






1 Department of Biochemistry, Biotechnology and Bioinformatics, Avinashilingam Institute for Home Science and Higher Education for Women, 641043 Coimbatore, India
2 Redox Regulation Laboratory, Department of Zoology, Odisha University of Agriculture and Technology, College of Basic Science and Humanities, 751003 Bhubaneswar, India
3 OrangeCross Home Health Private Limited, 751012 Bhubaneswar, India
Abstract
Molecular network-based studies have gained tremendous importance in biomedical research. Several such advanced technologies in molecular biology have evolved in the past decade and have contributed to building up enormous molecular data. These molecular networks gained much significance among researchers triggering widespread use of experimental and computational tools. This interest led researchers to compile data of biomolecules systematically and to develop various computational tools for analyzing data. In the present scenario, an enormous amount of molecular network databases are available which can be accessed freely by the public. This is the central focus of this article.
Keywords
- Drug discovery
- Online tools
- Networks
- Therapeutics
- Drug development
- Drug design
- Web tools
- Review
A drug is a chemical component whose structure is probably known, exerts a biological effect when it is administered to an organism [1]. The role of a drug is to prevent or cure a particular disease or disorder. In the past, health practitioners and people used plant extracts as a medicine to treat or cure diseases. However, currently, most of the drugs available commercially are either synthesized or produced using genetic engineering and scaling up techniques [2].
A pharmaceutical drug alters the structure and activity of molecular networks by changing the activity of biomolecules. The drug targets may be peptides, proteins, or nucleic acids. Drugs are classified into three categories (i) biological compounds that target membrane receptors and extracellular proteins (ii) nucleic acids which target mRNA [3, 4], (iii) low molecular weight compounds which target receptors or enzymes [5]. These small molecules are mostly preferred because of their low cost and can be delivered easily to the target site. But the major disadvantage of these small molecules is that they can target a very less number of proteins [6]. Many alternative forms of medicines are now also available for the treatment of several diseases albeit their scientific background is yet to be established [7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20]. Therefore, controversies are also seen in literature with such remedies [21]. Although both in silico and in vivo approaches are adopted to discover drugs with potential [22], drug, dose, compatibility, side effects are existing issues in biomedical sciences. Therefore, many nutraceuticals are suggested for consumption and championed to be equally important as medicines to handle diseases [13, 23, 24, 25, 26, 27].
In the field of biomedical research, identification of a drug target is a time consuming and laborious task. Moreover, it is not possible to screen every target for a drug in the laboratory for both direct and alternative medical approaches [13, 14, 15, 16, 18, 19, 26]. Starting from environmental health [7, 27], nutritional data [25], to complicated miRNA interaction in health and aging [3, 4], the generation of vast data is very common. For example, bio-medical pathway studies, not only provide new insights, but also vast data [13, 23, 24]. Although many in silico tools are used to study bio-phenomena in cells [28, 29, 30, 31], tools for handling data, as well as techniques for identification of drugs and their targets by in silico methods have gained momentum only lately. Most of the computational methods for analyzing drug-target interactions are based on receptor or ligand models. In the case of receptor-based methods, a target with a known structure is subjected to docking to analyze its drug binding efficiency [32]. On the other hand, the ligand-based prediction of Drug Target Interaction (DTI) involves the comparison of drugs with the target protein’s ligand. Singh et al. [33] studied some potential targets of drugs by using the ligand-based prediction method. In recent years, network-based methods play a major role in the identification of drugs and their targets. The major advantage of this method is that even if the 3D Structure of a drug or target is not known, their interaction can be predicted.
Analyzing the human genome by sequence homology to the known drug targets is one of the ways to identify an effective drug target. For this analysis, information from protein structural databases is very vital. Structural information plays a vital role because some homologs cannot be identified using sequence data alone, moreover, analysis of the effectiveness of drug-target binding can also be predicted through this. Side effect similarity is one of the methods which can also be used to identify drugs and their targets [34]. In this article, we have discussed Network-Based Drug Discovery (NBDD) and its use in health sciences.
Interaction of a drug with its target is the basis for drug discovery. Identifying drug-target interaction experimentally is a laborious and expensive undertaking. With the development of internet and data repositories, it is efficient and inexpensive to use computational methods for identifying the interaction of a drug with its target. Computational methods can be subdivided into different categories like molecular docking-based, pharmacophore-based, similarity-based, machine learning-based and network-based methods. Among these methods, network-based methods are highly reliable and have an advantage over others (Fig. 1).
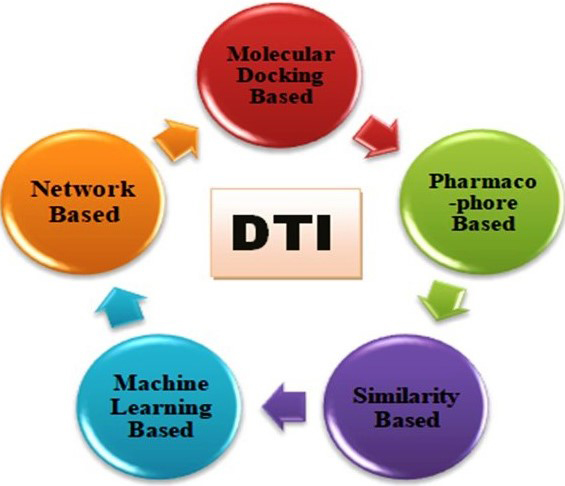 Fig. 1.
Fig. 1.Methods for the identification of drug target interaction.
Since there is an enormous development in network pharmacology and systems biology, there is a leap in the drug discovery paradigm from linear mode (single drug → single target → single disease) to a network mode (multiple drugs → multiple targets → multiple-diseases) [35, 36, 37]. The change in this paradigm means that there is a possibility for a drug can interact with multiple targets rather than with a single target [38, 39, 40]. The interaction of a drug with its target can lead either to a desired therapeutic effect or some undesired side effects [33, 37, 38, 41, 42, 43]. Knowledge of the above can help predict drug-target interaction during the discovery of a new drug and prevent unnecessary side effects thereby increasing the therapeutic efficacy of the drug.
Traditionally, researchers used to identify the drug-target interactions using some parameters like their dissociation and inhibition constant, half-maximum effective and inhibitory concentration via biochemical experiments both in vivo and in vitro. The major drawback of adopting these methods is the time and expense required to systematically test every drug for its interaction with the target through biochemical experiments. To overcome these problems, a lot of computational tools have been developed in the past decade which is cost-effective and highly efficient [44, 45, 46].
It is a traditional method for the prediction of a drug-target interaction. This method is mainly based on the three-dimensional structure of the targets and it is widely used [47, 48, 49, 50, 51, 52]. In this method, scoring functions are used to analyze the drug-target interaction, and a quantitative docking score can be obtained for a drug to its corresponding target. This docking score can be directly correlated with the binding affinity of a drug with its target [53, 54]. In another docking method called reverse docking, a potential target can be predicted for a known drug which is vice versa of the above method [47, 55, 56]. The major applications of molecular docking include polypharmacology, drug repositioning, analysis of adverse side effects, and target hunting [57]. Many web applications including TarFisDock [58] and DRAR-CPI [32] were designed to perform docking based on target hunting.
Pharmacophore modeling is a widely used method in the identification of drug-target interaction. Pharmacophore based methods are mainly categorized into two types namely structure-based and ligand-based methods (Fig. 2). Both the subtypes can be used efficiently in the prediction of drug-target interactions [47, 59]. The programs that are available for pharmacophore modeling includes Molecular Operating Environment (MOE) (Chemical Computing Group), Pharmer [60], LigandScout [61], Screen (ChemAxon Screen Suite), DiscoveryStudio (BIOVA Discovery Studio) and Phase [62]. Some of the web servers for pharmacophore modeling include PharmMapper [63], ZINCPharmer [64], CavityPlus [65], and Pharmit [66]. The interaction of a protein with the ligand normally exerts a pharmacological effect and this is the basis of ligand-based pharmacophore modeling. The web server that can be used to identify the pharmacophore of selected ligands is pharmagist. The major advantage of this web server is it is available free of cost and it will give the results in few minutes [67]. On the other hand, the structure-based pharmacophore modeling requires a ligand-binding structure which will analyze the interaction site and produce a suitable pharmacophore model. Zinc Pharmer is open-source software that is used mainly in this method. Usually, the structures are derived from the protein database bank and ZINC database and subjected to analysis in ZINC Pharmer for pharmacophore modeling [68, 69]. After analysis, a statistical score for the potential targets will be available which further enhances the method. More than 7000 ligand-based pharmacophore models are available in this software.
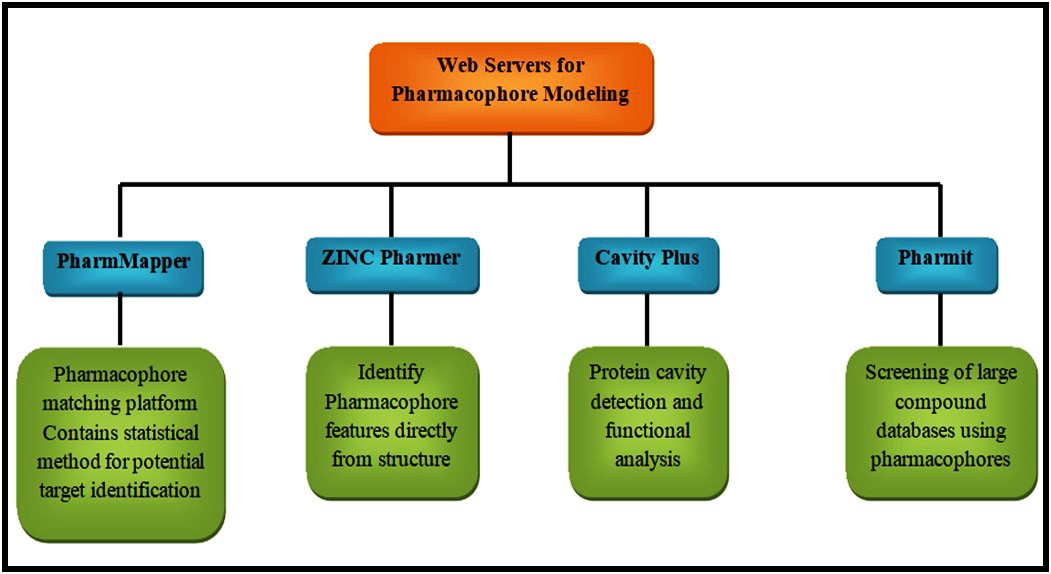 Fig. 2.
Fig. 2.Web servers for pharmacophore modeling.
It is also a traditional method for the prediction of drug-target interaction [70]. It purely depends on the assumption that similar drugs will have similar targets and vice versa. The similarity among two different drugs can be analyzed either using the structure of a drug or its profile, and the similarity between the targets can be identified using a sequence of the targets. The input data including the structure or profile of a drug or the target sequence should be given by the user. if the drug structure is given as an input data the webserver analyses for the similarity between the given data and the data available in the databases which can be used to predict the DTI from known drug target to the unknown one [71]. Different types of similarity-based methods are available including 3D shape-based similarity [72], two-dimensional (2D) fingerprint-based similarity [73], and phenotypic based similarity [34] methods. Many web applications including ChemMapper [74] and Similarity Ensemble Approach (SEA) [75] are available to which applies 2D and 3D similarity for the prediction of drug-target interaction (Fig. 3).
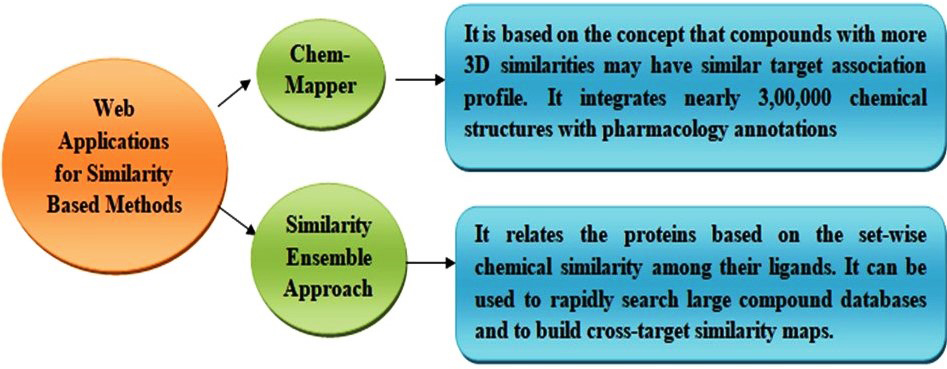 Fig. 3.
Fig. 3.Web applications for similarity-based method for the identification of drug target interaction.
Machine-learning based methods are generally used in drug-target interaction and are experiencing rapid development in recent years [76, 77]. When compared to the traditional machine learning techniques, some modern techniques like deep learning techniques are applied more recently for the prediction of drug-target interaction [78, 79]. Even though the three-Dimensional structure data of the target can be used for developing machine learning methods [80], of late protein sequence descriptors and molecular descriptors are mostly used [76, 81, 82].
Machine learning-based methods are broadly classified into two subtypes: supervised and semi-supervised methods. In the case of the supervised machine learning method, a training set requires both the positive and negative labeled samples for the prediction of drug-target interaction. Here the positive labeled sample indicates the known interaction of known drug targets and the others are labeled as negative. The supervised method is again subdivided into similarity-based methods and feature vector-based methods. Similarity-based methods mainly rely on the similarity between the different drugs or targets but in the feature vector based method, feature vectors are used as the training data. These feature vectors contain the different properties of targets and drugs. Huge number of supervised models have been proposed by the researchers and most of them are found to be feasible. A semi-supervised machine learning-based method requires a little labeled data and more unlabeled data for the prediction of drug-target interaction. The semi-supervised method uses the labeled data to identify the unlabeled data [83].
When compared to all other DTI prediction methods, the network-based method has gained much prominence recently. The network-based methods do not only depend based on 3 Dimensional structures of the targets. The algorithms used to derive the network-based method include recommendation algorithms and link prediction algorithms. In the past decade, a recommendation algorithm called network-based inference was derived. This algorithm is also called probabilistic spreading (ProbS) [84].
Network-based methods are one of the simplest methods and it does not need any other additional information like structure or sequence for the prediction of drug-target interaction. Several other methods were also developed based on this method [85, 86, 87]. Since these methods are not dependent on the 3-dimensional structure of the protein it can be useful for the targets whose 3D structures are not known. The major advantage of these techniques includes simple, fast, and accurate. It is based on very simple physical processes such as resource diffusion [86, 87, 88], collaborative filtering [88, 89], and random walk [90] on networks. When compared to the other drug-target interaction prediction methods explained earlier the calculation procedure for the network-based methods is very simple. So, network-based methods are preferred mostly, and they can run very fast on the computer.
Drug repositioning is a process which is used to find a new curative value for pre-existing drug, and it is quite closely related to drug target prediction [91]. Since an already existing drug is used it is very easy to identify the target when compared to working with a new drug. For drug repositioning, the above-mentioned methods for drug-target interaction can also be applied. Many software programs have already been developed for drug repositioning. Some examples of drug repositioning are the use of thalidomide for severe erythema nodosum leprosum, retinoic acid for acute promyelocytic leukemia and sildenafil for erectile dysfunction and pulmonary hypertension [92, 93].
In technical words, the process of identifying new indications for the already approved drugs is called drug repositioning [94]. Since this process uses existing drugs, some of the preapproval tests usually performed for the drugs are not needed since they are already proved to be safe for human consumption. Therefore, the drug discovery process is very short in the case of drug repositioning [95, 96]. For this reason, governmental, non-governmental agencies, and academic researchers are showing a great interest in drug repositioning. Drug repurposing, drug redirecting, drug re-tasking, drug re-profiling, and therapeutic switching are the synonyms for drug repositioning. New therapeutic uses for failed drugs (which are safe to use) can also be developed using drug repositioning [97]. More in silico tools have been developed for drug repositioning [98].
Drug repositioning has major advantages, first and foremost they are cost-effective and there is less risk for drug development since it is developed from an already existing drug. Repositioned drugs can easily pass through all the clinical trials and they have a very less possibility of developing any adverse effects thereby facilitating the development of a potential drug in a shorter span [99, 100, 101]. It has a wide range of applications in disease and related therapeutic areas. Evolving new anticancer drugs from available anticancer drugs is one of the leading fields in drug repositioning because the demand for anticancer drugs continues to increase [100].
Drug repositioning plays a vital role in developing drugs for diseases by which affect only a few people, the major reason behind this is that there is an insufficient financial benefit for developing a new drug by the pharmaceutical industry [102]. With the help of drug repositioning, it is possible to identify novel therapies for those diseases with limited development costs. The major problem for drug inefficiency is drug resistance. For the use of non-antibiotic drugs to overcome antimicrobial resistance, Drug repositioning will be very helpful [103]. Drug repositioning helps to improve the efficacy of a drug and to avoid drug failure [104].
Drug target interaction networks form the basis for network-based models. For the construction of a quality network enormous and sufficient data is needed [105, 106]. Large amounts of data are available from small molecules to macromolecules and they are available online for free (Table 1). These data include structures, properties, etc. [77].
| S.No. | Data base | Application |
| 1 | Binding DB | It is a publicly accessible database that contains measured binding affinities. It contains 1,794,819 binding data, for 7,438 protein targets and 796,104 small molecules. |
| 2 | Binding MOAD | It is a subset of the protein data bank and it contains many examples for ligand-protein binding. So it is called the Mother of All Databases (MOAD). The main aim of this database is to collect the data about protein crystal structures with their relevant ligands. These data are normally extracted from the literature. |
| 3 | ChEMBL | It is a database that contains biologically active molecules with drug-like properties. It contains information including chemical, genomic data, and bioactivity to translate the genomic information into an effective drug. |
| 4 | DrugCentral | Drug Central contains information about the mode of action of a drug, pharmacological action, chemical entities, and pharmaceutical products. Some information about discontinued drugs is also available in this database. |
| 5 | IUPHAR/BPS Guide to PHARMACOLOGY | This database has information about ligand-activity-target relationships; this information is taken from chemistry and medicinal literature which are high in quality. The main aim of this database is to provide maximum information about drug targets. |
| 6 | PDBbind-CN | It is a compilation of experimentally determined binding affinity data for all the biomolecules present in PDB. |
| 7 | PDSP Ki Database | This database contains information about the potentiality of the drugs to interact with the molecular targets. It contains experimentally derived and published affinity values for more number of drugs with their targets. |
| 8 | PubChemBioAssay | This database is a repository of biological activity of different compounds and it also contains descriptions of bioactivity assays that are used to screen chemical substances that are present in the PubChem Substance database. |
| 9 | RCSB Protein Data Bank | It has information about 3D shapes of nucleic acids, proteins, and complex molecules. This database helps students and researchers by creating tools and resources for molecular biology, structural biology, computational biology, and beyond. |
| 10 | SuperTarget | It is an extensive database that contains 332828 drug-target interactions. |
| 11 | STITCH | This database contains predicted interactions between a variety of proteins and chemicals. |
| 12 | TDR Targets | This Database helps to identify molecular targets for the drug discovery process, by focusing on pathogens responsible for a particular disease. It is an integration of pathogen-specific genomic information with functional data that are retrieved from various sources, including literature. |
Several ways are available for the construction of a DTI network. Drug target interaction data can be downloaded from databases such as Drug Bank [107] and Therapeutic Target Database [108] and these databases are available online for free access to the public. The drug-target pairs downloaded from these databases can be used for constructing a drug-target interaction network. But the disadvantage of these databases is that the drug-target interaction is not proven experimentally so it is not quantitative. Hence there may be some problems during the merging of drug-target interaction data from different sources. On the contrary, some drug target interaction databases provide experimentally determined DTI data with quantitative activity values such as EC50, Ki, IC50, and Kd values. For example BindingDB [109], Binding MOAD [110], ChEMBL [111], DrugCentral [112], IUPHAR/BPS Guide to PHARMACOLOGY [113], PDBbind-CN [114], PDSP Ki Database [115], PubChemBioAssay [116], RCSB Protein Data Bank [117], SuperTarget [118], STITCH [119], TDR Targets [120], Thomson Reuters Integrity, etc. Once the quantitative drug target interaction is retrieved from these databases, data filtering and merging can be done.
Numerous data from different databases are used by the researchers to enable DTI prediction. It includes chemical tool box such as Open Babel [121] to generate chemical sub-structures for the drugs and PaDEL-Descriptor [122]. Some databases like DrugBank [107], DrugCentral [112], and KEGG DRUG [123] can be used to obtain the Anatomical Therapeutic Chemical classification (ATC) codes of drugs.
Comparative Toxicogenomics Database (CTD) [124], SIDER [125], and OFFSIDES [126] are the databases that can be used to collect the Side effects of drugs. UniProt knowledgebase [127] can be used to download the Sequences of target proteins. From the information obtained from these databases construction of many types of data can be done. An example of this is that substructures of the drugs and targets can be used to calculate the chemical likeness of drug-drug pairs [88, 90]. ATC codes and side effects databases can be used to calculate curative and adverse-effect similarity networks of drug-drug pairs [128]. Protein sequence databases can be used to calculate the sequence similarity of target-target pairs [88, 90]. Once these similarity data are obtained from these similarity databases, this information can be used to construct networks.
When a drug binds to some other proteins in addition to their desired targets there is a major chance for it to induce some physiological side effects. Since these side effects are to be considered during drug trials, it is necessary to avoid drug interaction with proteins other than the target. This prevents the disapproval of the drug. In the SIDER database, all the information regarding side effects is available; this database contains side effect information for more than 1,000 drugs which are already available in the market [125, 129] correlated the side effects of a drug molecule with the proteins it binds to using the SIDER database. From this, it is concluded that the drug’s target can be predicted based on their side effects (Fig. 4).
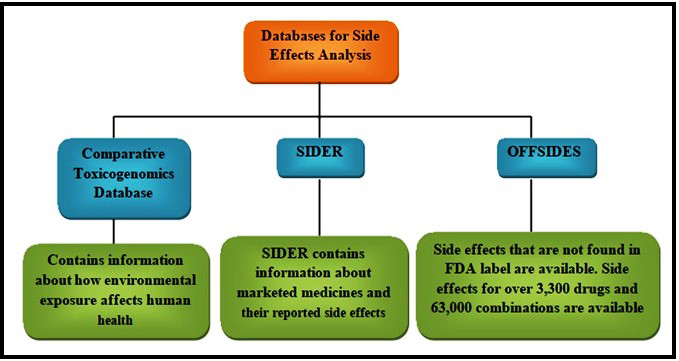 Fig. 4.
Fig. 4.Databases for side effect analysis.
Medicinal plants are complex, and they contain multiple active components which makes them highly efficient. The presence of these active components makes them a potential candidate for the preparation of network-based multipotent drugs [84, 130]. Active components of these herbs can be isolated using various isolation techniques and can be used with the chemical drugs which are commercially available to produce a synergistic effect. A suitable and well-known example of this kind of synergistic effect is the use of Human Immunodeficiency Virus (HIV) triple cocktail treatment along with the administration of tannin phytoconstituent isolated from a medicinal plant. This treatment is very effective against the propagation of HIV. Tannin is found to exert this effect by suppressing the activities of protease, reverse transcriptase, and integrase which are mainly responsible for the HIV propagation. Tannin also obstructs the viral fusion and entry into the host [131].
Another example of this is the artemisinin, an antimalarial drug extracted from the Qinghao, a Chinese medicinal herb. This artemisinin is effective when it is combined with some other chemical drugs. When artemisinin is combined with chloroquine it prevents drug resistance by Plasmodium falciparum in malaria [132, 133, 134]. Other chemical drugs like meflorquine, fansidar are also effective against malaria when they are combined with artemisinin [135].
The traditional method of treating diseases using herbal formulas will be the major source for the development of multi-target drugs. In traditional medicinal treatment, our ancestors developed herbal formulations on their own based on their experience and long-term practice. There are three major steps involved in the development of network-based multi-target drugs. They are i) analyzing the efficiency of original herbal formulation ii) isolation of potent bioactive compound from the formulation iii) development of a new multi-component drug from the formulation. This component will exhibit a cumulative effect [136, 137]. Traditional herbal drugs with multi-target potential and modern systems biology are the greatest milestone in the revolution of network-based drug discovery.
When a drug is administered to a host there are two possible outcomes one is a therapeutic effect and the other is undesirable side effects. The main application of network-based methods is analyzing the molecular mechanism of both these effects. The network namely Drug-Gene-Disease networks are commonly used to elucidate molecular mechanisms [86, 87, 128, 138, 139]. When the information about a drug, its target site so-called gene, and specific disease is known it is easy to construct a Drug- Gene- Disease network.
The association of gene and disease is available in the databases like CTD [124], HuGE Navigator [140], Online Mendelian Inheritance in Man [141], and PharmGKB [142]. Once the drug-gene-disease network has been constructed, the constructed network can be visualized using visualization tools including Cytoscape [143]. This can be used to see the network visually. Some other bioinformatics tools like gene set enrichment analysis [144] are used for the gene function analysis in the network. With the help of these analysis results and from the published data the molecular mechanisms can be elucidated (Fig. 5).
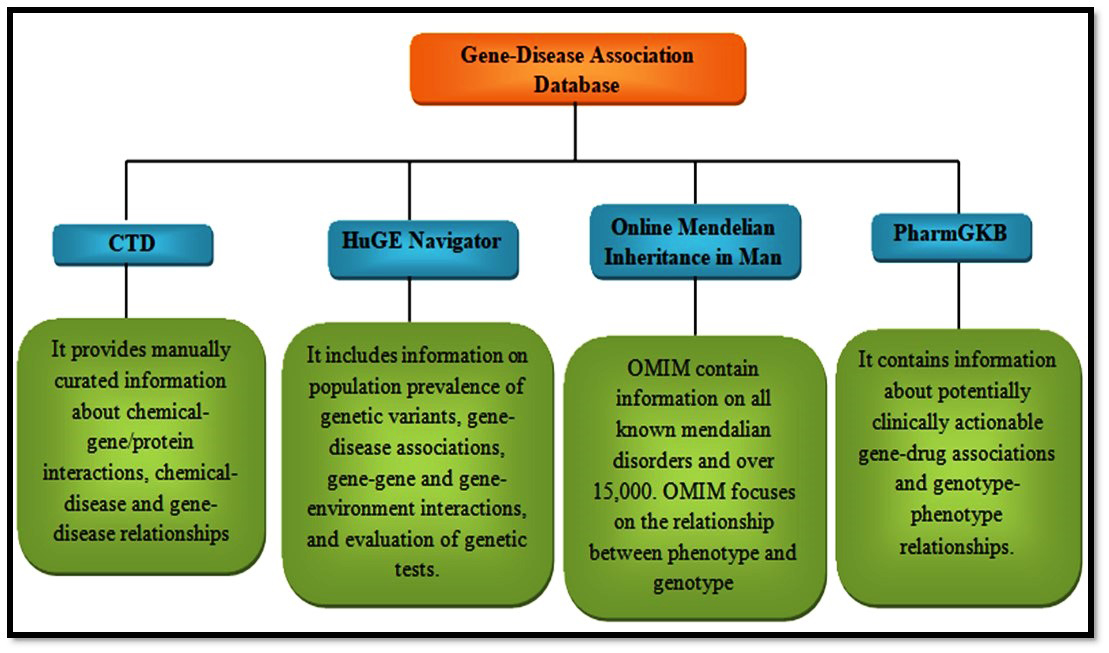 Fig. 5.
Fig. 5.Databases for gene-disease association.
Protein-protein interaction networks are responsible for the formation of enzymes, macromolecules that are needed for cellular processes. It plays a major role in maintaining the body’s homeostasis. When there is any dis-regulation in this network it results in cellular dysfunction and finally leads to various diseases. Protein-protein interaction networks are highly specific triggering researchers to consider them as targets for drug designing. Researchers specifically target disease-related pathways [145, 146, 147].
With the proper knowledge of the Protein-protein interaction network of an organism or a cell, it is possible to predict the relationship between its genotypes and phenotypes. Change in this network leads to abnormal conditions and diseases. The surface molecules present in protein-protein interaction sites are important for studying unnecessary interactions by molecular recognition mechanism. Because of these advantages, Protein-protein interaction networks have a great therapeutic application in the rational designing of drugs. Appropriate knowledge about the molecular recognition mechanism of PPIs and the interpretation of PPI networks can ease the experimental methods. Experimental methods are divided into two types. They are i) methods used to identify large scale PPIs ii) methods used to identify single PPIs [148]. Some of the high-end methods like phage display, affinity purification, and yeast two-hybrid system can explore more of PPIs by expression of an individual protein. Cryo-electron microscopy, X-ray crystallography, and nuclear magnetic resonance (NMR) spectroscopy are some of the analytical methods that can analyze a specific PPI. These analytical methods can determine PPI sites even at their atomic level. These analytical methods have some disadvantages due to some physicochemical factors including post-translational modification (PTM), transient dynamics [149, 150], and proteins with intrinsically disordered regions [151, 152, 153, 154].
Usually, proteins are expressed in a location and transported to another location to exert its role. These proteins will never interact unnecessarily with the other in vivo but there is a chance for them to undergo unnecessary interactions in vitro. The major disadvantage of the experimental methods is that they consume more time, need more manpower and also, they are expensive. So, in silico approaches are efficient in identifying PPIs and PPI sites.
For the understanding of complex biological systems, protein-protein interaction network-based analyses are extremely necessary since they play a major role in most of the cellular events. These protein-protein interaction data are not available in a single database. They are found in different databases. Therefore all this information available in different databases have to be gathered and a single repository should be created for easy access by the public (Table 2). Chel et al. [77] and Razick et al. [155] have attempted to integrate all this information for the creation of integrated repositories.
| S.No | Web Server | Function |
| 1 | PRISM PROTOCOL | Performs PPI by structural matching |
| 2 | Coev2Net | It contains interaction information collected from high throughput analysis. |
| 3 | InPrePPI | It is based on genomic content for predicting PPIs especially in prokaryotes. |
| 4 | TSEMA | It predicts the interaction between two protein families. |
| 5 | G-NEST | It uses chromosomal closeness/gene neighborhood to predict PPI. |
| 6 | InterPreTS | It uses the tertiary structure of the proteins to predict PPI. |
| 7 | MirrorTree | Predicts PPI based on taxonomic context |
| 8 | Struct2Net | Structure-based prediction of PPI networks |
| 9 | STRING | Used for the functional enrichment analysis of PPIs |
| 10 | PoiNet | It combines or correlates tissue-specific expression data and PPIs. |
| 11 | OrthoMCL-DB | It is used to align proteins based on their structural similarity. |
| 12 | PrePPI | It contains both experimentally determined and predicted PPIs of the human proteome. |
| 13 | COG | It is based on the Phylogenetic classification of proteins. |
| 14 | iWARP | It uses protein sequences to predict PPIs. |
| 15 | PHOG | It uses a novel algorithm for the identification of orthologs based on phylogeny. |
| 16 | PIPE2 | It predicts PPI based on the sensitivity and specificity of the selected proteins. |
| 17 | BLASTO | It uses ortholog group data to perform BLAST. |
| 18 | PreSPI | It is used to predict the interaction probability of proteins. It is a domain combination based prediction system. |
| 19 | SPPS | It is a sequence-based method for the prediction of PPI. |
| 20 | HomoMINT | It assigns proteins to orthology groups. It assigns proteins to ortholog groups applying human protein as the important ortholog. |
The major disadvantage of these PPI networks is that they may have some false positive or false negatives which make its quality poorer, so it is much necessary to evaluate the PPI network properly. Before constructing a PPI network, it is much advisable to collect the data from different analytical methods that are published in different publications. 3D structure of interacting proteins is essential for constructing PPI networks [156]. The important properties of PPI should be taken into consideration before constructing a network which includes post-translational modifications, interaction type, homologous associations, cellular/tissue environment, gene expression patterns, and subcellular localization [157].
In silico approaches for the prediction of PPIs has gained much importance since the experimental mapping of protein interactomes is not possible because vast numbers of proteins are available. This in silico approaches demonstrates whether two proteins interact or will not. An in silico docking between two proteins that rely on the physicochemical and structural properties of individual proteins is an alternative for PPI network construction [148, 158]. The main disadvantage of this docking technique is that it is difficult to interpret proteins with large conformational changes without proper knowledge about PPI interaction sites [159].
Various in silico approaches for the prediction of protein-protein
interaction network involves structure-based and sequence-based approaches, gene
fusion, phylogenetic tree, chromosomal closeness, and gene expression study-based
approaches. The main principle behind structure-based approaches is that if two
proteins have a similar structure then their interaction network also may be
similar. For example, if proteins 1 and 2 interacts with each other than protein
1
Several protein-protein interaction databases are available online for the researchers including PINA2.0, IntAct, BioGrid, APID, MINT, HitPredict, DIP, BIND [167].
All the networks will have their unique characteristics, for example, PPI networks provide knowledge about which kind of protein is involved in interaction, but it will not provide information about the reaction of these proteins to an external stimulus. This disadvantage can be overcome by the application of gene expression data as it provides the reaction of an organism to an external stimulus. Thus, a single network cannot give the entire information about an organism. It is necessary to integrate the different types of networks to make a repository with the entire information about an organism [169, 170].
A popular and well-known model for integrated networks is the ABC model. This model gives information about the relationship between two concepts. Consider that C is a disease; B is one of the properties of the disease which is mined from a database. Consider A is an individual drug and it has some specific effect on the characteristics of a disease which is obtained from another database. So, if we integrate these data and create a single repository, all the information about the specified disease can be accessed easily.
Two drug discovery types that have been proposed based on this ABC model. They are (i) open discovery model (ii) closed discovery model. As far as the closed discovery model is considered the components A and C are known but component B alone is unknown. So an attempt is made to find the relation of A and C to the component B. open discovery model undergoes two stages one is the identification of the relationship between A and B and the other is the identification of the relationship between B and C. In both the cases, multiple relationships between ABC can be studied [171].
Various other methods were also constructed successfully based on this method. One such kind CoPub [171] that gives a clear knowledge about the relationship between genes, diseases, drugs, and pathways. Yet another method which is similar to this is the method proposed by [172] that provides information about the relationship between disease-gene and gene-drug by applying the ABC model. The main objective of this ABC model is to identify anticancer drug candidates for repurposing.
Contemporary biomedical research is most meaningful with molecular network-based studies, especially NBDD studies because it has good potential for prediction of interaction between important biomolecules involved in the disease. The main advantage of NBDD is that it deals with enormous molecular data associated with medical science which are generated from several advanced molecular biology technologies. NBDD not only helps in simplifying access and understanding of data from the huge molecular networks, but it also helps to study biomedical phenomena deeply, employing both in silico and in vitro tools. The results help in silico biologists to develop new tools for target-oriented work precisely. NBDD data are crucial for achieving individual targets in bio-medicines through publicly available molecular data.
Not applicable.
PJ and RN conceived and designed the review outlines. PJ and SI originally wrote the paper. RN, BP and SB have critically evaluated, edited the original draft.
We thank the three anonymous reviewers for excellent criticism of the article.
This study was supported by grants from SERB, DST, Govt. of India, New Delhi (ECR/2016/001984 by and Department of Biotechnology, DST, Government of Odisha, Bhubaneswar (1188/ST, Bhubaneswar, dated 01.03.17, ST- (Bio)-02/2017).
The author declares no conflict of interest.
2D, two dimensional; 3D, three dimensional; CTD, Comparative Toxicogenomics Database; DTI, Drug Target Interaction; HIV, Human Immunodeficiency Virus; MOE, Molecular Operating Environment; NBDD, Network-Based Drug Discovery; PPI, Protein-Protein Interaction.