

Frontiers in Bioscience-Scholar (FBS) is published by IMR Press from Volume 13 Issue 1 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Laboratory of Preclinical Research of Natural Products, Paranaense University, Umuarama, PR, Brazil
2 Laboratory of Electrophysiology and Cardiovascular Pharmacology, Faculty of Health Sciences, Federal University of Grande Dourados, Dourados, MS, Brazil
Abstract
Atherosclerosis is an inflammatory, progressive, and chronic illness that involves several molecular and epigenetic factors. Despite treatment limitations, clinical and therapeutic approaches have undeniably changed radically in recent decades through better knowledge of the pathophysiological basis of the disease, which has considerably improved patients’ survival and quality of life. Some of these advances are attributable to basic biomedical research that provides insights into a better understanding and identification of new molecular and cellular targets for atherosclerosis treatment. Although rodent models have contributed substantially to a better understanding of the development of atherosclerosis, the accuracy of these models remains controversial. Research that utilizes genetic rodent models is well established, but the use of specific diets that are associated with other risk factors (e.g., hypertension, hormone deprivation, and pharmacological tools) is still debatable. The present review provides an update on non-genetic rat models of atherosclerosis and an overview of the main methodologies that are currently available.
Keywords
- Atherogenesis
- Cardiovascular Disease
- Diet
- Dyslipidemia
- Rodents, Review
Cardiovascular diseases (CVDs) comprise a set of heart and blood vessel disorders, including hypertension, heart failure, and atherosclerosis, among others. More than 17 million people die annually from CVD. Four of every five deaths are caused by heart attack and stroke (1). These end clinical events are frequently caused by atherosclerosis, one of the leading causes of morbidity and mortality worldwide (2-5). Atherosclerosis is an inflammatory, progressive, and chronic illness that involves several molecular and epigenetic factors. The disease, once considered a simple consequence of aging, can occur from the earliest stages of life, mainly affecting the population over 45 years old (6-8). Endothelial dysfunction is a key early trait in the development and progression of atherogenic plaques and their respective complications. This condition is characterized by a reduction of the bioavailability of vasodilators that are derived from the endothelium, such as nitric oxide (NO). However, elevated levels of low-density lipoprotein (LDL) in the blood are the most common risk factor for the development of atherosclerosis. Such high levels of LDL tend to accumulate in the subendothelial space and undergo oxidative modifications through the action of reactive oxygen species (ROS), becoming oxidized particles (oxLDL-C). These particles induce an inflammatory response that is mediated by interleukin-1 (IL-1), IL-6 and others inflammatory mediators, resulting in increasing in expression of adhesion of intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) by endothelial cells (9, 10). Consequently, monocytes infiltrate into the subendothelial space, become macrophages, and then become foam cells through the phagocytosis of oxLDL-C molecules and initiate plaque formation (11-13). The inflammatory response also causes the migration of smooth muscle cells that secrete extracellular matrix proteins and contribute to atheroma growth (14). More advanced plaques are characterized by a necrotic center, many foam cells, few smooth muscle cells, neovascularization within plaques with hemorrhages, and a thin fibrous cap that separates the plaques from the bloodstream. Plaques that form in arteries remain stable for years but may become rapidly unstable, rupture, and trigger the formation of thrombus, leading to a higher risk of acute cardiovascular events (5, 13). The main mechanisms that are involved in the pathogenesis of atherosclerosis are summarized in Figure 1.
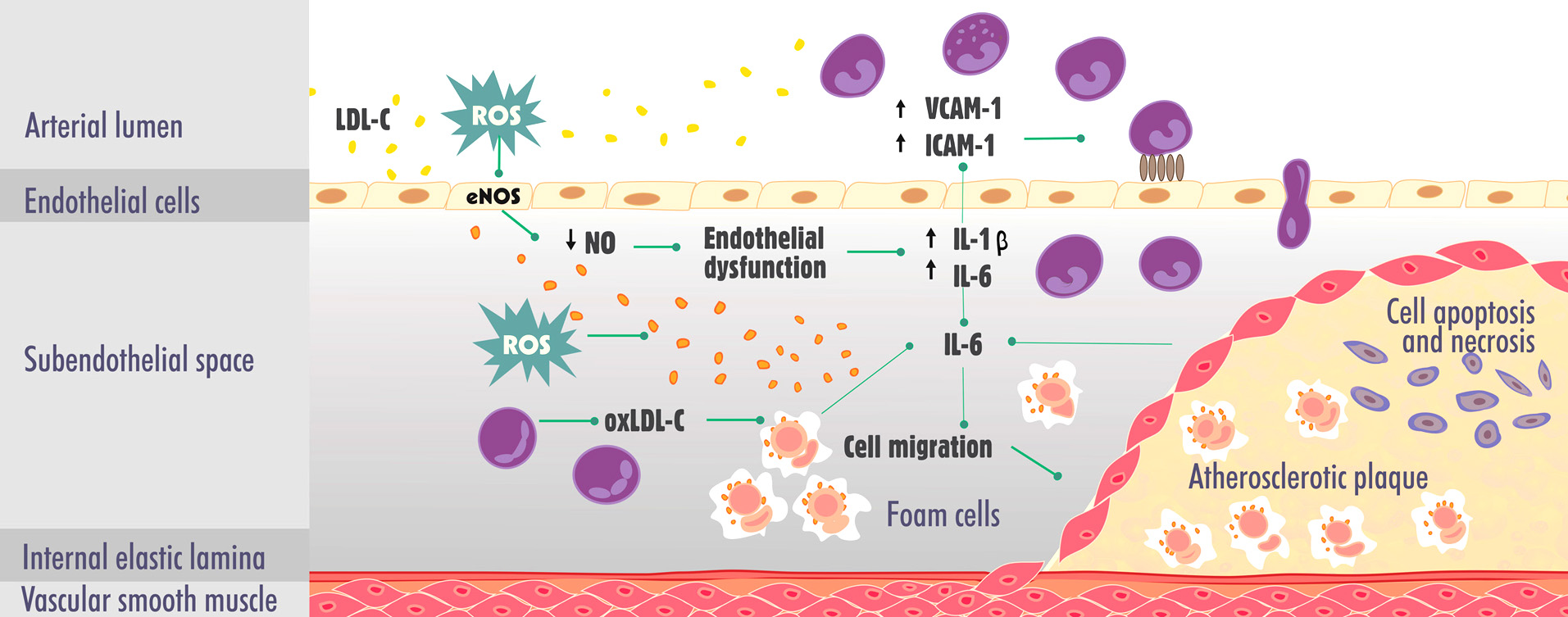 Figure 1.
Figure 1.Main mechanisms involved in the pathogenesis of atherosclerosis. eNOS, endothelial nitric oxide synthase; IL-1, interleukin-1; IL-6, interleukin-6; ICAM-1, intercellular adhesion molecule-1; LDL-C, low-density lipoprotein cholesterol; NO, nitric oxide; oxLDL-C, oxidized low-density lipoprotein cholesterol; ROS, reactive oxygen species; VCAM-1, vascular cell adhesion molecule-1.
The high incidence of CVD-related atherosclerosis is responsible for high expenditures in the healthcare system. Pharmacological options for the treatment of atherosclerosis include vasodilators, β-blockers, hypolipidemic drugs (mainly statins), and agents that are used for the prevention of thrombotic complications, including antiplatelet actions. However, some of these treatments have several side effects, and many therapeutic agents do not control all of the underlying mechanisms (e.g., inflammation) that lead to progression of the disease and the final stage of atherosclerosis (i.e., plaque rupture) (5, 15-17).
Despite treatment limitations, clinical and therapeutic approaches for the management and treatment of cardiovascular disease have undeniably changed radically in recent decades through better knowledge of the pathophysiological basis of the disease, which has considerably improved patients’ survival and quality of life (18-20). Some of these advances have come from basic biomedical research that has provided insights into a better understanding and identification of new molecular and cellular treatment targets. However, translating these advances in the laboratory into clinical application requires further translational research (21, 22). Greater interactions between basic researchers and clinicians are required, making translational research a two-way road that can lead to more applicable and innovative results (21, 23).
The central drawback of the use of animals in translational research involves difficulties in reproducing important behavioral aspects of the disease. Some models are promising and easily reproducible, but others fail to demonstrate similarity to disease progression in humans (e.g., face validity, construct validity, and predictive validity) (24, 25). However, the contributions of scientific studies that utilize animals are unquestionable. In fact, approximately 90% of Nobel prize research in physiology and medicine has described important discoveries through animal experimentation (26-29).
Despite the limitations of animal models with regard to transferability and predictability, their use for studying atherosclerotic disease is essential for elucidating the mechanisms that are involved in the development and progression of the disease, plaque rupture, and associated clinical events. Such models also allow investigations of the pharmacological potential of agents that can prevent or delay development of the disease (13, 30). The established models are generally based on accelerated plaque formation by cholesterol-rich diets, the manipulation of genes that are involved in cholesterol metabolism, and the introduction of additional risk factors (e.g., hypertension and diabetes) (13, 30-34). Indeed, although research that uses genetic models is well established, the use of diets that are associated with other risk factors in rodents is still controversial. The present review presents an update on non-genetic rodent models of atherosclerosis and provides an overview of the main methodologies that are available.
Animals have been used for research in scientific and didactic fields for centuries. There are reports of the use of vertebrate species for the study of anatomy and physiology since 500 BC, and the contribution of these studies to the advancement of science is unquestionable. They are responsible for the most evident advances in many biological fields to prevent, diagnose, and treat diseases (24, 35-38).
Experimental models that are used in research have the goal of representing a real situation or condition. These models must be precise and as similar as possible to the study objective because they are tools to understand the pathophysiological mechanisms of diseases (13, 38). The choice of animal species for experimentation is important for defining which parameters can be evaluated. The species is determined by the aim of the investigation and financial and technical factors (32, 39, 40). Several species, such as mice and rats (96%), guinea pigs (19%), rabbits (18%), hamsters (13%), and larger mammals (e.g., cats and non-human primates (< 1%)) are currently used in the development of scientific studies (41).
The use of rodents for biomedical research has many advantages compared with other species. They are small, easy to handle, and relatively inexpensive. They have a relatively short lifespan, low maintenance cost, and high rates of reproduction and are highly resistant to successive inbreeding (31, 42, 43). Domesticated rats (Rattus norvegicus) were the first rodents species that was used in preclinical research. After the first standard rat strain was developed in 1909, Wistar rats became the preferred tool in basic research. To date, this species accounts for half of all rats that are used in research laboratories (35, 44). Mice (Mus musculus) have also been used in research since the 19th century. The first transgenic mouse model was produced in 1988. In 2002, this species became the second mammal, after humans, to have its whole genome sequenced (45). This discovery, associated with other technological advances, has allowed unlimited potential to understand genetic and non-genetic factors that are involved in the pathogenesis of diseases. Mice have since become the most used species in rodent biomedical research (35, 45, 46).
With regard to CVD, research has undergone considerable expansion. Several animal models are widely accepted, and many are still under development. The common consensus is that the animal model must be matched to the experimental design (31, 32, 47). Investigations of arrhythmia, cardiac failure, embolism, and cardiopulmonary bypass are scarce. There are well-described animal models of hypertension, myocardial infarction, and atherosclerosis (32, 47). Several research groups worldwide have investigated the underlying mechanisms of the atherogenic process. The most widely used animal species for atherosclerosis research are guinea pigs, hamsters, rabbits, rats, and mice. In contrast to larger mammals, the development of complex atherosclerotic lesions in rodents usually involves a shorter time period (31). However, none of these models are ideal. They have both advantages and disadvantages with regard to the atherogenic process, lipid profile, and evolution of the disease in humans (3, 31).
Mouse models are limited and fail to fully replicate atherosclerosis in humans. Research has provided insights into a better understanding of the biological processes and underlying mechanisms of the disease. Because of the relative ease of genetic manipulation and short time of atherogenesis evolution, murine genetic models are currently the most widely used species in research because they can reveal the factors that are involved in different stages of the disease through the activation or inhibition of cells that are involved in the atherosclerotic process (48-53). The most common genetic models for studying atherosclerosis are the apoE −/− and LDLR −/− models (31, 54). However, these models do not accurately reproduce the pathophysiological conditions that determine evolution of the disease in humans. Thus, associations between multiple risk factors (i.e., genetic, environmental, and dietary factors) appear to be a plausible alternative for mimicking the prevailing conditions that are shared with humans (13, 31).
Although rodent models have contributed substantially to a better understanding of atherosclerosis, the accuracy of these models remains an object of great controversy (55). The factors that generate this lack of consensus with regard to rodent models of atherosclerosis are many: (i) absence of the manifestation of unstable atherosclerotic plaques with overlying thrombosis (i.e., an event that leads to acute clinical manifestations in humans), (ii) lack of development of the characteristically tick fibrous cap, (iii) the number of lamellae in the normal arterial media layer is smaller in rodents than in humans, (iv) the absence of medial vasa vasora that is observed in the large arteries in humans, (v) the absence of plaque development in the coronary arteries but rather in the aortic root, likely because of the high heart rate of rodents, (vi) the atheroresistance of wildtype mice, which are considered “high-density lipoprotein” (HDL) animals and not exhibit the same range of HDL subsets and cholesteryl ester transfer protein (CETP) that is present in humans and related to atheroprotection, and (vii) lesions that contain lipids in rodents are generally considered a residual lesion that results from acute arteritis (31, 47, 55-60).
The great challenge of investigators is to develop a model whereby intra-plaque microvessels, hemorrhages, spontaneous atherosclerotic plaque ruptures, myocardial infarction, and sudden death can be observed (13). In the search for the ideal model of atherosclerosis, many research groups have proposed different non-genetic rodent models to study the disease. These models include several alterations of the animals’ diet, pharmacological ad surgical manipulations, and combinations of these factors (i.e., mixed models).
The most common non-genetic rat models for atherosclerosis research involve the combination of a high-fat diet with surgical procedures or the administration of vitamin D3 (Table 1). A high-fat diet (88% standard diet plus 2% cholesterol and 10% lard) alone is incapable of inducing more advanced stages of atherosclerosis. In fact, it leads only to earlier events of the disease (e.g., mild intimal thickening and cellular hypertrophy in the intima and media of the aorta), such as a model with Wistar rats that are treated with a high-fat diet for 16 weeks (61). A viable alternative has been the co-administration of high doses of vitamin D3 (cholecalciferol). Data show that this procedure facilitates the induction of significant vascular and soft tissue calcifications (62).
| Animal | Duration of experiment | Diet | Intervention (pharmacological or surgical) | References |
| Wistar rats | 16 weeks | 88% standard diet plus 2% cholesterol and 10% lard | — | 61 |
| Sprague-Dawley rats | 17 weeks | 81.3% basic feed, 10% lard, 3% cholesterol, 0.5% sodium cholate, 0.2% propylthiouracil, 5% sugar | Vitamin D3 (600,000 IU/kg, i.p.) | 63 |
| Wistar rats | 12 weeks | 13% fat, 3% cholesterol, 0.7% sodium cholate, 0.4% propylthiouracil, 7% sucrose | Vitamin D3 (600,000 IU/kg, i.p.) every 4 weeks for 12 weeks | 64 |
| Wistar rats | 10 weeks | 80.3% normal diet, 11% animal oil, 4.5% cholesterol, 1.5% sodium cholate, 0.7% propylthiouracil, 4% refined sugar | Vitamin D3 (70 U/kg, continuous administration) for 3 days | 65 |
| Wistar rats | 9 weeks | 83.8% normal diet, 10% animal oil, 3.5% cholesterol, 0.5% sodium cholate, 0.2% propylthiouracil, 5% refined sugar | Vitamin D3 (70 U/kg, continuous administration) for 3 days | 66 |
| Wistar rats | 12 weeks | 10% lard, 1% cholesterol, 0.2% pig bile salts, 10% egg yolk powder, 78.8% basal diet | Vitamin D3 (600,000 IU/kg, i.p.) plus ovalbumin (3 mg/kg, hypodermic route) for 3 consecutive days | 67 |
| Sprague-Dawley rats | 12 weeks | 78.3% basal diet, 10% lard, 1% cholesterol, 5% egg yolk powder, 0.5% sodium cholate, 0.2% propylthiouracil, 5% sucrose | Vitamin D3 (700,000 IU/kg, i.p.), four injections every 2 days | 68-69 |
| Sprague-Dawley rats | 15 weeks | 75 g lard, 25 g cholesterol, 5 g sodium cholate, 50 ml of propylene glycol, 50 ml of Tween-80 and distilled water (500 ml) | Vitamin D3 (600,000 IU/kg, i.p.) plus additional i.p. injection of 300,000 IU/kg every 30 days | 70 |
| Sprague-Dawley rats | 15 weeks | 4% cholesterol, 1% cholic acid, 0.5% PTU, salad oil | Vitamin D3 (600,000 IU/kg, i.p.) plus additional i.p. injection of 300,000 IU/kg every 30 days | 71 |
| Wistar rats | 80 days | Intragastric administration of 15 mg/ml lard, 45 mg/ml cholesterol, 7.5 mg/ml sodium cholate, 3 mg/ml propylthiouracil, 75 mg/ml sugar | Vitamin D3 (200,000 IU/kg, i.p.) for the first 3 days, followed by 150,000 IU/kg every 20 days for 80 days | 72 |
| Sprague-Dawley rats | 9 weeks | 80.8% normal diet, 10% animal oil (sheep ghee), 3.5% cholesterol, 0.5% sodium cholate, 0.2% propylthiouracil, 5% refined sugar | Vitamin D3 (100,000 IU/kg) by gavage in the first 4 days of the experiment | 73 |
| Sprague-Dawley rats | 16 weeks | 2% cholesterol and repeated heated soy oil | Ovariectomy | 74 |
| Wistar rats | 6 weeks | 82.3% regular rat chow, 10% lard, 2% cholesterol, 0.5% cholic acid, 0.2% 6-methyl 2-thiouracil, 5% sucrose | Aortic endothelial denudation (by scraping the aortic lumen with a balloon catheter inserted into the left femoral artery) and/or 2 kidney-1 clip (2K1C) surgery | 75-76 |
Another apparently viable association, in addition to vitamin D3, is the inclusion of a drug that can reduce metabolism in animals. Thus, use of the antithyroid drug propylthiouracil has shown promising results. Fu et al. (63) induced atherosclerosis in male Sprague-Dawley rats by intraperitoneal (i.p.) vitamin D3 administration (600,000 IU/kg) combined with a high fat diet (81.3% basic feed, 10% lard, 3% cholesterol, 0.5% sodium cholate, and 5% sugar) and 0.2% propylthiouracil. After 17 weeks, the inner wall of the arteries markedly increased and contained ridged spots and visually obvious plaques. Histopathological analyses revealed a significant increase in inner membrane thickness of the aortic arch, some damaged endothelial cells, the calcification of smooth muscle cells, and disordered smooth muscle cells. Furthermore, a significant increase in total cholesterol (TC) and LDL-C was observed.
Administration of the same dose of vitamin D3 (given intraperitoneally every 4 weeks for 12 weeks) and 0.4% propylthiouracil but with some modifications of the high-fat diet (13% fat, 3% cholesterol, 0.7% sodium cholate, and 7% sucrose) induced atherosclerosis in male Wistar rats (64). Atherogenesis was also observed in rats with a shorter exposure time (10 weeks) with modifications of the vitamin D3 dose (70 U/kg, continuous administration for 3 days) and propylthiouracil (0.7%) and a high-cholesterol diet (80.3% normal diet, 11% fat, 4.5% cholesterol, 1.5% sodium cholate, and 4% refined sugar) (65). The same vitamin D3 administration protocol was used by Hu et al. (66). With a few adjustments of the diet (83.8% normal diet, 10% fat, 3.5% cholesterol, 0.5% sodium cholate, and 5% refined sugar) and propylthiouracil concentration (0.2%), a model of atherosclerosis was established in Wistar rats at 9 weeks. The alterations that were described in this model included morphological abnormalities of the aortic intima (i.e., fatty streaks, the accumulation of foam cells, and disarranged endothelial cells), increases in TC, triglycerides (TG), and LDL-C, and higher levels of IL-6 and tumor necrosis factor-α (TNF-α).
Zhang et al. (67) developed a model of atherosclerosis in Wistar rats during 12 weeks using a high-fat diet (10% lard, 1% cholesterol, 0.2% pig bile salts, 10% egg yolk powder, and 78.8% basal diet) in association with vitamin D3 (600,000 IU/kg, i.p.) and an antigen emulsion that comprised complete Freund's adjuvant and ovalbumin (3 mg/kg, hypodermic route on the animals’ back). Three weeks later, booster injections of ovalbumin (2.5 mg/kg, i.p.) were repeated every week for 3 consecutive weeks. After 12 weeks, increases in TC, TG, and LDL-C levels were observed, with increase in the concentrations of C-reactive protein (CRP), IL-1β, IL-8, and IL-18. Furthermore, the elastic layers in the intima and media were damaged, and extensive atherosclerotic plaques that contained foam cells, inflammatory cells, cholesterol crystals, and tissue calcifications were observed.
Jiang et al. (68) produced a valid model of atherosclerosis in rats after 12 weeks of diet exposure. However, some modifications of the experimental design were implemented. Male Sprague-Dawley rats were fed a high-fat diet (78.3% basal diet, 10% lard, 1% cholesterol, 5% egg yolk powder, 0.5% sodium cholate, and 5% sucrose) plus 0.2% propylthiouracil and vitamin D3 (700,000 IU/kg, i.p.) for 6 weeks, which was divided into four injections every 2 days. This protocol was previously described by Yang et al. (69). This model increased serum lipid, TC, TG, and LDL levels, caused redox status imbalance, induced severe hepatic steatosis, and caused extensive atherosclerotic plaques that contained foam cells, cholesterol crystals, and tissue calcification.
In the study by Li et al. (70), male Sprague-Dawley rats were first treated with vitamin D3 (600,000 IU/kg, i.p.), and an additional injection at a lower dosage (300,000 IU/kg) was repeated every 30 days. Additionally, 10 ml/kg of a fat emulsion (75 g lard, 25 g cholesterol, 5 g sodium cholate, 50 ml of propylene glycol, and 50 ml of Tween-80) was administered daily for 15 weeks. Serum levels of TC, TG, and LDL significantly increased. Histopathological analyses revealed thickening of the arterial wall, internal elastic membrane damage, the proliferation of smooth muscle cells, and fibrous tissue with many areas of calcification. This model appeared to be based on a previous study by Pang et al. (71). The only differences were the adjustments of the high-fat diet (4% cholesterol, 1% cholic acid, and vegetable oil) associated with 0.5% propylthiouracil.
The atherogenic effects of a high-fat diet plus vitamin D3 were also evaluated by Gou et al. (72). Male Wistar rats received vitamin D3 (200,000 IU/kg, i.p.) daily for the first 3 days, followed by 150,000 IU/kg (i.p.) every 20 days for 80 days. During this period, the rats were fed daily with 1.5 ml/kg of a high-lipid emulsion (15 mg/ml lard, 45 mg/ml cholesterol, 7.5 mg/ml sodium cholate, 3 mg/ml propylthiouracil, and 75 mg/ml sucrose, intragastric administration). After 80 days, alterations of the lipid profile were observed, including increases in TC, TG, and LDL-C levels and a decrease in HDL-C levels. The authors also observed an inflammatory process (i.e., increases in IL-6 and CRP levels and a decrease in NO levels), redox status imbalance, and obvious signs of atherosclerosis in histopathological sections of the aorta (i.e., thickening of the arterial intima, the disordered arrangement of smooth muscle cells and elastic fibers, red foam cells, fatty plaques, and calcium deposition).
In the model that was proposed by Namazi et al. (73), vitamin D3 (100,000 IU/kg) was administered by gavage in the first 4 days of the experiment in male Sprague-Dawley rats. In addition to vitamin D3, atherosclerosis was induced by a high-fat diet (80.8% normal diet, 10% animal oil (sheep ghee), 3.5% cholesterol, 0.5% sodium cholate, and 5% refined sugar) plus the antithyroid drug propylthiouracil (0.2%). The experiment lasted 9 weeks. The authors observed marked hypercholesterolemia, redox status imbalance, degeneration of the tunica intima, lipid deposition, and the formation of atheromatous plaques.
Some studies have investigated the combination of surgery plus a high-fat diet. Exposure to 2% cholesterol and repeated heated soy oil associated with ovariectomy increased serum parameters that are related to atherosclerosis in female Sprague-Dawley rats. After 16 weeks, serum levels of thiobarbituric acid reactive substances (TBARS), TC, TG, LDL, and homocysteine significantly increased (74). Another mixed model that was proposed by Sasaki et al. (75) evaluated the combination of a high-fat diet and renovascular hypertension in male Wistar rats. The animals were subjected to aortic endothelial denudation (by scraping the aortic lumen with a balloon catheter that was inserted in the left femoral artery) and/or 2 kidney-1 clip (2K1C) surgery (i.e., a classic model of renovascular hypertension). The animals were fed a diet that contained 82.3% regular chow, 10% lard, 2% cholesterol, 0.5% cholic acid, 0.2% 6-methyl 2-thiouracil, and 5% sucrose. The diet alone did not induce significant atherosclerosis, but the combination of the diet with endothelial denudation caused slight thickening of the intimae with smooth muscle cell proliferation. The complete mixed model (i.e., diet, endothelial denudation, and 2K1C) caused marked atheromatous plaque formation with prominent cellular proliferation. The same protocol was used by Chen et al. (76), which provided a global transcriptomic network of atherosclerosis development in male Sprague-Dawley rats.
Although no ideal animal model is able to fully reproduce all stages of human atherosclerotic disease, the contributions of the existing models to elucidating the pathophysiological mechanisms of the disease and identifying new therapeutic targets are undeniable. Moreover, these models can be further refined and exploited to explore the etiopathogenesis and regression of atherosclerosis. Rodents are more resistant to the development of atherosclerosis compared with larger animals. In fact, unlike rabbits, for example, a high-cholesterol or high-fat diet alone is insufficient to induce the formation of atherosclerotic plaques. The majority of models that are described in the literature involve combinations of high-cholesterol or high-fat diets with the administration of suprapharmacological doses of vitamin D3 and/or antithyroid drugs. Although most studies have utilized such mixed models, there is a lack of consensus with regard to the exact ingredients of the diet and exposure time (e.g., 6-17 weeks). Mixed models that employ other risk factors, such as hypertension, endothelial dysfunction, and sexual hormone deprivation, are encouraging. This may open new avenues for the inclusion of new risk factors or pharmacological tools that can better standardize the lesions or the model as a whole. There is no consensus in the literature with regard to the ideal age of the animals (i.e., old or young), cholesterol or fat content, time of diet exposure, pharmacological adjuvants (e.g., cholecalciferol and propylthiouracil), the presence or absence of sex hormones, and hypertension, among other factors. Future studies should seek to standardize these variables, including new models of hypertension, endothelial dysfunction that is caused by chemical or physical methods, and specific rodent lineages (e.g., spontaneously hypertensive or obese rats). The future of research in this field is promising. The standardization of new non-genetic rodent models of atherosclerotic disease will likely contribute to the prevention, staging, or regression of this important pathology.
The authors declare that there is no conflict of interest with regard to publication of this paper.. This work was supported by grants from the Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT, Brazil; no. 59/300.046/2015 and 59/300.351/2016), Paranaense University, and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil; no. 449464/2014-8).
Abbreviations: CETP, cholesteryl ester transfer protein; CRP, C-reactive protein; HDL, high-density lipoprotein; IL-1, interleukin-1; IL-6, interleukin-6; ICAM-1, intercellular adhesion molecule-1; LDL-C, low-density lipoprotein cholesterol; NO, nitric oxide; oxLDL-C, oxidized low-density lipoprotein cholesterol; ROS, reactive oxygen species; TBARS, thiobarbituric acid reactive substances; TC, total cholesterol; TG, triglycerides; TNF-α, tumor necrosis factor-α; VCAM-1, vascular cell adhesion molecule-1.