



 , Luciano Saso 3, Silvia Chichiarelli 4, Catalina Rojas-Solé 1, Víctor Pinilla-González 1, Juan Carlos Prieto 4,5,6, Abraham I. J. Gajardo 2,6, Ruben Aguayo 7, Ramón Rodrigo 6,8,*
, Luciano Saso 3, Silvia Chichiarelli 4, Catalina Rojas-Solé 1, Víctor Pinilla-González 1, Juan Carlos Prieto 4,5,6, Abraham I. J. Gajardo 2,6, Ruben Aguayo 7, Ramón Rodrigo 6,8,*
1 Faculty of Medicine, Universidad de Chile, 8380000 Santiago, Chile
2 Critical Care Unit, Hospital Clínico Universidad de Chile, 8380000 Santiago, Chile
3 Department of Physiology and Pharmacology “Vittorio Erspamer”, Faculty of Pharmacy and Medicine - “Sapienza” - University of Rome, 00185 Rome, Italy
4 Now with Department of Biochemical Sciences “A. Rossi Fanelli”, Faculty of Pharmacy and Medicine -“Sapienza” - University of Rome, 00185 Rome, Italy
5 Cardiovascular Department, Hospital Clínico Universidad de Chile, 8380000 Santiago, Chile
6 Institute of Biomedical Sciences, Faculty of Medicine, 8380000 Santiago, Chile
7 Internal Medicine Department, Hospital San Juan de Dios, 8380000 Santiago, Chile
8 Now with Molecular and Clinical Pharmacology Program, Institute of Biomedical Sciences, Faculty of Medicine, University of Chile, 8380000 Santiago, Chile
Abstract
Acute myocardial infarction (AMI) is one of the main causes of mortality worldwide. Currently, the most effective treatment is percutaneous coronary angioplasty (PCA). However, paradoxically, the restoration of blood flow induces myocardial reperfusion injury (MRI), contributing up to 50% of the final infarct size. Oxidative stress, characterized by a burst of reactive oxygen species (ROS) following reperfusion, plays a fundamental role in its pathophysiology, causing inflammation, endothelial dysfunction, and cell death mainly through autophagy, apoptosis, ferroptosis, necroptosis, and pyroptosis. To mitigate these injury mechanisms, numerous antioxidant strategies have been evaluated using both in vitro and in vivo models with promising results, but limited benefit when tested in humans. Several antioxidants have biological properties that counteract ROS-induced damage by acting as ROS scavengers, metal chelators, and antioxidant enzyme enhancers. In this review, we focus on the mechanisms by which oxidative stress induces cell death after AMI and highlight the most promising therapeutic antioxidant agents that could provide comprehensive protection against MRI. A multitarget cardioprotective strategy, combining interventions with strong preclinical evidence, could provide a more effective approach for reducing MRI. Our study aims to bridge the gap between basic and clinical research and explore the potential clinical applications of antioxidants.
Keywords
- acute myocardial infarction
- oxidative stress
- antioxidants
- pharmacological cardioprotection
Acute myocardial infarction (AMI) is a leading cause of mortality worldwide, causing 9 million deaths globally each year and more than 10% of the annual loss of disability-adjusted life-years [1]. Rapid and effective reperfusion therapy, primarily through percutaneous coronary intervention (PCI), has improved the treatment of AMI and improved survival rates [2]. However, paradoxically, following the restoration of blood flow, myocardial reperfusion injury (MRI) ensues, accounting for up to 50% of the final infarct size [3].
The pathophysiology of MRI is complex and multifactorial, involving oxidative stress, calcium overload, mitochondrial dysfunction, and inflammation, among other cellular mechanisms, that ultimately lead to programmed cell death pathways such as autophagy, apoptosis, necroptosis, ferroptosis, and pyroptosis [4].
Despite extensive research efforts, there are currently no effective therapies that can completely reduce MRI. Numerous pharmacological and mechanical interventions have been explored, including ischemic conditioning strategies and antioxidant therapies, but their translation into clinical practice has been largely unsuccessful [5]. One of the critical challenges in developing cardioprotective strategies is the multifactorial nature of MRI, which may require a multitarget approach to address the involvement of various mechanisms of injury [6].
In this review, we explore the current knowledge of MRI, focusing on the potential of combined antioxidant therapies to provide enhanced cardioprotection. We will also discuss the underlying mechanisms of MRI and programmed cell death, recent advances in therapeutic strategies, and the current challenges in translating preclinical findings into effective clinical treatments. Finally, we will present a novel multitherapy approach based on antioxidant cardioprotection.
Ischemia initiates a cascade of cellular events characterized by hypoxia, which arrests oxidative phosphorylation, leading to mitochondrial membrane depolarization, adenosine triphosphate (ATP) depletion, and inhibition of myocardial contractile function [3]. A burst of reactive oxygen species (ROS) is generated early upon reperfusion. When the rate of ROS production exceeds the antioxidant potential of the heart, the increased concentration of these small reactive molecules, including oxygen free radicals such as superoxide and hydroxyl radicals, as well as non-radical species such as hydrogen peroxide, will exacerbate tissue damage [7].
As the duration of ischemia increases, maladaptive processes emerge, notably the accumulation of ROS from sources such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and mitochondrial reverse electron transport (RET) at complex I, triggered by the accumulation of succinate [8]. ROS production exacerbates mitochondrial dysfunction, leading to mitochondrial swelling and rupture, primarily through the opening of the mitochondrial permeability transition pore (mPTP). This event releases cytochrome-c into the cytosol, initiating apoptotic signaling [9].
The ischemic-induced proton accumulation within the cell further reduces intracellular pH, activating the Na+/H+ exchanger (NHE), which, combined with inhibition of the Na+/K+ ATPase due to lack of ATP, leads to excessive intracellular sodium. This sodium overload reverses the action of the Na+/Ca2+ exchanger, resulting in an accumulation of intracellular calcium, which exacerbates mitochondrial dysfunction and promotes further ROS generation. The resulting calcium overload and mitochondrial and DNA damage trigger programmed cell death pathways [10]. Once cytosolic membranes rupture, cellular contents are released into the extracellular space, acting as damage-associated molecular patterns (DAMPs) that can propagate cellular injury across neighboring cells [3].
When blood flow is suddenly restored to the ischemic myocardium, the heart undergoes a complex series of damaging processes collectively called MRI. This phenomenon occurs due to several interconnected mechanisms, including oxidative stress, calcium overload, pH changes, and inflammation, all of which exacerbate the original ischemic damage [11].
Excessive ROS generation is a key driver of reperfusion injury, promoting inflammation, endothelial dysfunction, and programmed cell death through lipid peroxidation and protein damage, ultimately leading to greater infarct size and impaired cardiac function. Mitochondria are the main source of intracellular ROS, mainly through the mitochondrial electron transport chain. RET at complex I plays a significant role in ROS generation, especially in the early stages of reperfusion [12]. In addition to mitochondria, NOX is activated during reperfusion and contributes to ROS production [13]. Moreover, the activity of xanthine oxidase (XO), uncoupled nitric oxide synthase (NOS), and activation of NOX in infiltrating leukocytes, such as neutrophils and macrophages, further exacerbate ROS accumulation and tissue damage [14]. Iron dysregulation also plays a critical role, primarily through increased intracellular free iron during reperfusion, which participates in the Fenton reaction. This reaction involves ferrous iron (Fe2+) reacting with hydrogen peroxide (H2O2), generating hydroxyl radicals (•OH) that exacerbate oxidative damage [15] (Fig. 1).
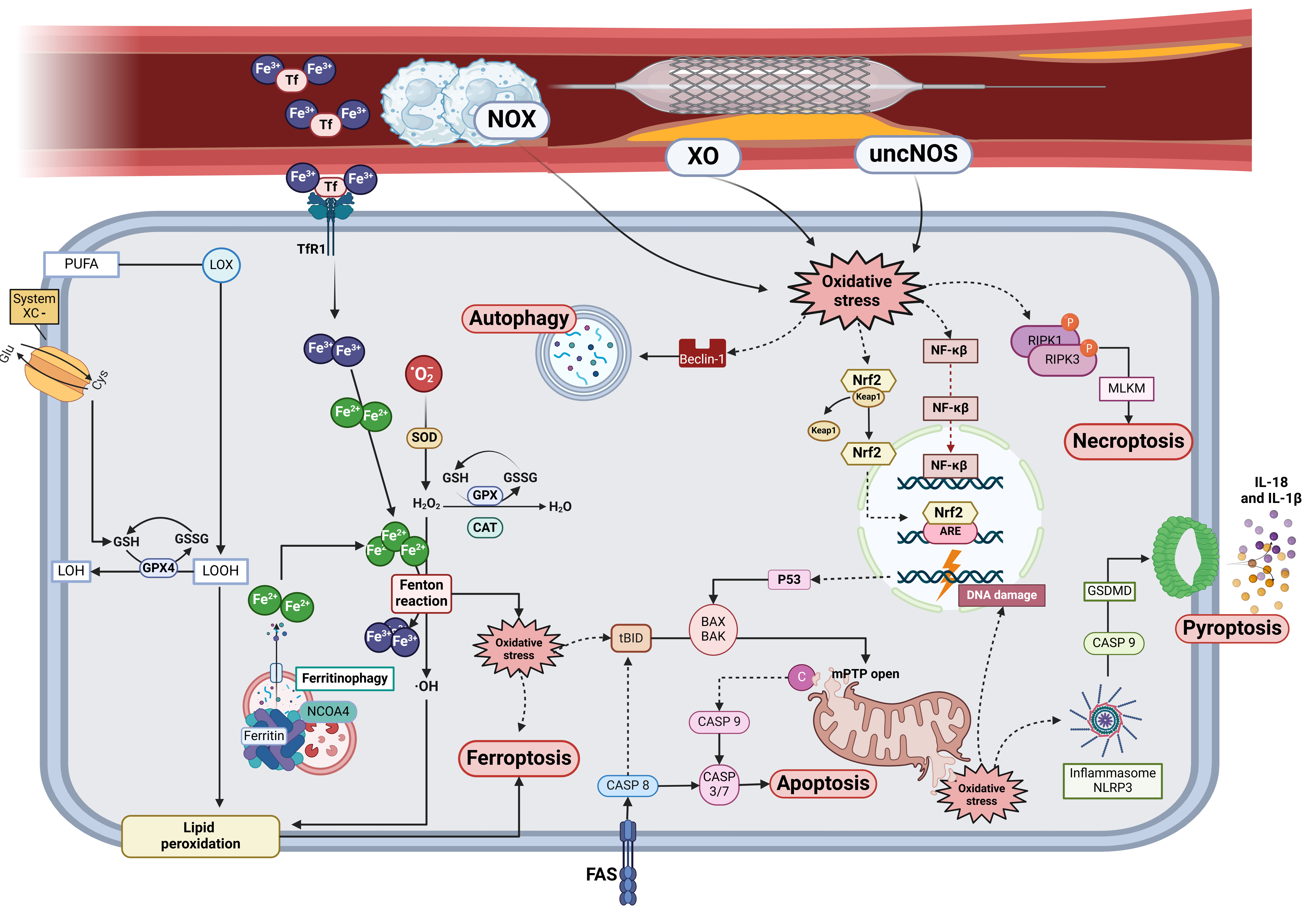 Fig. 1.
Fig. 1.
Molecular mechanisms of generation of reactive oxygen species
(ROS) and programmed cell death in myocardial reperfusion injury after
percutaneous coronary angioplasty. ARE, antioxidant response element; BAX,
Bcl-2 associated X protein; BAK, Bcl-2 homologous antagonist/killer; CAT, catalase;
CASP, Caspase; Fe3+, ferric iron; Fe2+, ferrous iron; GPX, glutathione
peroxidase; GSH, reduced glutathione; GSSG, oxidized glutathione; GSDMD,
gasdermin D; GST, glutathione transferase; HO-1, heme oxygenase-1; I
Oxidative stress is not solely driven by oxygen radicals; it also involves reduced bioavailability of nitric oxide (NO), a molecule with cardioprotective roles, including inhibition of neutrophil accumulation, superoxide scavenging, and enhancement of coronary blood flow through vasodilation [16]. However, NO’s therapeutic potential is controversial, as it can form peroxynitrite (ONOO-), a reactive oxidant from NO and superoxide that oxidizes protein and non-protein thiols [17] and increases mitochondrial protein tyrosine nitration [18]. NO may either mitigate oxidative stress and prevent cell death [19] or, at higher concentrations, contribute to cellular damage [20].
In addition to oxidative stress, reperfusion leads to a phenomenon known as the calcium paradox. Normally, calcium is necessary for oxidative phosphorylation in the mitochondria. Ischemia and ATP depletion lead to intracellular calcium accumulation in cardiomyocytes and mitochondria primarily through the voltage-dependent anion channel (VDAC) and the mitochondrial calcium uniporter complex [21]. During reperfusion, increased ROS levels and mitogen-activated protein kinase (MAPK) activation [22] contribute to a feedback loop through phosphorylation of the sodium-hydrogen exchanger (NHE), raising intracellular sodium and, consequently, intracellular and mitochondrial calcium [10]. This calcium overload triggers a cascade of events, including mitochondrial dysfunction, cardiomyocyte hypercontracture, and the opening of the mPTP with the release of cytochrome-c, leading to further cell death. Although experimental evidence suggests that blocking this mechanism may protect against reperfusion injury [23, 24], it has yet to be successfully translated to clinical trials [25, 26].
Metabolic alterations add another layer of complexity to MRI. During ischemia, the myocardium switches from fatty acid metabolism to glucose metabolism due to limited oxygen availability [27], contributing to acidosis. After reperfusion, mPTP opens in the mitochondria and generates depolarization its membrane, which contributes to the uncoupling of oxidative phosphorylation, aggravating ATP depletion [28]. This insight has led to therapeutic approaches such as glucose-insulin-potassium (GIK) therapy, which will be discussed later.
Inflammation also plays a critical role in reperfusion injury. Within hours of reperfusion, neutrophils are recruited to the damaged myocardium, adhering to the endothelium, releasing ROS, and secreting degradative enzymes. This inflammatory response exacerbates tissue damage by clogging capillaries and increases oxidative stress [29]. Therefore, the goal would be to diminish the initial proinflammatory phase. While experimental interventions aimed at reducing neutrophil activity, such as using leukocyte-depleted blood, blocking cell adhesion molecules, or inhibiting complement, have shown potential [30, 31, 32], these strategies have not been consistently translated into clinical success [33, 34].
Normally, 2 to 5 percent of the oxygen consumed by mitochondria is converted to superoxide and neutralized by the cell’s antioxidant machinery, which involves catalase, glutathione peroxidase, and superoxide dismutase, among others [35]. In MRI, excessive ROS production also activates multiple second messenger pathways, besides all the previously described mechanisms of cell injury, which have also been the target of direct pharmacological interventions or are aimed to be regulated if oxidative stress is controlled.
MAPK is a group of enzymes that includes extracellular regulated kinases (ERKs),
p38 kinases, and c-Jun N-terminal kinases (JNKs). The two latter are activated by
oxidative stress and are pro-apoptotic [36]. The phosphoinositide-3
kinase/protein kinase B (PI3K/Akt) pathway is upregulated after MRI [37, 38].
Downstream effectors of Akt include the endothelial Nitric Oxide Synthase (eNOS),
mammalian target of Rapamycin (mTOR), glycogen synthase kinase 3
The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is a signaling mechanism involving tyrosine kinases associated with the intracellular domains of membrane-bound receptors. This pathway facilitates communication between cell surface receptors and the nucleus, which regulates cell survival and apoptosis. Specifically, STAT1 promotes apoptosis, while STAT3 exerts an anti-apoptotic effect [41].
The loss of cardiomyocytes is the direct consequence of cell death. Clinically, this translates into a greater infarct size, a significant determinant in the prognosis of these patients [2]. Understanding the mechanisms of cell death and their interactions is crucial for developing targeted therapies aimed at mitigating damage. Such insights could help design synergistic treatments to improve outcomes in patients with AMI. The connection between the oxidative stress generated by ischemia/reperfusion (IR) and programmed cell death is outlined in Fig. 1.
Autophagy is a component of cellular homeostasis responsible for degrading and recycling damaged proteins and cytoplasmic organelles. In response to ischemic stress, autophagy is activated mainly by inhibiting mTOR [42]. This process degrades macromolecules and organelles by transporting them to lysosomes in double-membrane vesicles termed autophagosomes. Autophagy is regulated by complexes involving beclin-1 and autophagy-related genes (ATGs), ensuring that damaged proteins and organelles are compartmentalized and removed before they trigger pro-apoptotic pathways [43]. However, autophagy can be double-edged. Excessive autophagy can cause autodigestion and cell death, while its inhibition could prevent the removal of damaged organelles, pushing cells towards apoptosis or necroptosis.
Beclin-1 is essential for autophagosome formation. Discordantly, its deletion ameliorates cardiac dysfunction resulting from severe pressure overload. Its overexpression leads to increased autophagy and an exacerbation of cardiac dysfunction [44], and increased apoptosis due to decreased beclin-1 leads to a reduction in infarct size [45]. In contrast, deletion of ATG5 in adult cardiomyocytes caused heart failure under baseline and pressure overload conditions. These opposing results highlight the complex role of autophagy in cardiac pathology [46].
Apoptosis is a regulated form of cell death characterized by cell shrinkage, chromatin condensation, and plasma membrane blebbing without rupture [47]. This pathway is initiated through two main mechanisms: the extrinsic pathway, which involves the activation of cell surface receptors (e.g., Tumor necrosis factor receptor 1 [TNFR1], Fas receptor, TNF-related apoptosis-inducing ligand [TRAIL]) leading to caspase-8 activation, and the intrinsic pathway, which is triggered by mitochondrial signals, such as cytochrome-c release and the formation of the apoptosome complex (Apoptotic protease activating factor 1, caspase-9) that ultimately activates caspase-3. Besides ROS production and mitochondrial calcium overload, the B-cell lymphoma 2 (Bcl-2) protein family proteins contribute to pore formation and mitochondrial outer membrane permeabilization allowing the release of cytochrome-c. Bcl-2 associated X protein (BAX)/Bcl-2 homologous antagonist/killer (BAK) promote apoptosis through mitochondrial membrane permeabilization, while survival pathways upregulate anti-apoptotic members such as Bcl-2. Also, VDAC increases mitochondrial matrix calcium concentration, contributing to ROS generation [48].
Apoptosis in myocardial reperfusion injury is notable for its non-inflammatory nature, as macrophages clear apoptotic cells without inducing an inflammatory response. However, excessive apoptosis can interfere with cell survival mechanisms, including autophagy, and may lead to unnecessary cell death. By reducing apoptosis, broad caspase inhibitors have been shown to limit MRI settings, supporting the therapeutic potential of targeting apoptotic pathways to mitigate damage [49].
Another key apoptosis regulator in cardiomyocytes is the apoptosis repressor with caspase recruitment domain (ARC). ARC inhibits apoptosis by interacting with both death receptors and mitochondrial pathways. ARC also interacts with p53, preventing its pro-apoptotic transcriptional activities [50]. Overexpression of ARC in cardiomyocytes reduces infarct size in animal models, while global ARC deletion exacerbates myocardial damage [51]. Furthermore, caspase inhibitors have shown variable success in reducing infarct size and cardiac dysfunction post-AMI [52]. Through caspase inhibition, the preservation of contractile proteins such as troponin T may mitigate some of the functional losses associated with reperfusion injury.
Programmed necrosis, also called necroptosis, leads to cell swelling, formation of a necrosome, and plasma membrane rupture, releasing cellular content and promoting an inflammatory response. The process is triggered through death receptors such as TNFR1 and Fas, activating receptor-interacting serine/threonine-protein kinase (RIPK) 1 and RIPK3. When caspase 8 activity is suppressed, RIP1 and RIP3 interact, forming the necrosome complex with mixed lineage kinase domain-like (MLKL), which subsequently oligomerizes and translocates to the plasma membrane, causing rupture [53].
Additionally, RIPK3 can phosphorylate Ca2+/calmodulin-dependent protein kinase (CaMKII) and Phosphoglycerate mutase, contributing to mPTP opening and mitochondrial dysfunction, which enhances ROS production and cell death. Also, extracellular RIPK3 has a role as a DAMP, binding to the receptor for advanced glycation end-products (RAGE), activating CaMKII and exacerbating myocardial injury [54]. Notably, it has been shown that necrostatin-1, an inhibitor of RIPK1, reduces IR injury (IRI), suggesting potential for therapeutic intervention [55].
Iron dysregulation has emerged as a key player in MRI and the pathophysiology of other cardiovascular diseases such as heart failure, cardiac hypertrophy, diabetic cardiomyopathy, and septic heart injury, positioning iron as a substrate for cardiovascular disease [56, 57, 58].
Ferroptosis, a form of regulated cell death driven by iron and ROS, is increasingly recognized as a critical mechanism in MRI and occurs when imbalances in iron and ROS lead to lipid peroxidation, mitochondrial damage, and overall organelle dysfunction [59].
Transferrin receptor 1 (TfR1) is a membrane protein that facilitates iron transfer from the extracellular environment into cells, thereby contributing to the intracellular iron pool necessary for ferroptosis. It regulates iron uptake and ensures iron is stored in a non-toxic form within ferritin [60]. Notably, TfR1 is highly expressed in the ischemic myocardium, leading to an influx of iron ions into cardiomyocytes during this phase [61].
Ferritin is a cellular iron storage protein that regulates iron efflux [62]. Its heavy subunit (FTH) has ferroxidase activity that converts Fe2+ to Fe3+ for storage inside the shell, being the main iron storage protein [63]. Ferritinophagy is an autophagy process that degrades ferritin [64]. This process is mediated by the nuclear receptor coactivator 4 (NCOA4), an autophagy cargo receptor that binds FTH1 and is delivered into the autolysosome for degradation, releasing free iron [65]. Notably, levels of FTH1 are decreased in myocardial tissues that undergo IR [66]. Subsequently, it was shown that glutaminolysis inhibition attenuated myocardial IRI by blocking ferroptosis [67].
The inhibition of the cystine/glutamate antiporter (system Xc⁻) depletes intracellular reduced glutathione (GSH), leading to the inactivation of glutathione peroxidase 4 (GPX4), which, under normal conditions, plays a key role in neutralizing lipid peroxides [68]. Down-regulation of GPX4, oxidizing GSH, and generating ferric iron from the Fenton reaction lead to lipid peroxidation and ferroptotic mitochondrial injury. Ferroptotic ROS generation contributes to myocardial cell death, and interestingly, iron chelators improve myocardial survival [69]. The substrates for peroxidation are phospholipids with polyunsaturated fatty acids (PL-PUFAs) [70]. ROS that originate from the Fenton reaction are catalyzed by iron-dependent lipoxygenases (LOXs). The obtained PL-PUFAs are then oxidized into lipid hydroperoxides (PL-PUFAs-OOH), which trigger ferroptosis [71].
Ferroptosis is primarily initiated during the reperfusion phase rather than during ischemia [72]. This observation underscores the importance of post-ischemic iron homeostasis, as clinical data indicate that iron overload is an independent risk factor for adverse left ventricular remodeling following reperfusion [73, 74].
Although ferroptosis is closely related to other types of programmed cell death, it is distinct from other forms of cell death, such as apoptosis, as it primarily involves oxidative membrane damage without significant nuclear involvement. Its regulation is influenced by key enzymes such as acyl-CoA synthetase long-chain family member 4 (ACSL4), which promotes the incorporation of PUFA into phospholipids, making membranes more susceptible to peroxidation [75]. Indeed, ACSL4 overexpression transfection blocked cardiomyocyte protective effects by an antioxidant substance [76]. Other molecules are the ferroptosis suppressor protein 1 (FSP1), which works independently of GPX4 and helps reduce lipid peroxides, providing an alternative mechanism of ferroptosis resistance [77] and the Coenzyme Q10 (CoQ10), which is another antioxidant involved in preventing lipid peroxidation. FSP1 uses CoQ10 to counteract oxidative damage [78]. A specific system, Xc-SLC7A11, is tightly involved in ferroptosis regulation, and modulates different pathways inhibiting ferroptosis [79]. Nuclear factor-erythroid 2-related factor 2 (Nrf2) has been widely investigated as a negative ferroptosis regulator, as it inhibits ROS production and reduces intracellular iron uptake. A previous study identified GPX4 and SLC7A11 as transcriptional downstream targets of Nrf2 [80].
So far, there is some monotherapy in human studies of several compounds focused on targeting ferroptosis in MRI in vitro and in vivo, in which different mechanisms have been proposed as potential pathways and strategies to mitigate the MRI. While most compounds are studied in the form of monotherapy, some that act on different pathways such as apoptosis and ferroptosis—referred to by some authors as the “strategy of killing two birds with one stone”—greatly enhance the therapeutic effect on MRI without the need for additional pharmaceutical excipients, offering strong potential for clinical application [61]. Iron chelators like Deferoxamine and Ferrostatine-1 have been effective in reducing cardiac dysfunction, cell death, and infarct size [15, 67].
Pyroptosis is a regulated form of cell death characterized by the permeabilization of the plasma membrane and the release of inflammatory cytokines into the extracellular space, exacerbating inflammation and tissue damage. This process is mediated by gasdermin D (GSDMD), a protein that in its inactive form is auto inhibited. Upon activation by caspases, such as caspase-1 or caspase-11, GSDMD undergoes proteolytic cleavage, which then embeds into the cell membrane to form pores. These pores lead to cell swelling, membrane rupture, and the release of inflammatory signals [81].
The NLR family pyrin domain containing 3 (NLRP3) inflammasome is a key activator
of pyroptosis, mainly through the activation of caspase-1, which not only cleaves
GSDMD but also processes the proinflammatory cytokines interleukin-1
Preclinical studies have shown numerous promising cardioprotective agents, some of which will be mentioned in the following sections. In particular, oxidative stress plays a central role in MRI, prompting extensive research into natural and synthetic antioxidant compounds. While many antioxidant agents have shown promising results in preclinical models, few have progressed to clinical trials. This section highlights the results from clinical studies evaluating that have assessed antioxidant agents that have been tested in humans. Rather than providing an exhaustive list, we have focused on the largest trials and recent meta-analyses that have assessed the potential cardioprotective effects of these agents (Table 1, Ref. [84, 85, 86, 87, 88, 89, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102, 103, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125, 126, 127, 128, 129, 130, 131, 132, 133, 134, 135, 136, 137, 138]). Additionally, we provide a brief overview of the mechanisms of action of these antioxidants, along with the challenges and limitations that have hindered their translation into routine clinical practice. This perspective aims to offer a clearer understanding of both their therapeutic potential and the barriers to their widespread adoption.
| Compound | Pathway/Mechanism involved | Results in humans |
| Melatonin | Free radical scavenger [85]. | Lowered CK-MB levels on postoperative days 2 and 3 after CABG surgery, and shortened ICU stay, without significant changes in other inflammatory markers [88]. |
| Nrf2/ARE [86]. | Improved LVEF and reduced HR. Reduced levels of cTnI, IL-1 | |
| SIRT1 [84]. | Early administration significantly reduced infarct size, while late administration was associated with a larger infarct size [90]. | |
| Decreases apoptosis (JAK2/STAT3) [87]. | Did not improve the myocardial salvage index or levels of troponin T, CK-MB, or oxidative stress biomarkers compared to placebo [91]. | |
| N-acetylcysteine | ROS scavenger and precursor for glutathione biosynthesis [116]. | Decreased plasma MDA levels, LVEF was higher, there was no difference in reduction of infarct size according to CK-MB levels [93]. |
| Reduced infarct size in patients with STEMI undergoing PCI [92]. | ||
| Reduced early cardiac remodeling by diminishing levels of MMP-2 and MMP-9, reduced hospital stays and reduced the incidence of MACE [94]. | ||
| Allopurinol | Inhibits XO. Free radical scavenger [95]. | Treatment improves the TIMI flow. No improvement in cardiovascular events, complications, troponin levels and ECG ST-elevation regression [96]. |
| Reduces urinary isoprostanes levels. Higher recovery of LVEF [96]. | ||
| More effective ST-elevation recovery and lower cTnI, CK and CK-MB peak values. Lower incidence of MACE, with no significant differences in LVEF [97]. | ||
| Edaravone | Free radical scavenger [98]. | Reduced incidence of ventricular tachyarrhythmias [101]. |
| Decreases apoptosis (JAK2/STAT3) [99]. | Reduction of reperfusion arrhythmias, myocardial stunning, and lethal reperfusion injury [100]. | |
| Improved left ventricular systolic function immediately after reperfusion. | ||
| Higher LVEF and fewer heart failure-related rehospitalizations at 12 months [100]. | ||
| Sodium thiosulphate | Inhibits mitochondrial complex IV, increases SOD activity [102]. | No clinical benefit in reducing reperfusion injury in a sample with low risk for large infarction [104]. |
| Activates Nrf2 [103]. | ||
| Nicorandil | Stimulates mitochondrial ATP-sensitive potassium channels [105]. | Reduced the incidence of the no-reflow phenomenon and MACEs, A combination of intracoronary and intravenous administration further reduced the incidence of MACEs [107]. |
| Decreases pyroptosis (TLR4/MyD88/NF-κB/NLRP3) [106]. | No significant difference in infarct size, cardiac function, or MACEs between the nicorandil and placebo groups over 6 months [108]. | |
| Quercetin | SIRT1 [109]. | Lower CK-MB AUC, suggesting a smaller infarct size. Reduced the incidence of reperfusion-induced intramyocardial hemorrhage. No significant differences were noted in LVEF or LV remodeling indicators [110]. |
| Ascorbic Acid | Free radical scavenger. Nrf2/ARE [117]. | Meta-analysis of seven controlled trials (872 patients) examining peak cTn and CK-MB levels post-procedure and oxidative stress biomarkers. AA reduced peak cTn levels by 43% and peak CK-MB levels by 14% and decreased oxidative stress biomarkers [120]. |
| Inhibits NADPH oxidase synthesis [118]. | Randomized study with 532 patients receiving either a 3-g AA infusion or normal saline before PCI. The incidence of myocardial injury was significantly lower in the AA group. AA use was an independent predictor of reduced myocardial injury [121]. | |
| Prevents the oxidation of BH4 [119]. | ||
| Statins | Nrf2/ARE [115]. | Early high-dose rosuvastatin therapy did not improve myocardial perfusion or reduce infarct volume compared to conventional low-dose therapy in STEMI patients undergoing primary PCI [116]. |
| Decreases the activity of NADPH oxidase [114]. | High-dose atorvastatin pretreatment followed by continued treatment for 5 days did not reduce infarct size in STEMI patients undergoing primary PCI [122]. | |
| Reduces the expression of LOX-1 [111, 112, 113]. | ||
| Dexmedetomidine | AMPK/GSK-3 |
Decrease in cTnI and TNF- |
| Dapagliflozin | MAPK signaling inhibition [125]. | The primary hierarchical composite outcome showed significantly more wins for DAPA compared to placebo. No significant difference in the composite of cardiovascular death or hospitalization for heart failure. |
| No significant reduction in cardiovascular death or hospitalization for heart failure compared to placebo [126]. | ||
| Empagliflozin | AMPK/Nrf2 [127]. | No significant difference of either a first hospitalization for heart failure or death from any cause compared to placebo [128]. |
| Puerarin | AMPK [129]. | Fewer angina pectoris attacks and ST segment changes during the balloon dilation stage of PCA compared to the conventional group. |
| Reduced blood levels of vWF and ET-1, while increasing NO levels, compared to the conventional group [130]. | ||
| Sevoflurane | Not yet clear [131]. | No reduction in the incidence of cardiac and non-cardiac events during the 6 months following cardiac surgery involving extracorporeal circulation. Trend towards reduced treatment needs and fewer hospital admissions, when experiencing any events [131, 132]. |
| Propofol | Akt/p53 [133]. | After a CPB, compared with SEV, propofol reduced incidence of AKI, decreased inflammatory markers such as IL-6, CRP and segmented neutrophil counts [134]. |
| Deferoxamine | Chelates non-transferrin bound iron (free iron), iron in transit between transferrin and ferritin (labile chelating iron pool), hemosiderin, and ferritin [135]. | Reduced post-PCI serum iron. No differences in serum ferritin, soluble transferrin receptor, F2-isoprostane levels, CRP levels, infarct size, creatine kinase, cTnI, the mean ST-segment resolution, the degree of wall motion abnormality, LVEF [136]. |
| No significant difference between preischemia and reperfusion in the indirect measure of ROS activity. Better preservation of myocardial cells. Reduction in the number of severely damaged mitochondria [137]. | ||
| Reduction in thiobarbituric reactive substances (TBARS) levels, LVEF improvement, better myocardial recovery reflected by wall motion score index improvement [138]. |
AA, ascorbic acid; AMPK, AMP-activated protein kinase; AKI, acute kidney injury;
ARE, antioxidant response element; ATP, adenosine triphosphate; AUC, area under
curve; BH4, tetrahydrobiopterin; cTnI, cardiac troponin I; CABG, coronary artery
bypass graft; CPB, cardiopulmonary bypass; CK-MB, creatine kinase-MB; CRP,
C-reactive protein; ECG, electrocardiogram; ET-1, endothelin-1; GSK-3
Melatonin is a lipophilic hormone predominantly produced predominantly by the pineal gland in most living organisms, primarily responsible for regulating circadian rhythms, particularly the sleep-wake cycle. Clinically, melatonin has been employed for decades to treat conditions related to altered biological rhythms and sleep disorders [84]. Over time, additional therapeutic properties of melatonin have been identified, including its potent anti-inflammatory and antioxidant effects, leading to its investigation in various conditions [84].
More recently, the potential of melatonin to mitigate IRI has attracted
increasing attention. Its antioxidant and cardioprotective effects are mainly
attributed to its ability to directly scavenge free radicals, an action also
shared by several of its metabolites [85]. Furthermore, melatonin enhances the
activity of endogenous antioxidant enzymes by upregulating the transcription
factor Nrf2 and sirtuin 1 (SIRT1) [86, 139]. SIRT1, a protein deacetylase, is
stimulated by melatonin to enhance the stability and nuclear translocation of
Nrf2, boosting the production of antioxidant enzymes and suppressing
pro-inflammatory signaling pathways, such as nuclear factor
kappa-light-chain-enhancer of activated B cells (NF-
It also exerts anti-apoptotic effects by activating the JAK2/STAT3 signaling pathway [87]. Additional proposed mechanisms include inhibiting autophagy and suppressing quinone oxidoreductase two activity, although these remain debated within the scientific community [84, 87].
These properties have provided the foundation for randomized controlled trials (RCTs) evaluating the efficacy of melatonin in preventing IRI in patients with AMI with mixed results. In patients with ST-segment elevation myocardial infarction (STEMI) melatonin administration during reperfusion did not significantly improve the myocardial salvage index or reduce cardiac biomarkers (hs-cTnT, creatine kinase (CK)-MB) and oxidative stress markers compared to placebo [88]. However, a post-hoc analysis from the Melatonin Adjunct in Acute Myocardial Infarction Treated with Angioplasty trial suggested a time-dependent effect, with early melatonin administration (within 136 minutes of symptom onset) associated with smaller infarct size, whereas delayed administration (249 minutes) increased infarct size [89]. In coronary artery bypass graft (CABG) surgery patients, melatonin demonstrated dose-dependent benefits, including improved left ventricular ejection fraction (LVEF), reduced heart rate, cardiac injury biomarkers, inflammatory markers, and apoptosis, with higher doses (20 mg) yielding stronger effects [90]. Another CABG trial reported reduced postoperative CK-MB levels and shorter intensive care unit (ICU) stays with melatonin supplementation, though without significant changes in other biomarkers [91].
A meta-analysis of several trials concluded that melatonin did not improve cardiac function or reduce infarct size [143]. The variability in trial methodologies, including differences in melatonin dosages (most studies used 50 mg intravenous (IV), nonetheless, dosages other treatment regimens 2 mg intracoronary (IC), and 3 mg per os (PO) routes of administration (PO, IV, and IC) could have explained the heterogeneity in infarct size reduction measured by troponin levels, since there is no consensus of the optimal clinical plasma concentration and mode of administration [144], and timing of treatment, may have influenced these findings. Notably, early administration, whether IC or IV, was associated with reductions in infarct size and improvements in LVEF, while delayed administration showed the opposite effect [143]. Regarding the measurement of the MRI, few trials have used cardiac magnetic resonance imaging (CMRI) to measure infarct size, while most of them rely on troponin levels [88, 89]. From a safety perspective, melatonin administration has generally been well tolerated, with no significant adverse effects reported [145].
So far, the combination therapies with melatonin in the heart is not well
studied. A recent study evaluated the synergistic effects of melatonin and
captopril in a rat model of chronic heart failure. This combination improved
systolic blood pressure, left ventricular function, while reducing myocardial
injury markers and oxidative stress. The study demonstrated that the treatments
effectively inhibited apoptosis by reducing caspase-3 activity and alleviated
fibrosis by lowering the levels of transforming growth factor-beta 1
(TGF-
Future research should focus on optimizing the timing and mode of melatonin administration to maximize its cardioprotective effects. Additionally, more extensive clinical trials are needed to account for patient-related factors such as age, comorbidities, and gender. Finally, further investigation is required to elucidate the precise mechanisms underlying melatonin’s therapeutic actions, as many remain incompletely understood.
N-acetylcysteine (NAC) is a compound derived from the amino acid L-cysteine,
which has antioxidant properties and therapeutic uses in different clinical
settings. It replenishes glutathione, a crucial antioxidant that helps protect
cells from oxidative stress and damage [147]; it is also a reductant of disulfide
bonds, a scavenger of reactive oxygen species, and its mechanism involves the
inhibition of NF-
Moreover, NAC has shown potential benefits in treating AMI. A study showed that oral supplementation of 600 mg NAC every eight hours for 72 hours can reduce high-sensitivity C-reactive protein, myeloperoxidase and Galectin-3 levels in AMI patients receiving fibrinolytic therapy [149]. Another study in patients who received fibrinolytic therapy plus NAC demonstrated that, compared to controls, plasma malondialdehyde (MDA) levels decreased, and LVEF was higher. However, there was no difference in infarct size according to CK-MB levels [150]. Furthermore, NAC may help prevent early cardiac remodeling by reducing levels of metalloproteinase (MMP)-2 and MMP-9. Additionally, it has been shown to shorten hospital stays in patients after AMI and also reduce the incidence of major adverse cardiovascular events (MACE) [151].
Despite these positive findings, evidence on the effect of NAC in reducing MRI in humans remains inconclusive [152, 153]. One possible explanation is that the antioxidant effects of other agents used in reperfusion interventions, such as halogenated anesthetics or propofol, may diminish the observable benefit of NAC. Additionally, variability in NAC dosage (ranging from 4 to 300 mg/kg), treatment duration (1 hour to 5 days), and administration routes (PO, IV, or a combination of both) may have influenced the results. Also, clinical trials have been performed in different populations. Although they present MRI, differences between patients undergoing CABG, PCI, or thrombolysis make it difficult to draw broadly applicable conclusions.
In addition, NAC has been studied in combination with other treatments, such as nitroglycerin, to improve blood flow in patients experiencing AMI. Some trials have shown that when administered intravenously, high-dose NAC can enhance the effects of low-dose intravenous nitroglycerin, leading to better blood vessel dilation and increased oxygen delivery to heart tissue. This is related to a reduced infarct size in patients with STEMI undergoing PCI [92]. However, there are no recent studies in humans and the high incidence of side effects limits the clinical applicability of this therapeutic strategy [154]. In rats, a treatment combining NAC and allopurinol synergistically enhanced cardiac adiponectin (APN) content, an adipokine anti-ischemic properties, and reduced IRI. NAC alone increased cardiac APN levels and AdipoR2 expression, while allopurinol amplified NAC’s effects, restoring key signaling pathways. Both NAC and allopurinol independently reduced myocardial infarct size and CK-MB release, with their combination demonstrating synergistic cardioprotective effects [93].
Regarding biomarkers, comparisons between levels of TGF-
While research is promising, more large-scale studies are needed to fully confirm the benefits of this safe drug in reducing damage and improving recovery in patients with AMI.
Allopurinol is a medication primarily used to treat gout, some kidney stones, and cardiovascular diseases [94]. It is a purine base analog and works by inhibiting the enzyme xanthine oxidoreductase (XOR), which exists in two isoforms in vivo: xanthine dehydrogenase (XDH) and XO. XDH produces NADH and uric acid, the latter acting as a free radical scavenger, while XO catalyzes the formation of both uric acid and superoxide anion. Under conditions of ischemia and hypoxia, ATP depletion induces a reversible conversion of XDH to XO through the oxidation of sulfhydryl groups. Upon reperfusion, increased oxygen availability and elevated XO activity result in excessive ROS generation, exacerbating oxidative stress. In this context, allopurinol effectively inhibits XO activity and mitigates ROS production [155].
In the context of MRI in patients with STEMI, there is methodological heterogeneity. Further supporting its antioxidant role, when given before reperfusion in patients with AMI, the group treated with allopurinol showed reduced urinary isoprostane levels, a biomarker produced when free radicals cause lipid peroxidation in cell membranes. Also, there was a recovery of LVEF [156]. In a different RCT, allopurinol pre-treatment in 140 patients undergoing fibrinolysis reported improved ST-segment resolution at 90 minutes, reduced infarct size (as measured enzymatically), and a lower incidence of in-hospital MACE [95]. It has also shown an improvement in thrombolysis in myocardial infarction (TIMI) flow following PCI. This effect was expected considering the localization of ROS in capillary endothelial cells, along with cell swelling, activated neutrophils, and aggregated platelets, all of which contribute to the no-reflow phenomenon in STEMI patients undergoing emergency PCI. Nevertheless, other outcomes were not improved, such as cardiac adverse events, troponin levels, and electrocardiogram (ECG) ST-elevation regression [96]. Other RCT demonstrated that allopurinol resulted in a more effective ST elevation recovery and lower cardiac troponin I (cTnI), CK, and CK-MB peak values. In this same study, after a 1-month follow-up period, patients had a lower incidence of MACE, but no differences in LVEF were detected [97].
These findings suggest that while allopurinol may benefit coronary blood flow and specific inflammatory markers, its overall impact on cardiovascular outcomes in AMI remains limited.
Edara Edaravone is a lipophilic drug with potent free radical scavenging activity by quenching •OH and inhibiting both •OH-dependent and •OH-independent lipid peroxidation [98]. It was initially studied for its neuroprotective effects, particularly in the context of stroke and, more recently, neurodegenerative conditions such as amyotrophic lateral sclerosis (ALS) [98, 157]. In recent decades, edaravone has also been recognized for its cardioprotective potential, mainly due to its capacity to attenuate cardiomyocyte apoptosis via activation of the JAK2/STAT3 signaling pathway [99].
Clinically, it has demonstrated efficacy in reducing IRI in AMI. When administered prior to reperfusion, it reduces infarct size, lowers the incidence of reperfusion-induced arrhythmias, and improves LVEF by decreasing oxidative stress and free radical generation [100]. Additionally, edaravone has been shown to lower plasma levels of monocyte chemoattractant protein-1 (MCP-1), which is associated with improved long-term cardiac recovery and a reduced incidence of heart failure [101].
Furthermore, reductions in biomarkers of tissue damage and oxidative stress, such as thioredoxin, have been observed, suggesting that edaravone may positively influence long-term survival and prognosis in AMI patients treated with this antioxidant [100]. Its ability to inhibit lipid peroxidation and prevent endothelial damage, combined with its high tissue accessibility due to its low molecular weight, further strengthens its potential as a cardioprotective agent against reperfusion injury [100].
Edaravone, in combination with obeticholic acid, has shown promising
cardioprotective effects in an animal model of cardiotoxicity. The treatment
significantly reduced levels of alkaline phosphatase (ALP), aspartate
aminotransferase (AST), lactate dehydrogenase (LDH), CK-MB, and cTnI, while
ameliorating histopathological cardiac abnormalities. It also decreased oxidative
stress markers such as MDA, increased antioxidant defenses (superoxide dismutase
[SOD] and GSH), and upregulated Nrf2, peroxisome proliferator-activated receptor
gamma (PPAR-
Despite these promising findings, the clinical evaluation of edaravone in this context remains limited, with few studies assessing its long-term benefits. Additional challenges include its rapid elimination from the body and the need for early, controlled administration to achieve optimal therapeutic effects [159]. Moreover, the absence of large-scale RCTs further limits the generalizability of these results.
Sodium thiosulphate (STS) is a water-soluble compound derived from hydrogen sulfide, employed for treating acute cyanide poisoning and carbon monoxide toxicity and to mitigate cisplatin-related toxicity in chemotherapy. Recently, STS’s anti-inflammatory and antihypertensive antioxidant effects have been identified [160].
The antioxidant activity of STS is linked to its capacity to inhibit mitochondrial complex IV, which reduces the production of mitochondrial free radicals. Moreover, STS allosterically binds to the enzyme SOD and positively regulates Nrf2 enhancing its antioxidant activity [102, 103].
The clinical application of STS in the context of AMI is relatively novel, with only one clinical trial conducted in 2022. This trial did not demonstrate a clinical benefit from STS administration, as treatment during reperfusion failed to reduce infarct size or improve LVEF [104, 161]. Nevertheless, this study establishes a foundation for future research to realize the therapeutic benefits observed in preclinical models.
Nicorandil is an antianginal drug distinguished by its dual mechanism of action. It functions as a nitric oxide donor, inducing vasodilation and as an agonist of ATP-sensitive potassium channels. These properties make it a potent vasodilator, reducing both ventricular preload and afterload and improving oxygen delivery under ischemic conditions [162]. However, nicorandil’s potential extends beyond these effects. It attenuates oxidative stress by stimulating the opening of mitochondrial ATP-sensitive potassium channels, which optimizes oxidative phosphorylation, reduces free radical generation, and decreases the opening of the mitochondrial permeability transition pore [105]. Furthermore, preclinical studies suggest that nicorandil may positively regulate eNOS activity and reduce cardiomyocyte pyroptosis [106, 163].
Given these properties, nicorandil has been proposed as a promising cardioprotective agent. A meta-analysis of 18 RCTs demonstrated that nicorandil administration improved coronary no-reflow phenomenon and ST-segment resolution after PCI, and reduced MACE [107]. The cardioprotective effects were more pronounced when nicorandil was administered IC or IV. Another meta-analysis analyzed 14 studies with 1762 patients to assess the effectiveness of nicorandil in reducing periprocedural myocardial injury (PMI) and MACE during PCI. The results showed that it significantly reduced both PMI and MACE, likely due to its dual action as a nitrate and potassium channel opener, which improves coronary blood flow and myocardial preconditioning. Specifically, it also reduced myocardial injury, oxidative stress, and ferroptosis markers. LV end-diastolic diameter and the incidence of unstable angina and heart failure 12 weeks post-PCI was also reduced although LV function parameters remained similar between groups [164]. However, limitations such as variability in administration methods and lack of subgroup data indicate the need for further research on optimal dosing and comparative effectiveness,
Despite these promising results, the benefits of nicorandil have not been consistently observed. For example, a recent RCT with 83 patients found no significant improvement in infarct size, cardiac function, or the incidence of MACE [108]. One RCT reported that nicorandil treatment significantly reduced infarct size and edema compared to nitrate therapy, although the study’s small sample size limited its generalizability [165]. Additionally, Ilyas et al. [166] reported that nicorandil enhanced ST-segment resolution, lowered cTnI levels 6 hours post-PCI, and reduced the incidence of MACE, although CK-MB levels were not significantly affected.
Therefore, while nicorandil shows potential cardioprotective effects, the evidence remains inconsistent across studies. Larger, well-designed RCTs are necessary to establish its efficacy and clarify nicorandil’s efficacy in preventing IRI.
Quercetin is a natural polyphenol found in plants and is recognized for its
broad spectrum of biological activities [167]. Its antioxidant capacity and
potential as a cardioprotectant in the context of IRI have garnered attention.
The primary mechanism proposed for these cardioprotective effects is the positive
modulation of SIRT1 [109]. Activation of SIRT1 reduces oxidative stress by
enhancing antioxidant defenses, promoting transcription factors Nrf2 and FOXO1,
both of which are critical for cellular protection against oxidative damage
[168]. Moreover, SIRT1 has been shown to attenuate inflammation and cardiomyocyte
apoptosis by inhibiting the NF-
Despite its promising cardioprotective profile in preclinical studies, the clinical use of quercetin in patients with AMI has been hindered by its unfavorable pharmacokinetics. Quercetin is poorly soluble in water, exhibits low bioavailability, and is rapidly metabolized [167]. However, the recent development of nanoformulations has helped to mitigate some of these limitations. In this context, an RCT recently investigated a nanoformulation of quercetin in patients with AMI to evaluate its ability to attenuate IRI. The study showed a reduction in infarct size and intramyocardial hemorrhage, although no significant effect on LVEF was observed [110].
This trial faced limitations, including using the area under the CK-MB curve to estimate infarct size. Additionally, the small sample size reduced the study’s statistical power. Despite these constraints, the trial provides a foundation for future RCTs with larger sample sizes to better assess quercetin nanoformulations’ efficacy, optimal dosage, and duration to maximize their cardioprotective potential.
Statins are a class of medications primarily known for inhibiting the enzyme 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase), a key regulator of cholesterol synthesis. By lowering cholesterol levels, statins address a significant risk factor for the development of cardiovascular disease. However, their cardioprotective properties extend beyond cholesterol reduction, as they have been shown to exert protective effects at the molecular level through various mechanisms. These include the interruption of RhoA-mediated ventricular remodeling pathways, elevation of adenosine levels, reduction of mPTP opening, enhancement of mitophagy via AMP-activated protein kinase (AMPK) pathway activation, and increased antioxidant activity [169, 170].
Preclinical studies indicate that statins may also decrease the activity of NADPH oxidase, enhance the activation of the Nrf2/antioxidant response element (ARE) pathway, and reduce the expression of the lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1), which is upregulated in cardiomyocytes under oxidative stress [111, 112, 113, 114, 115]. In this context, in vivo studies using swine models have demonstrated that early intravenous administration of atorvastatin prior to revascularization can significantly reduce IRI, leading to a decrease in infarct size [116, 171].
Translating these findings to clinical practice has proven challenging, as results observed in preclinical animal models are not consistently replicated in human subjects. The prospective ROSEMARY study exemplifies this issue; it compared high-dose rosuvastatin administration (40 mg before primary PCI and 40 mg/day for 7 days thereafter) with low-dose administration (placebo before primary PCI and 10 mg/day for 7 days post-PCI) [116]. The results indicated that infarct size and clinical outcomes were comparable between both groups, with no significant differences observed.
A potential explanation for the inconclusive results of this study may be related to the use of specific antiplatelet agents commonly included in standard therapy, such as aspirin. These agents may diminish the cardioprotective effects of statins by inhibiting cyclooxygenase-2, leading to reduced levels of adenosine, which mediates some of the cardioprotective benefits [172, 173]. In light of this, research has explored the use of alternative antiplatelet agents, such as cilostazol, which appear to enhance the effects of statins. However, human studies evaluating this combination remain limited [174, 175, 176].
There is substantial evidence supporting the role of oxidative stress in IRI following AMI. Given its complexity, the development of multi-targeted, antioxidant-based combination therapies presents a promising avenue for mitigating such damage. Multitherapy has been extensively studied as a strategy to combat IRI across various organs, including the liver, kidneys, brain, and myocardium. In this section, we will focus on the multitherapy approaches that have proven to be most effective in the context of MRI (Table 2, Ref. [177, 178, 179, 180, 181, 182, 183, 184, 185, 186, 187, 188, 189]).
| Compound | Pathway/Mechanism involved | Results in humans |
| GIK | Insulin inhibits NF-κB (PI3K/Akt). Upregulates eNOS expression (PI3k/Akt). Inhibits the opening of mPTP (PI3k/Akt/hexokinase) [177]. | A meta-analysis indicates that high-dose GIK in ACS patients undergoing reperfusion may enhance myocardial protection and long-term cardiac function, especially in STEMI cases. However, it also shows increased risks of phlebitis, hyperglycaemia, and hypoglycaemia, warranting cautious use in high-risk patients [180]. |
| Counteracts glucose toxicity [178]. | High-dose GIK therapy in ACS patients receiving reperfusion therapy reduced MACEs and exhibited good oxidative stress-lowering efficacy, but did not improve survival outcomes [179]. | |
| GIK therapy in ACS patients generally resulted in lower serious endpoint rates compared to placebo, with significant benefits observed in STEMI patients regarding mortality and cardiac arrest within 1 year [181]. | ||
| AA + Vit E | Vitamin E (alpha-tocopherol) prevents lipid peroxidation of PUFAs [185]. | Included 800 patients randomly assigned to receive either antioxidants or a matching placebo for 30 days. Patients treated with antioxidants had a lower incidence of in-hospital cardiac mortality and non-fatal myocardial infarction (14% vs. 19%, OR 0.82) [186]. |
| The LVEF of the high ascorbate (HA) group was significantly higher than that of the low ascorbate (LA) group, with values on day 84 in the HA group being 33% higher. 95% of HA patients achieved a Thrombolysis In Myocardial Infarction (TIMI) myocardial perfusion grade of 2–3, while only 79% of LA patients reached this grade [184]. | ||
| AA + Vit E + Vit A + beta-carotene | Unknown. | Mean infarct size (measured by CK and CK-MB) was significantly lower in the antioxidant group compared to the placebo group. |
| Lactate dehydrogenase levels increased slightly in the antioxidant group (88.6 IU/dL) compared to the placebo group (166.5 IU/dL). | ||
| Angina pectoris, total arrhythmias, and poor left ventricular function occurred less frequently in the antioxidant group. | ||
| Cardiac events were significantly lower in the antioxidant group (20.6% vs 30.6%) [185]. | ||
| Curcumin + Piperine | Curcumin acts in the upregulation of PPAR |
In patients with acute myocardial infarction, daily 500 mg of curcumin with piperine for 8 weeks improved lipid profiles by lowering LDL and raising HDL, enhanced glycemic control (reduced HbA1C), and decreased liver enzymes. No significant effects were noted on ejection fraction, cTnI, renal function, or electrolytes [183]. |
| Piperine acts in PPAR |
In post-CABG patients, a 5-day treatment with 500 mg of curcumin and 5 mg of piperine reduced C-reactive protein (CRP) and increased antioxidant capacity. There was a slight decrease in CK-MB, but no significant changes in cTnI, LDH, ejection fraction, atrial fibrillation incidence, or renal markers [182]. |
CK, creatine kinase; GIK, glucose-insulin-potassium; NF-
GIK therapy has been studied since the 1960s as a potential strategy to mitigate IRI [190]. The rationale behind GIK therapy is to improve myocardial metabolism while leveraging the cardioprotective properties of insulin. Glucose and potassium are included primarily to counteract adverse effects, such as hypoglycemia and hypokalemia. Insulin plays a pivotal role by reducing glucose toxicity, acting as a positive inotrope, reducing oxidative stress, and activating anti-apoptotic and anti-inflammatory cell survival pathways [177]. Furthermore, insulin promotes glucose utilization as the primary energy source, thereby decreasing free fatty acid oxidation, reducing oxygen consumption, and attenuating oxidative stress [178]. However, the evidence remains contradictory, with some studies showing no change in post-infusion oxidative stress parameters [191], while others have reported alterations in SOD enzyme activity [192].
Since its introduction, numerous clinical trials have investigated GIK therapy, mostly yielding unfavorable outcomes. A 2010 meta-analysis, which included nine RCTs with a total of 28,000 patients, demonstrated no significant reduction in mortality with GIK therapy in cases of STEMI [193]. Among the most notable studies, the CREATE-ECLA trial, which enrolled over 20,000 patients, found no significant differences in in-hospital mortality or cardiovascular complications following GIK infusion. This was likely influenced by the average administration time, which exceeded three hours after symptom onset [194].
However, the 2012 IMMEDIATE trial examined the early administration of GIK by emergency medical services, with an average time of 90 minutes from symptom onset. The primary outcomes (myocardial infarction biomarkers) were neutral, but secondary analyses revealed a significant reduction in infarct size at 30 days in 80% of patients with STEMI compared to placebo [195]. This finding has spurred further investigation, and the IMMEDIATE-2 trial is currently underway to explore these hypotheses [196].
More recent meta-analyses have sought to clarify the controversies surrounding GIK therapy. Yang et al. [197] reported that high-dose GIK reduces oxidative stress, improves LVEF, and decreases the risk of MACE. Similarly, Liu et al. [198] found that GIK improves coronary blood flow and cardiac function but is associated with an increased incidence of adverse events, such as phlebitis and hypoglycemia. Both meta-analyses underscore the need for large-scale clinical trials with long-term follow-up to more definitively assess the efficacy and safety of this therapy.
Curcumin is the active compound found in turmeric (Curcuma longa), a bright yellow spice commonly used in cooking, especially in Asian cuisine. It is a natural polyphenol known for its anti-inflammatory, antioxidant, and anti-cancer properties [199]. One of curcumin’s primary mechanisms of action is its ability to enhance the expression, stability, and nuclear translocation of Nrf2 [179]. It also has a direct role in IRI. In vitro studies have demonstrated its capacity to modulate apoptosis, autophagy, and, more recently, ferroptosis in cardiomyocytes [180, 200]. Some proposed mechanisms are through positive modulation of the Hes1 protein, an intermediary in these cell death pathways. Specifically, curcumin enhances Bcl-2 protein activity to inhibit apoptosis, reduces beclin-1 and SIRT1 activity to mitigate autophagy, and increases GPX4 activity to suppress ferroptosis. However, its low bioavailability remains a significant limitation, hindering its clinical application.
On the other hand, piperine is an active compound found in black pepper
(Piper nigrum), which also possesses anti-inflammatory,
antihypertensive, anti-cancer, and antioxidant properties [181]. These effects
are primarily attributed to the activation of the PPAR-
Despite the promising cardioprotective effects observed in preclinical studies, clinical evidence for this combination remains limited [203]. For instance, a study involving patients undergoing CABG surgery reported reduced in inflammatory parameters and increased total antioxidant capacity. However, no changes were observed in levels of cTnI, LDH, LVEF, or the incidence of atrial fibrillation [182]. Another study assessed the effects of the curcumin-piperine combination following AMI. The results demonstrated significant improvements in glycemic control and lipid profile. However, the treatment did not impact on LVEF or cTnI [183]. A recent review suggested that the curcumin doses used in clinical studies may have been too low to achieve meaningful cardioprotective effects. It proposed that higher doses, which have demonstrated safety, could more closely replicate the efficacy observed in animal models [184].
One of the most extensively studied approaches in this field is the combination of vitamins C (ascorbic acid, L-ascorbic acid or L-ascorbate) and E (alpha-tocopherol) [7]. Ascorbic acid (AA), a hydrophilic vitamin, functions primarily as a potent reducing agent, directly scavenging ROS such as superoxide anion [117]. Furthermore, it inhibits NOX synthesis and prevents the oxidation of tetrahydrobiopterin (BH4), thus avoiding the uncoupling of eNOS [118, 119]. Pharmacokinetic and pharmacodynamic studies have indicated that a minimum concentration of 10 mM is required to observe these effects in tissues under oxidative stress, a critical consideration for future studies evaluating its antioxidant efficacy [117].
Vitamin E is a fat-soluble micronutrient that accumulates in cell membranes, where it serves as both a membrane stabilizer and an antioxidant [204]. Its antioxidant capacity is primarily focused on preventing lipid peroxidation of PUFAs, thereby reducing damage to cell membranes [205]. The rationale behind combining vitamins C and E lies in their complementary actions: vitamin C operates in aqueous environments, while vitamin E functions in lipid environments [7]. Moreover, vitamin C plays a critical role in recycling alpha-tocopherol by reducing its oxidized form, thus enhancing its antioxidant activity [206].
Several clinical trials have been conducted to assess the efficacy of this combination therapy. One of the earliest studies, the MIVIT trial in 2005, investigated the clinical outcomes of patients treated with the combined therapy and observed a reduction in adverse events, although no specific clinical parameters were measured to explain these results [207]. The PREVEC trial demonstrated that patients who achieved elevated blood AA levels prior to reperfusion (via PCI) and continued oral supplementation with vitamins C and E showed improved ventricular function, as evidenced by increased LVEF and enhanced microvascular flow [208, 209]. However, a key limitation of this study was the lack of assessment of the therapy’s effects on infarct size and myocardial remodeling, both of which are critical factors in reperfusion injury [209]. A recent meta-analysis including eight RCTs that evaluated AA in PCI reported a reduction in myocardial damage biomarkers, without an improvement in myocardial function parameters [209]. As in other studies, there was considerable variability among trials, including differences in patient populations (AMI vs. stable angina) and timing of administration. Some studies implemented a continuation strategy extending weeks or months post-reperfusion. Additionally, most trials included relatively young patient cohorts, which may limit the generalizability of the findings [210, 211, 212].
Prior to the exclusive use of AA and vitamin E, combinations that included vitamin A and beta-carotene were tested and yielded favorable results in treated groups [185]. However, over time, the antioxidant and cardioprotective roles of vitamin A and beta-carotene have been questioned, leading to a decline in further studies on this combination [186, 213, 214].
Several authors have proposed combination therapy as a promising strategy for
MRI [215]. However, most of the evidence supporting the synergistic effects of
multi-target antioxidant therapies comes from preclinical studies [216]. A
combination of AA, NAC, and deferoxamine (DFO) has shown encouraging results and
is currently being evaluated in early-stage clinical trials. AA acts as a potent
antioxidant with cardioprotective properties, while NAC replenishes GSH stores,
chelates metal ions, and modulates inflammation via NF-
Several clinical trials are currently investigating novel therapeutic approaches for AMI. One of the most studied is supersaturated oxygen (SSO₂) therapy, which has demonstrated a significant reduction in infarct size and improved microvascular function. The AMIHOT I and II trials reported a 26% reduction in infarct size in patients treated within six hours of symptom onset [221], prompting further evaluation in the AMIHOT III trial (NCT04743245). Other pharmacological strategies under investigation include colchicine (NCT05734612), which mitigates inflammation by inhibiting NLRP3 inflammasome activation, and nicorandil (NCT04665648), a nitric oxide donor with cardioprotective effects. Additionally, left ventricular unloading prior to reperfusion has been proposed as a strategy to minimise myocardial damage, with its clinical impact currently under investigation in the STEMI-DTU trial (NCT03947619). Despite promising findings in phase II studies, many phase III trials have struggled to demonstrate significant clinical benefits, particularly in terms of reducing mortality and heart failure incidence, likely due to patient heterogeneity and methodological limitations [215].
One of the major challenges in translating preclinical findings into clinical practice lies in the multifaceted nature of MRI, which involves oxidative stress, inflammation, mitochondrial dysfunction, and various cell death pathways. While several cardioprotective interventions have failed to demonstrate a substantial clinical impact, recent studies have identified novel therapeutic targets. Among these, advancements in paracrine signaling modulation have shown promise, with microRNAs, peptides, and extracellular vesicles exhibiting regenerative potential in experimental models [222]. Additionally, extracellular matrix modulation, particularly through agrin, has emerged as a potential strategy for myocardial repair [222].
Given the multifactorial nature of MRI, the most effective therapeutic approaches are likely to involve a combination of antioxidant therapy, pharmacological interventions, mechanical strategies, and regenerative medicine. The integration of these modalities may represent the most viable approach to improving clinical outcomes in AMI patients. Ongoing research will be critical in overcoming current barriers and facilitating the implementation of more effective combination therapies in routine clinical practice.
MRI is a complex process characterized by oxidative stress, metabolic dysregulation, calcium overload, mitochondrial dysfunction, and alterations in cell signaling pathways, ultimately leading to membrane and/or DNA damage and inflammation. Oxidative stress plays a central role in driving cell death through these mechanisms.
Recent advances in understanding programmed cell death, including autophagy, apoptosis, necroptosis, ferroptosis, and pyroptosis, have revealed novel therapeutic targets for MRI. Strategies targeting oxidative stress, either through standalone antioxidants or combination therapies addressing multiple pathways, have shown promise in vitro and in vivo. For instance, iron chelators help regulate iron homeostasis, while insulin mitigates glucose toxicity. Compounds such as NAC, edaravone, melatonin, and allopurinol have demonstrated potential in reducing oxidative stress and infarct size (Fig. 2). However, their clinical efficacy may improve with multitarget approaches. Notably, the combination of AA, NAC, and DFO has reduced infarct size and provided myocardial protection in preclinical models, with initial clinical studies confirming its safety.
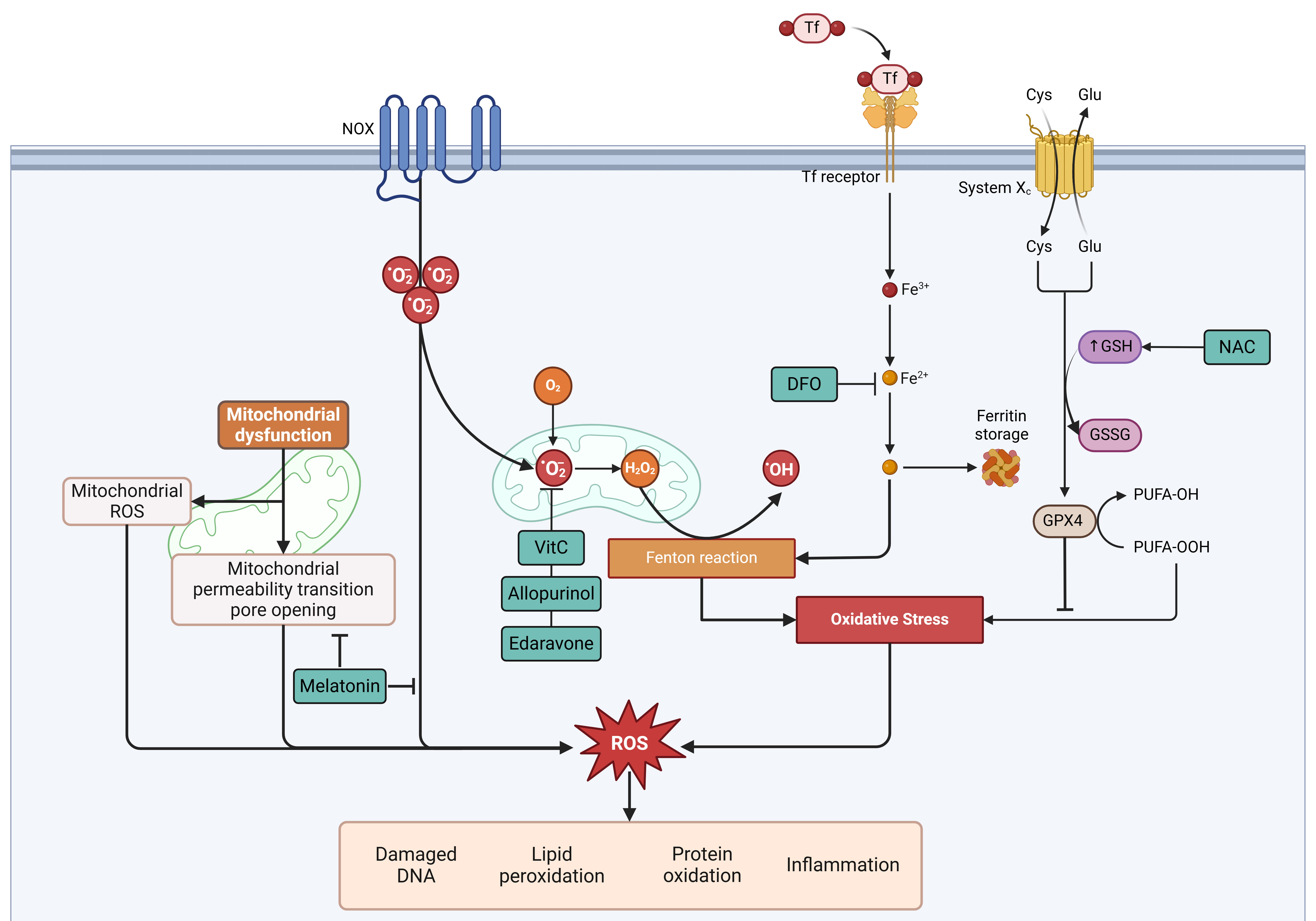 Fig. 2.
Fig. 2.
Sites of action of different drugs and their modulation in relation to oxidative stress and cell death. Cys, cysteine; DFO, deferoxamine; Fe3+, ferric iron; Fe2+, ferrous iron; Glu, glutamate; GPX4, glutathione peroxidase 4; GSH, reduced glutathione; GSSG, oxidized glutathione; H2O2, hydrogen peroxide; NAC, N-acetylcysteine; NOX, NADPH oxidase; •OH, hydroxyl radical; •O2⁻, superoxide anion; PUFA-OH, hydroxylated polyunsaturated fatty acid; PUFA-OOH, hydroperoxylated polyunsaturated fatty acid; System Xc⁻, cystine/glutamate antiporter; ROS, reactive oxygen species; Tf, transferrin; VitC, Vitamin C. Created with BioRender.com.
Despite significant advances in the clarification of the mechanisms of MRI, translating cardioprotective therapies from preclinical research to clinical practice remains challenging. While antioxidants have shown efficacy in animal models and some human trials, most studies are small-scale RCTs with limitations affecting generalizability. Emerging therapies, such as supersaturated oxygen (SSO2), have demonstrated a 26% infarct size reduction in phase II trials, warranting further investigation in phase III studies. Additionally, colchicine, which targets the NLRP3 inflammasome, and nicorandil, a nitric oxide donor, are being explored for their cardioprotective effects in AMI patients. Left ventricular unloading before reperfusion has also been proposed to enhance myocardial salvage, with ongoing trials assessing its clinical impact. However, heterogeneity in study populations, disease severity, comorbidities, and concomitant medications remains a barrier to clinical translation [215, 222].
Variability in therapeutic approaches further complicates implementation. Factors such as timing, dosage, and administration route (e.g., intracoronary vs. intravenous) vary across studies. Mechanistically, antioxidants are expected to be most effective when administered before and during reperfusion, a hypothesis supported by some trials, though rigorous evaluations remain limited. Outcome measures also differ, with infarct size widely regarded as the most robust indicator of myocardial injury, while functional outcomes such as LVEF, heart failure-related hospitalizations, and recurrent ischemic events provide additional insights into long-term therapeutic impact. Given the limited clinical data and methodological constraints, larger, well-powered, and mechanistically driven clinical trials are essential for a comprehensive evaluation of these strategies.
MRI remains a major therapeutic challenge due to its multifactorial nature and the complex interplay between oxidative stress, inflammation, and mitochondrial dysfunction. While significant progress has been made in identifying potential therapeutic targets, the translation of these findings into clinical practice has been hindered by limitations in study design, patient heterogeneity, and variability in outcome measures. Emerging multitarget approaches, such as combination antioxidant therapy, SSO2 administration, ischemic conditioning, and NLRP3 inhibition, represent promising strategies, but their efficacy needs to be validated in well-designed, large-scale clinical trials.
Future research should focus on refining experimental models that better replicate real-world patient populations, optimizing timing and dosing strategies for interventions, and standardizing clinical endpoints to facilitate meaningful comparisons across studies. Ultimately, a combination of pharmacological, mechanical, and regenerative strategies may be necessary to achieve significant improvements in MRI outcomes, underscoring the need for continued innovation and multidisciplinary collaboration in this field.
EV and RR led the conceptualization of the study. EV, CRS, and VPG were responsible for writing the manuscript and for the creation of the figures. EV, CRS, VPG, LS, SC, JCP, AIJG, and RA made substantial contributions in the acquisition and analysis of literature, provided supervision throughout the study and critically reviewed and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This study was supported by Agencia Nacional de Investigación y desarrollo (ANID), Fondo Nacional de Desarrollo Científico, Tecnológico y de Innovación Tecnológica (FONDECYT) grant number 1211850.
The authors declare no conflict of interest. Luciano Saso is serving as Guest Editor of this journal. We declare that Luciano Saso had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Ioanna-Katerina Aggeli.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.