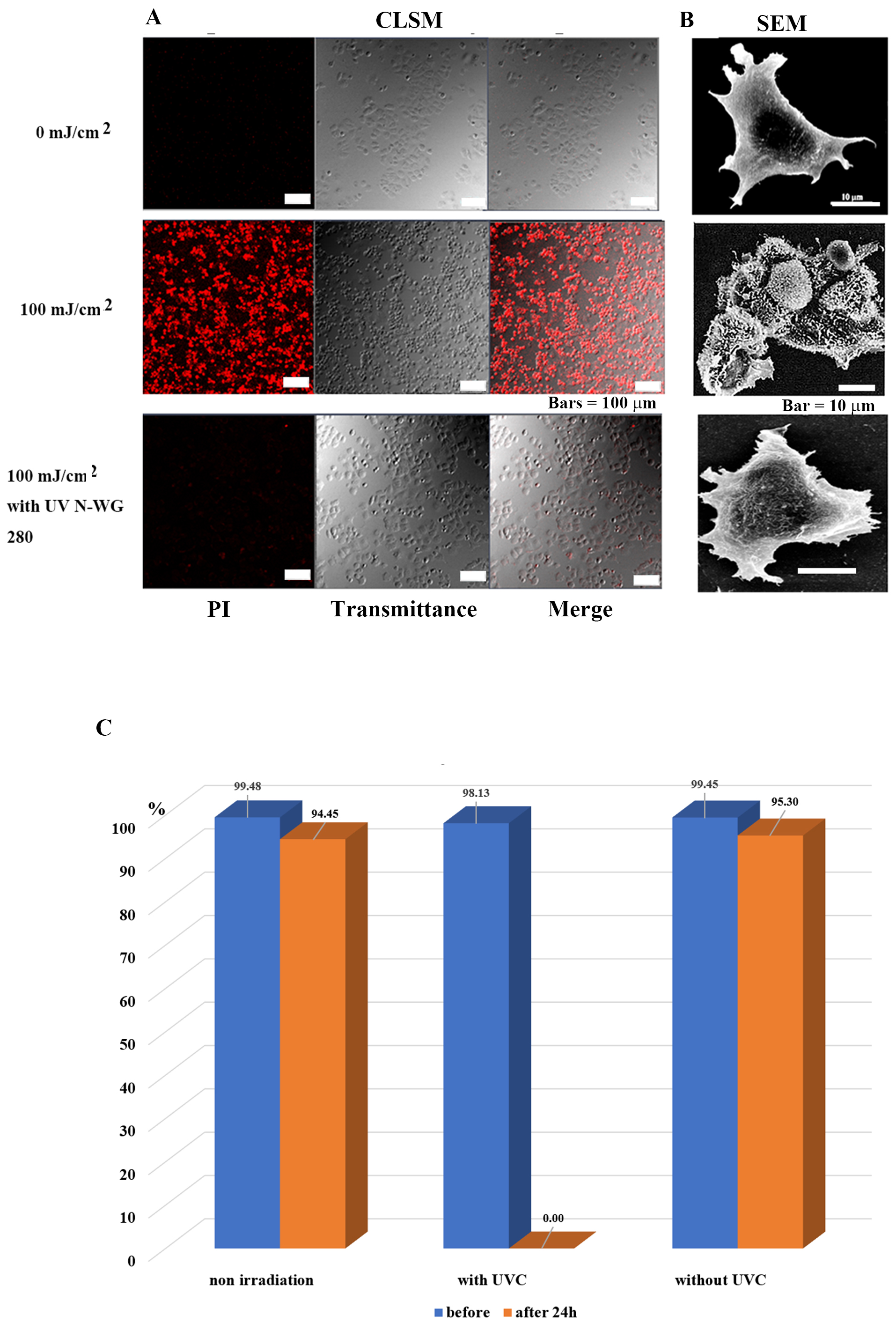 Fig. 3.
Fig. 3.Selective tumor cell (MCF-7) injury and death is determined by
the presence or absence of UV pulse irradiation. (A) Images of MCF-7 tumor cells
observed by confocal laser scanning microscopy after 24 h of irradiation. The
tumor cells in the control group survived, while nearly all treated tumor cells
died. Tumor cells survived following irradiation exposure after removing UV-C
with a long-pass filter UV N-WG280. (B) Scanning electron microscopy images of
MCF7 cells after 24 h of irradiation. In the control group, the nucleus, and
subcellular organelles were clearly observed; in the 100 mJ/cm
Pulsed UV (including UVC) irradiation killed tumor cells at 100 mJ/cm
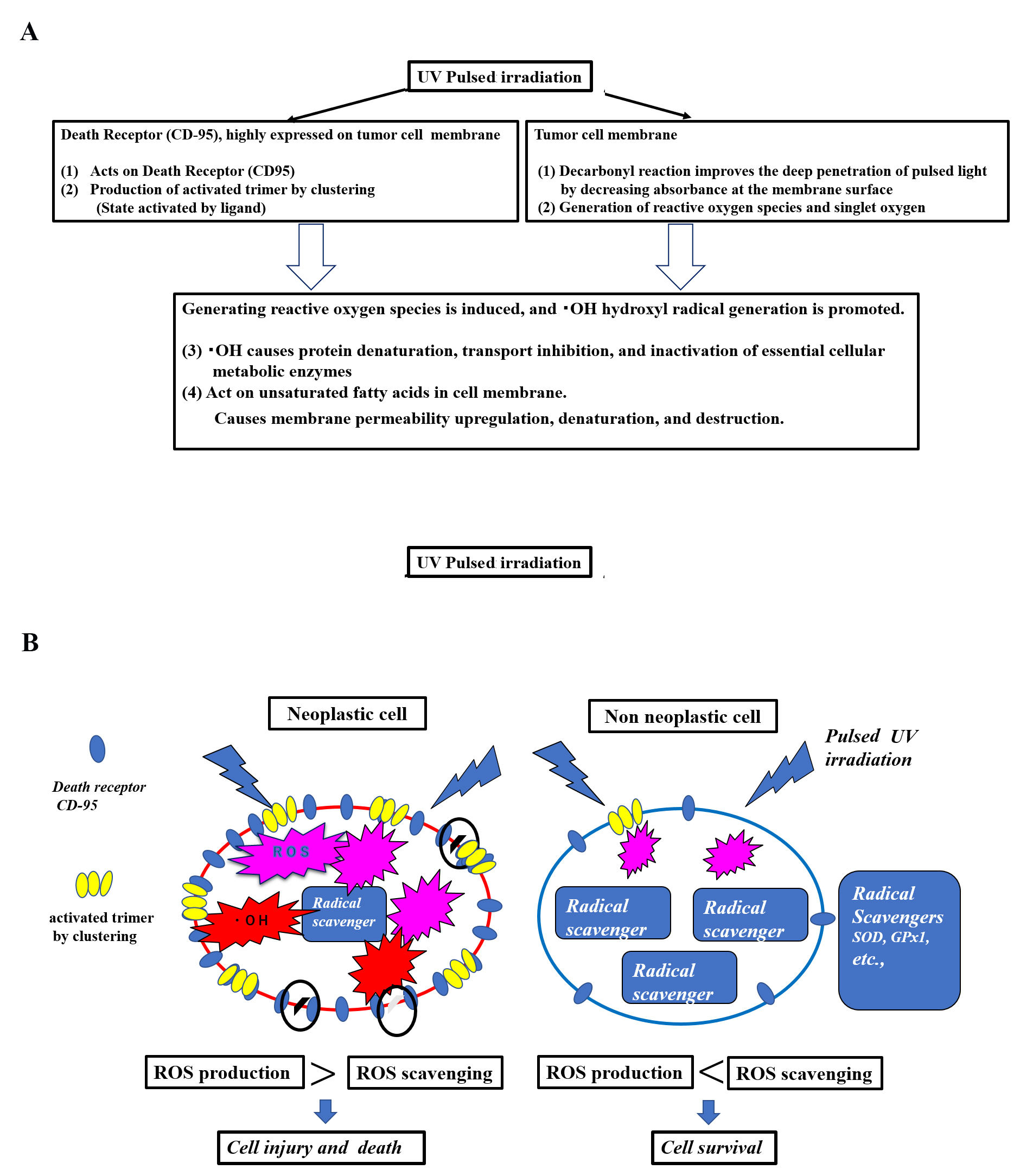 Fig. 4.
Fig. 4.Hypothesis of a mechanism of selective tumor cell injury/killing in pulsed light irradiation. (A) Hypothesis: Cellular injury and death due to pulsed UV irradiation membrane disruption. (B) Hypothesis: Cellular injury and death due to scheduled UV membrane disruption in neoplastic cells vs. non-neoplastic cells. This hypothesis involves selective tumor cell death by pulsed UV irradiation. Functional differences exist between tumor and non-tumor cells. Death receptors, such as CD95, are highly expressed by tumor cells, while being lowly expressed by non-tumor cells. Additionally, radical scavenging is low in tumor cells and high in non-tumor cells. Pulsed UV irradiation directly activates death receptors, such as CD95, thereby inducing receptor trimerization and clustering, primarily in tumor cells. It then induces the production of ROS and further production of •OH radicals. As tumor cells have a lower radical scavenging function, the production of ROS and •OH radicals occurs in a chain reaction, causing membrane destruction of subcellular organelles. Conversely, non-tumor cells express fewer death receptors and have a higher radical scavenging function; therefore, the removal of ROS and •OH radicals is prioritized, and the cells typically survive.
The death receptor (CD-95) is highly expressed on tumor cell membranes, and its localization was observed by immunocytochemical assay (Fig. 5) [15, 17, 18]. In tumor cells MCF7, CD95 was found to be highly localized on the plasma membrane, while in non-tumor Cos7 cells, CD95 was observed at low levels. The IMARIS analysis of positive counts in the same field of view showed MCF7:17418 and Cos7:497 positive grain, indicating that CD95 was approximately 37 times more localized in tumor cells (Fig. 5A). At the individual cell level, the localization of CD95 was clearly greater in tumor cells than in non-tumor cells (Fig. 5B).
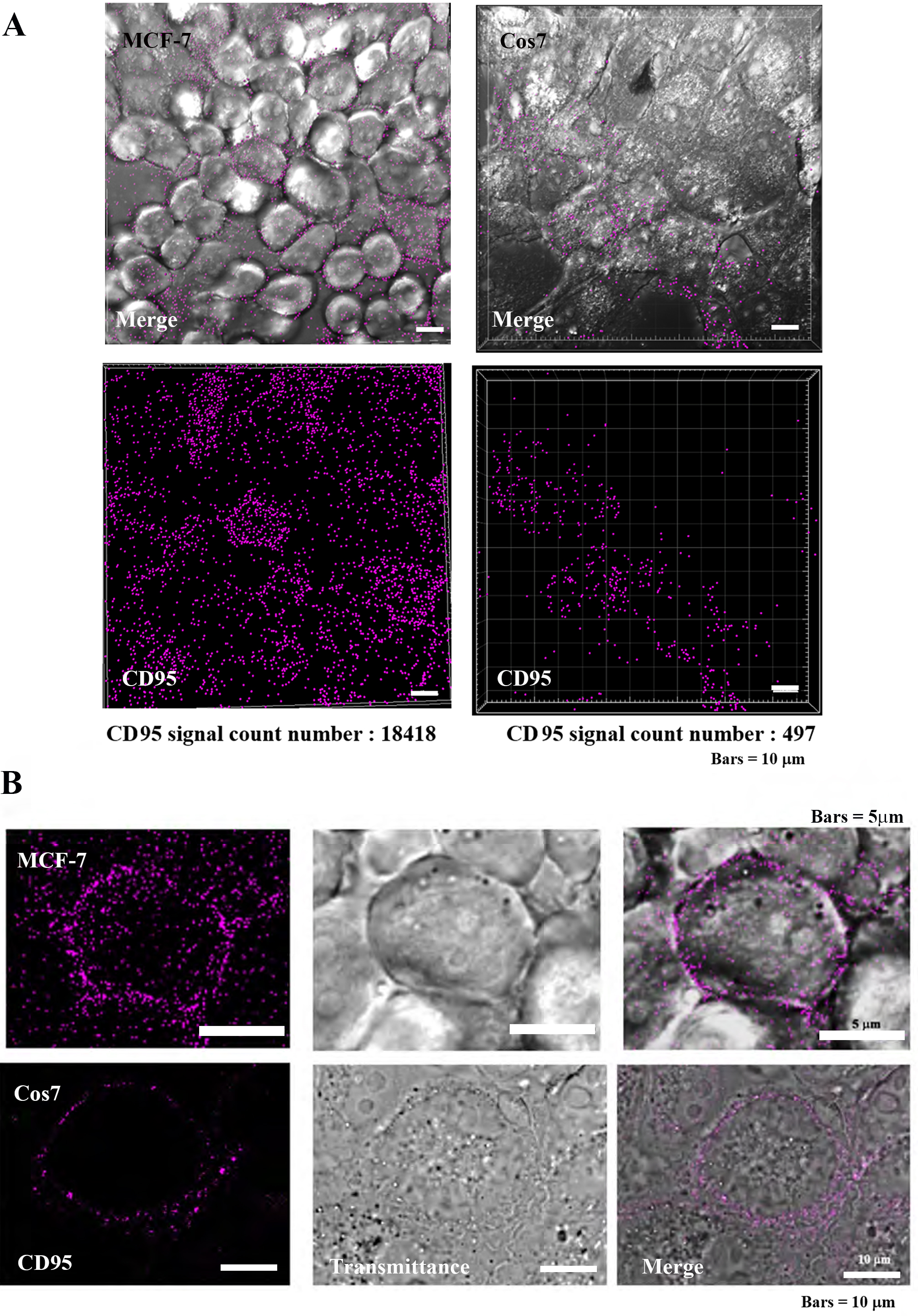 Fig. 5.
Fig. 5.Localization of CD95 in tumor cells (MCF-7) and
non-tumor cells (Cos7). (A) Confocal laser scanning microscopy (CLSM) images
(low power magnification, Bars = 10
Each cell was irradiated with a UV pulse flash at a predetermined irradiation
dose, as described above, and the effect on early apoptosis (DNA damage) was
examined [19, 20]. Using annexin V as a marker, early apoptosis was analyzed by
confocal laser scanning microscopy (CLSM) in a similar manner to that of cell
viability. In early apoptosis, phosphatidylserine (PS) is expressed on the outer
layer of the cell membrane and binds to annexin V, which has a high affinity for
PS. Annexin V is a 35–36 kDa protein that binds to phospholipids in a
Ca
MCF-7 and Cos7 were irradiated with 0 and 100 mJ/cm
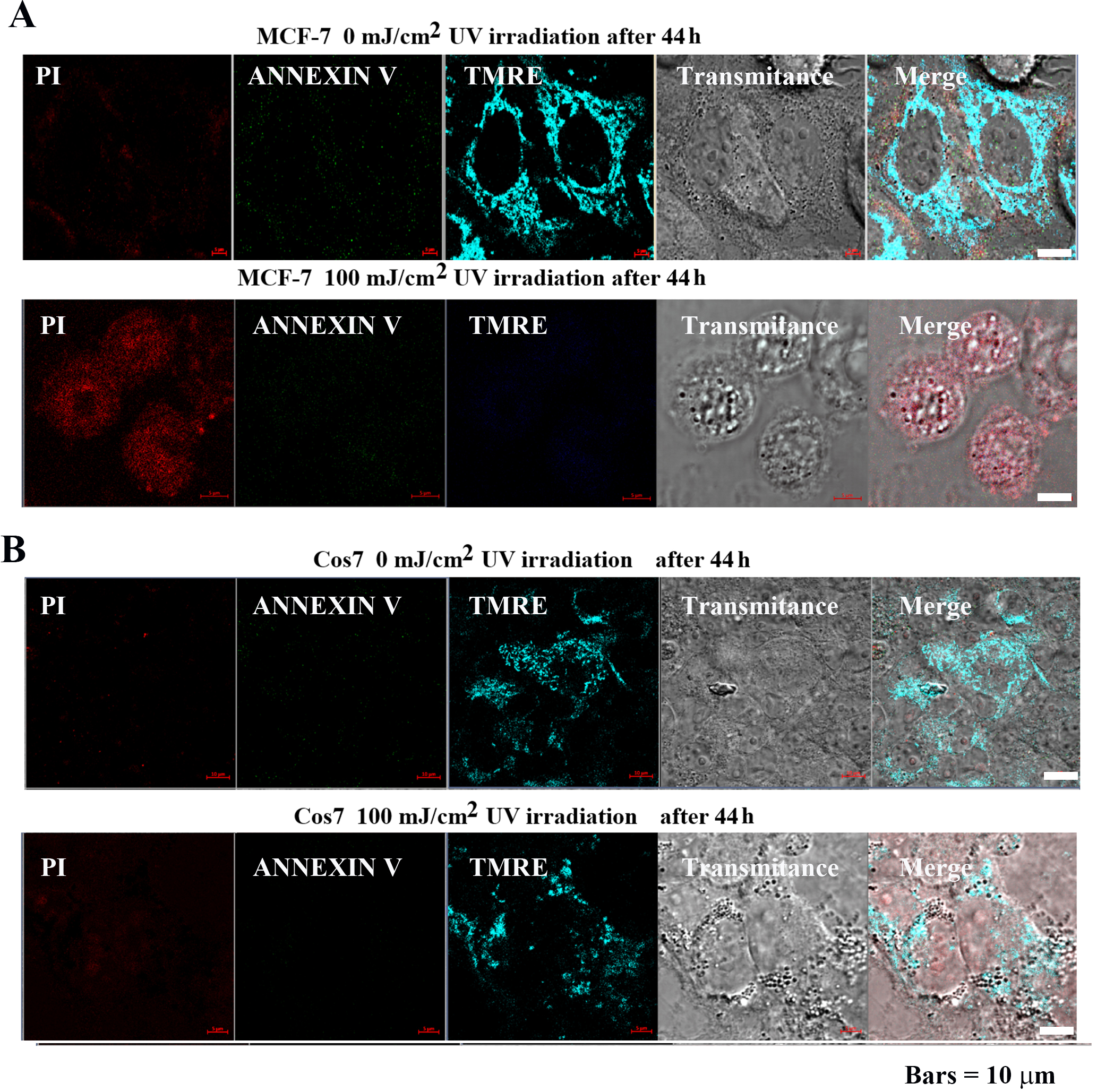 Fig. 6.
Fig. 6.Impact of pulsed UV irradiation on MCF-7 and Cos7
apoptosis and mitochondrial membrane potential. (A) In MCF-7 cells, the control
group exhibited low PI, Annexin V signal intensity, and the tetramethylrhodamine,
ethyl ester (TMRE) signal was highly localized in the mitochondria. In contrast,
the PI signal intensity increased after 100 mJ/cm
Mitochondrial membrane potential was analyzed using TMRE to confirm the effect of pulsed UV irradiation on mitochondria. TMRE is a cationic fluorophore of the ethyl ester of rhodamine. Owing to the lipophilic structure of the fluorescent dye, it easily penetrates lipid bilayers, albeit at a slow rate. In mitochondria with membrane potential, it accumulates at high concentrations up to the saturation level and emits red-orange fluorescence. However, when mitochondria lose their membrane potential due to apoptosis or metabolic stress, the fluorescent dye diffuses throughout the cell, and the fluorescence intensity decreases markedly [22].
In MCF-7 cells (Fig. 6A), TMRE signal intensity was high following 0 mJ/cm
Cell morphology, atrophy, cytoplasmic lysis, disruption of the cell membrane,
and destruction of subcellular organelles were observed. In contrast, in Cos7
(Fig. 6B), TMER fluorescence intensity remained high at both 0 mJ/cm
The CellROX™ Deep Red Reagent was used to confirm the response of
tumor cells (MCF-7) and non-tumor cells (Cos7) to oxidative stress (OS) induced
by pulsed UV irradiation. In MCF-7 cells, PI and OS signal intensities were
extremely low, and cell morphology was normal after 0 mJ/cm
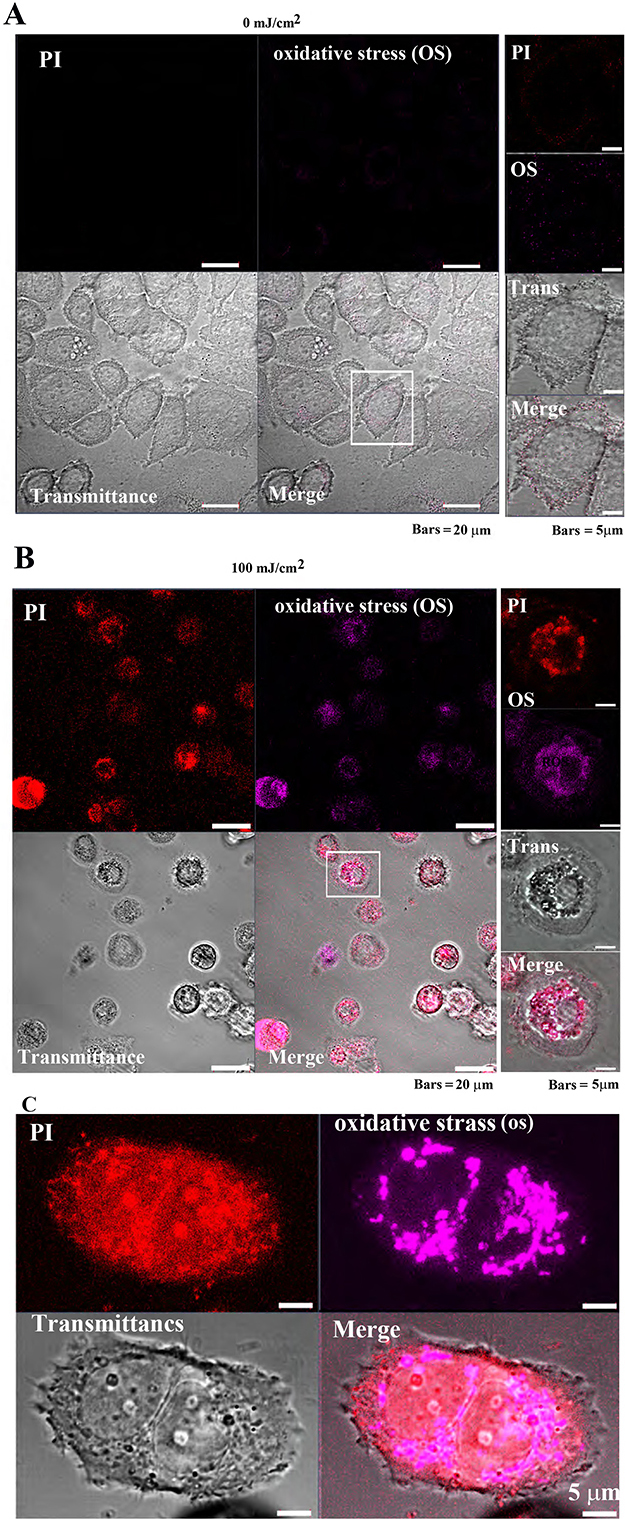 Fig. 7-1
Fig. 7-1Oxidative stress detection in MCF-7 cells following
pulsed UV irradiation. (A) Control MCF-7 cell’s (after 24 h of pulsed UV
irradiation) PI and oxidative stress (OS) signal intensity are weakly distributed
in the cytoplasm. (B) Twenty-four-hour UV-irradiated MCF-7 exhibited cell
atrophy, cytoplasmic loss, and enriched nuclei. Most cells were PI-positive (cell
death), and OS signals were also detected. Right panels: increased magnification
of the white square in the left panel. (C) OS detection in a single MCF-7 cell.
Observed images after 4 h of pulsed UV irradiation. Detection of OS in MCF-7
cells: Images were observed after 4 h of irradiation with pulsed UV at 100
mJ/cm
Additionally, following 100 mJ/cm
In Cos7 cells, PI and OS fluorescence intensities remained weak at both 0
mJ/cm
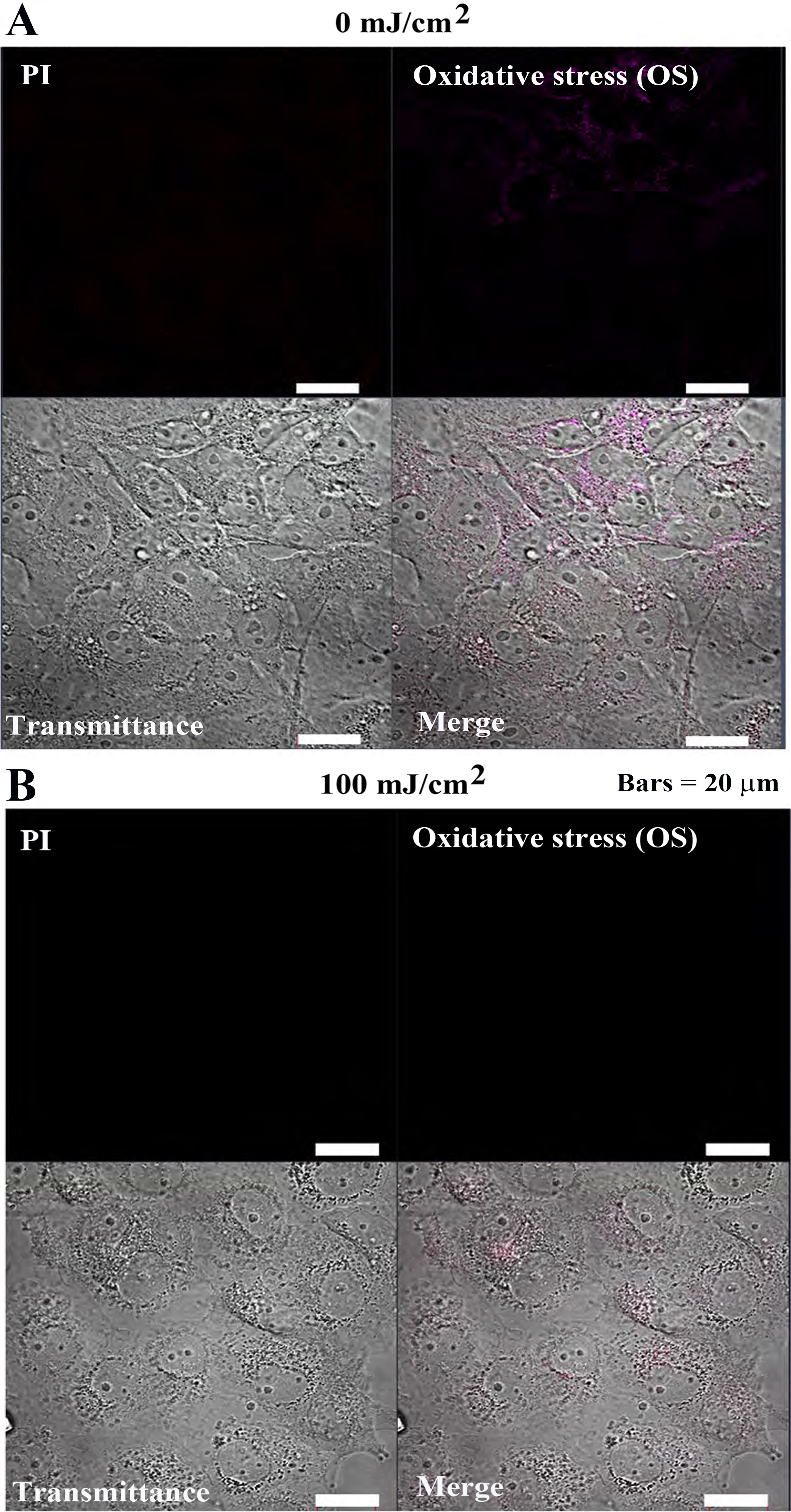 Fig. 7-2.
Fig. 7-2.Oxidative stress detection in Cos7 cells after 24 h of pulsed
UV irradiation. (A) 0 mJ/cm
Time-lapse observation of PI and Caspase-3/7 activity detection was performed
for 10 h after irradiating MCF-7 with 100 mJ/cm
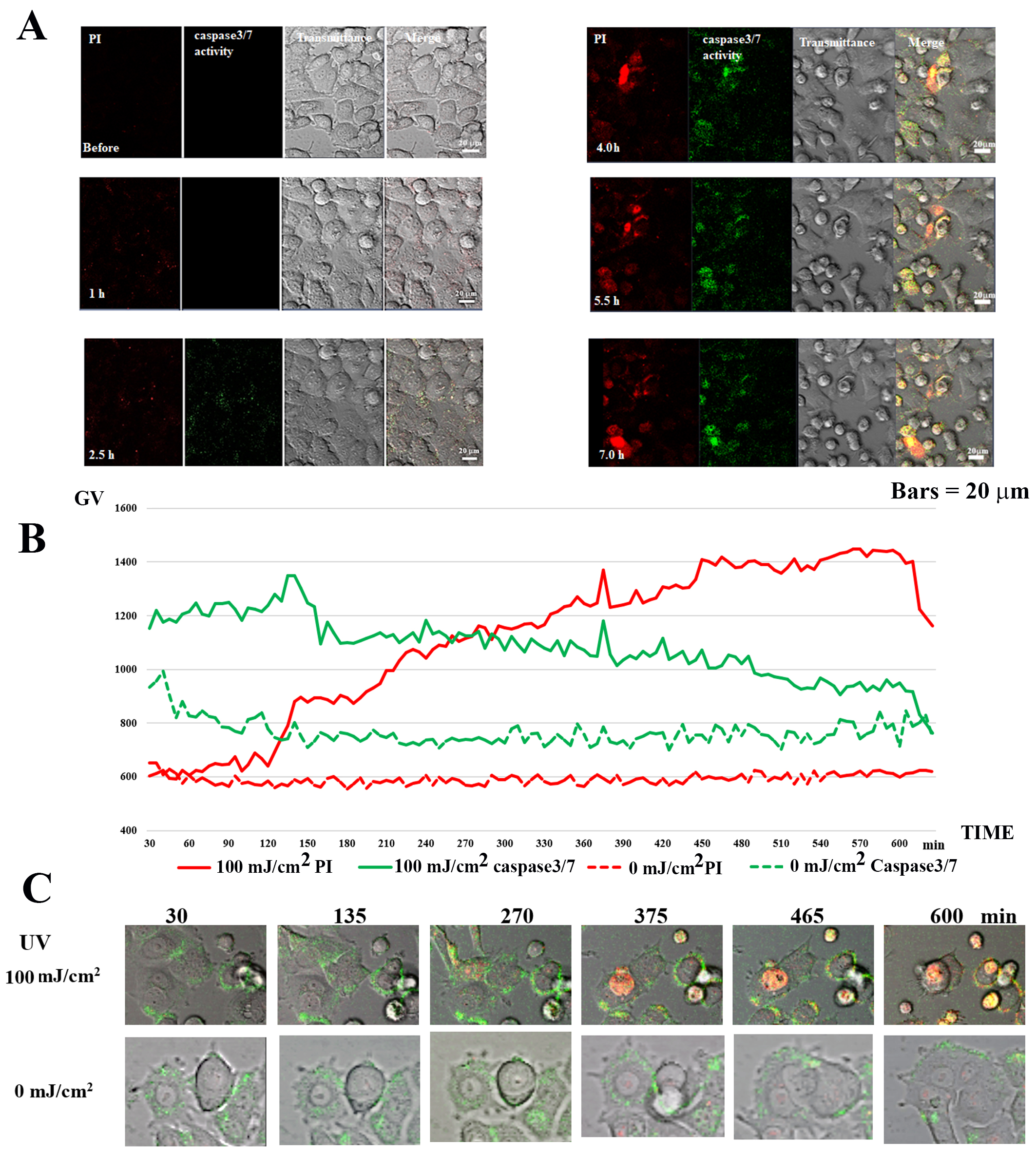 Fig. 8.
Fig. 8.Caspase 3/7 activity in UV-irradiated MCF-7 cells. (A) Caspase
3/7 and PI staining in 100 mJ/cm
In the case of 100 mJ/cm
To determine the ROS activity in the cells, HPF—an indicator of high ROS
levels—was used. After 40 h at 0 mJ/cm
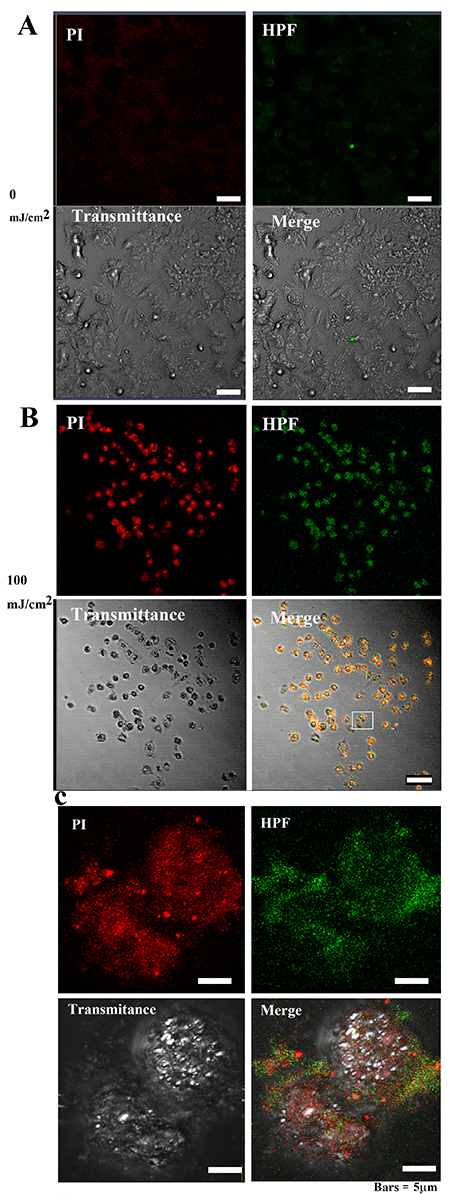 Fig. 9-1.
Fig. 9-1.•OH and 3-(p-hydroxyphenyl) fluorescein (HPF)
abundance in MCF-7 cells after 40 hours of irradiation. (A) The PI and OS signal
intensities in MCF-7 cells were weakly distributed in the cytoplasm of control
cells. Bar = 50
Following 0 mJ/cm
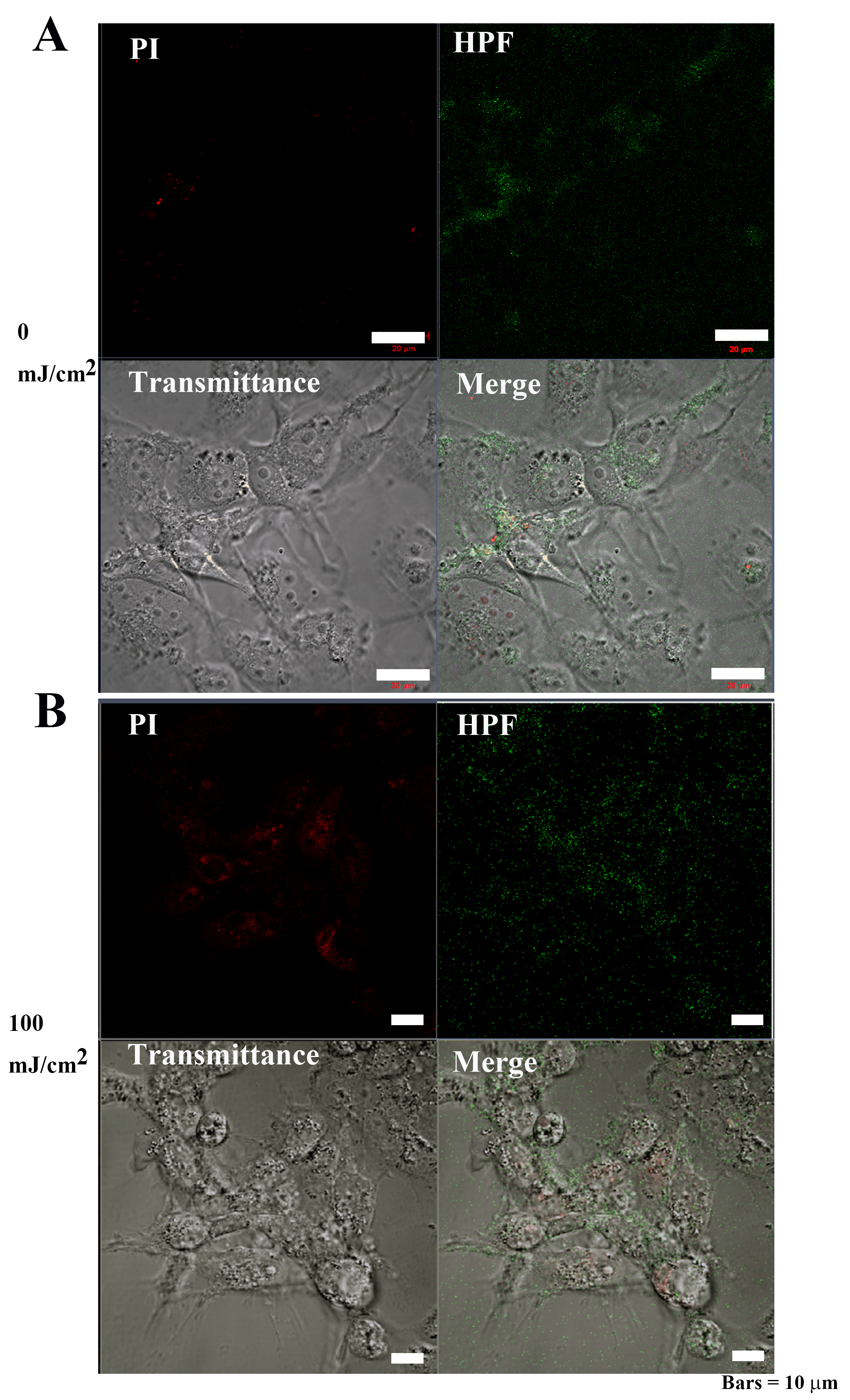 Fig. 9-2.
Fig. 9-2.OH and 3-(p-hydroxyphenyl) fluorescein (HPF)
abundance in Cos7 cells after 40 hours of irradiation. (A) The signal
intensities of PI and OS were weakly distributed in the cytoplasm of control Cos7
cells. Bar = 50
Observation of MCF-7 cells following UV irradiation for 24 h revealed that the
control cells exhibited weak PI and •OH, fluorescence signals (Fig. 10A), while cells treated with 100 mJ/cm
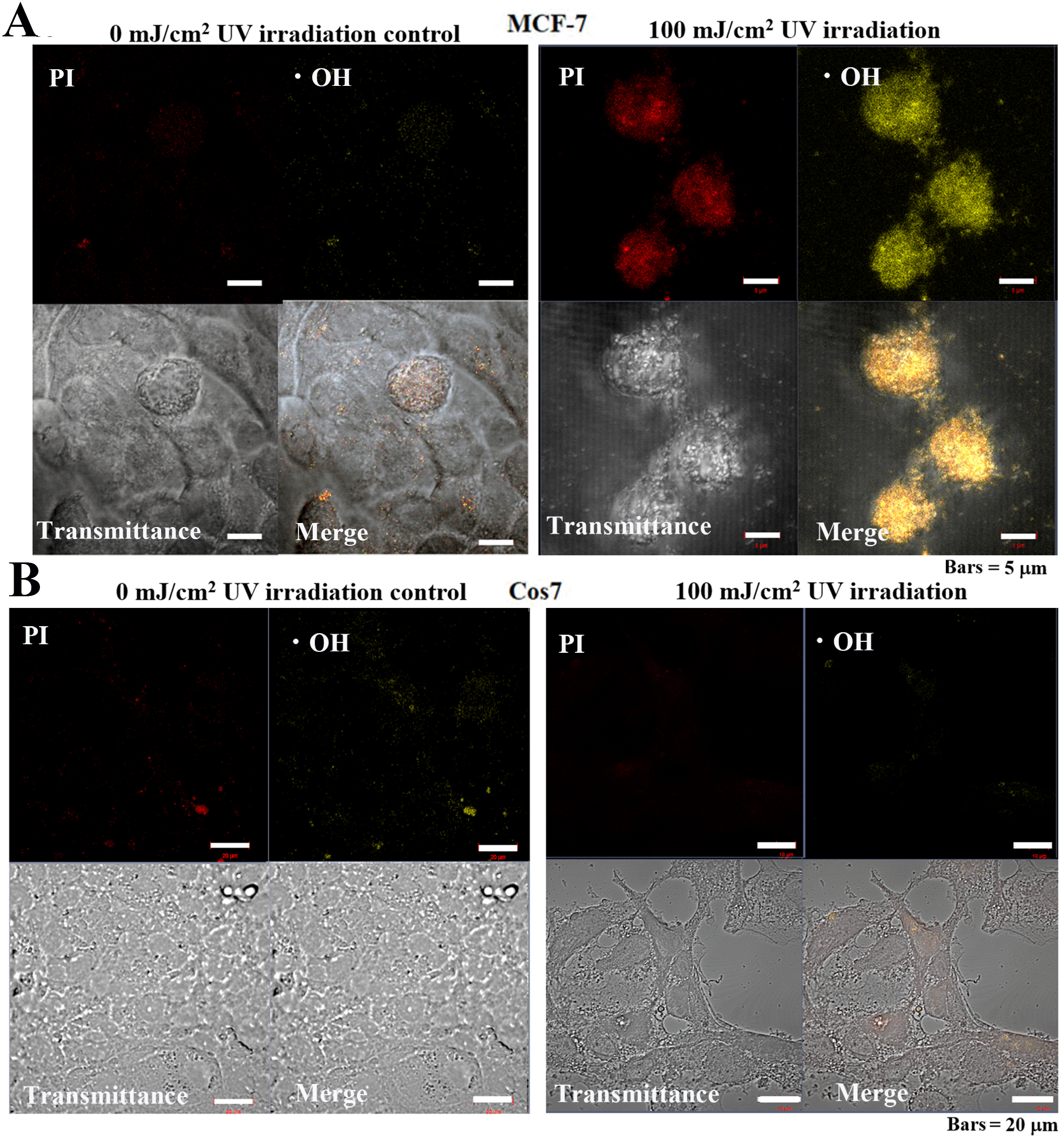 Fig. 10.
Fig. 10.Cellular mitochondrial ROS-OH580 activity under 24 h pulsed UV
irradiation in MCF-7 and Cos7 cells. (A) The signal intensities of PI and
ROS-OH580 in control MCF-7 cells were weakly distributed in the cytoplasm and
nucleus. Bar = 5
Continuous UV-C irradiation has long been known to be effective in killing a wide range of bacterial species and has been used as a means of sterilization [22, 23, 24]. However, continuous irradiation with UV rays in such an environment requires a long irradiation time of 10–50 min. In recent years, pulsed UV light and food sterilization equipment have become increasingly popular. Pulsed UV sterilization can kill a wide range of bacteria, viruses, yeasts, and protozoa within a few seconds of irradiation [9, 10, 11, 12, 13].
In the current study, we attempted to apply a method of pulsed UV light, which
has excellent bactericidal effects, for killing tumor cells. The pulsed UV
irradiation method was demonstrated to be tumor effective when irradiated with
100 mJ/cm
Hence, we confirmed the phenomenon of selectively injuring and killing only
tumor cells while negligible impacts on non-tumor cells (Table 1). Based on these
findings, we proposed a new drug-free UV pulse irradiation cancer therapy method
[PDT (PhotoDynamic Therapy)
The death receptors (Fas: CD95) on the cell membrane surface are expressed disproportionately by tumor cells compared to non-tumor cells [25, 26, 27, 28]. Application of pulsed UV irradiation causes clustering of these death receptors, resulting in their trimerization and activation, similar to that following activation by a ligand [29]. Meanwhile, the pulsed light causes decarbonylation of the surface of living tissue cells, thereby improving light penetration into the body. However, pulsed light generates ROS and singlet oxygen only during pulsed irradiation [30].
Intracellularly, generation of ROS is induced, and •OH generation is promoted [31, 32, 33]. Subsequently, •OH causes protein denaturation, transport inhibition, and inactivation of essential cellular metabolic enzymes. Additionally, •OH acts on unsaturated fatty acids in the cell membrane. Collectively, this causes upregulation, denaturation, and destruction of membrane permeability.
As mentioned above, tumor cells express higher levels of CD95 on their cell membrane compared to non-tumor cells. Upon pulsed UV irradiation, CD95 forms a cluster and undergoes trimerization, thereby stimulating the production of ROS. This phenomenon causes a domino-like chain reaction resulting in increased ROS production. However, the number of radical scavengers present in tumor cells is low, thus limiting the scavenging of ROS produced in the tumor cells, resulting in a further increase in ROS [34]. A subsequent chain reaction of ROS and •OH radical production proceeds, resulting in protein denaturation, transport inhibition, inactivation of essential metabolic enzymes, renewal, denaturation, and destruction of membrane permeability by radicals [16].
In contrast, given that non-tumor cells have fewer death receptors (CD95) [27], the presence of radical scavengers is high [34]. Hence, pulsed UV irradiation causes clustering of CD95 and activation of its trimer; however, ROS is produced in small quantities and readily scavenged, allowing the cells to return to their normal state without experiencing membrane destruction by OH radicals.
Our immunochemistry results support this hypothesis as CD95 localization was observed to be significantly greater in tumor cells versus non-tumor cells. More specifically, observations at the cellular level showed that the CD95 signals were localized at the plasma membrane surface (Fig. 5).
Apoptosis is cell injury induced by continuous-wave light UV irradiation
[20, 21]. Moreover, for each cell, the effect of irradiation with a UV pulse flash
at a prescribed frequency in the same manner as described above for early-stage
apoptosis (DNA disorder) was studied. Using Annexin V as a marker, early-stage
apoptosis was analyzed by CLSM. At an early stage of apoptosis, PS, found inside
the cell membrane, is expressed on the outside, where it binds with Annexin V.
Annexin V is a protein of 35–36 kDa that bonds with phospholipid in a
Ca
Tumor cells irradiated with 0 mJ/cm
Time-lapse observation of pulsed UV irradiation at 100 mJ/cm
When the oxidative damaging power of ROS and free radicals generated in the body outweighs the antioxidant power (radical scavengers) in the body, oxidative stress rises and leads to the production of OH radicals [37, 38].
In control tumor cells exposed to 0 mJ/cm
In contrast, within control non-tumor cells, HPF fluorescence was observed in some cells, and cell morphology was normal, while for the irradiated cells, weak PI and HPF fluorescence were observed in the cytoplasm. Taken together, these findings indicate that pulsed UV irradiation showed strong HPF fluorescence intensity in tumor cells and negligible fluorescence intensity in non-tumor cells (Figs. 9-1,9-2).
In control tumor cells, negligible levels of UV PI or •OH fluorescent signals were detected. Alternatively, in irradiated tumor cells, strong fluorescent signals were detected in the nuclei for both PI and •OH. Meanwhile, in non-tumor cells, no PI nor •OH fluorescent signals were observed in the control or irradiated cells (Fig. 10).
In tumor cells, the intracellular chain reaction of radical production induced by UV irradiation seems to exceed the scavenging function, leading to cell death. In contrast, in non-tumor cells, although UV-induced intracellular radical reactions occurred, the radical scavenging function was superior, suggesting that the cells underwent repair and survived [32, 39, 40, 41, 42]. However, when the same energy irradiation was performed with a continuous UV mercury lamp, both tumor and non-tumor cells were killed, and the phenomenon of selective tumor cell death did not occur (data not shown). Hence, as hypothesized, the observed phenomenon only occurred with pulsed UV irradiation.
The current method shows that tumor cells are selectively killed at a range value by different irradiation doses of pulsed UV light. Hence, only tumor cells can be selectively killed. Without the need for drug injection pretreatment (drug-free), only tumor cells can be easily eliminated in a short time without causing cell death of non-tumor cells simply via irradiation with pulsed ultraviolet light. It is believed that this strategy can be applied to identify drug-free treatments for tumor cancer tissues. In particular, we hope to use this principle to develop endoscopic treatment, intraoral treatment, and body-surface tumor irradiation devices. Collectively, this study suggests that the application of PDT (PhotoDynamic Therapy) to PPT (Pulsed Photon Therapy) is possible and effective.
PDT, PhotoDynamic Therapy; PPT, Pulsed photon therapy; UV, Ultraviolet; ROS, reactive oxygen species; NIR-PIT, near-infrared photoimmunotherapy; SUPR, super-enhanced permeability and retention effects; DMEM, Dulbecco’s modified Eagle’s medium; PFA, paraformaldehyde; PI, propidium iodide; TEM, transmission electron microscope; SEM, scanning electron microscope; PBS, phosphate-buffered saline; EDTA, ethylenediaminetetraacetic acid; BSA, bovine serum albumin; TMRE, tetramethylrhodamine ethyl ester; DEVD, a four-amino acid peptide; HPF, 3,3-(p-Hydroxyphenyl) fluorescein; DMF, imethylformamide; CLSM, Confocal Laser Scanning Microscopy; PS, phosphatidylserine; •OH, hydroxyl radical; OS, oxidative stress; TMQ, trimethrexate.
JI and YI performed the experiments and analyzed the data. JI designed the study and wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
We thank Thunderlight Corporation for providing the S-BP60 ultraviolet light irradiation system. This study was conducted using facilities of the Tokai University School of Medicine Office.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.