





1 School of Health Services Management, Xi’an Medical University, 710021 Xi’an, Shaanxi, China
2 Second Clinical Medical College of Shaanxi University of Chinese Medicine, 712046 Xianyang, Shaanxi, China
3 The Third Department of Neurology, The Second Affiliated Hospital of Xi’an Medical University, 710038 Xi’an, Shaanxi, China
†These authors contributed equally.
Abstract
Ischemic stroke ranks among the leading global causes of death and disability, driven by intricate mechanisms such as neuronal injury, inflammation, and oxidative stress. Emerging as a pivotal player in ischemic stroke progression is ferroptosis—an iron-dependent form of regulated cell death. Its hallmarks—iron metabolic dysregulation and lipid peroxidation—trigger cell membrane disruption and irreversible neuronal damage. Beyond that, ferroptosis intensifies inflammation and compromises the blood–brain barrier (BBB), substantially increasing the impact of ischemic injury. Research indicates that modulating ferroptosis-related molecular pathways could significantly mitigate the pathological progression of ischemic stroke. Based on a systematic search of PubMed, Web of Science, Embase, and Cochrane Library databases (as of April 30, 2025), this review focuses on the progress of research on the mechanisms and treatments of ferroptosis in ischemic stroke over the past five years, aiming to investigate the underlying mechanisms, pathological roles, cross-disease associations, and targeted therapeutic strategies, to lay a theoretical foundation for the development of advanced therapies, and to outline the challenges and future directions of the field.
Keywords
- ischemic stroke
- ferroptosis
- lipid peroxidation
- iron metabolism
- therapeutic targets
Ischemic stroke, an acute cerebrovascular disease caused by cerebral artery stenosis or occlusion leading to cerebral ischemia and hypoxia, constitutes approximately 70% of all strokes and is among the world’s deadliest and most disabling conditions [1, 2]. Notably, acute ischemic stroke exhibits marked heterogeneity. Clinical studies typically categorize them into different subtypes based on etiology and pathogenesis, such as cardiogenic embolic stroke, lacunar infarction, cryptogenic stroke of undetermined origin, and atherosclerotic thrombotic infarction. These subtypes differ significantly in terms of risk factors, pathophysiologic processes, stroke severity, and clinical prognosis [3, 4]. Its pathological hallmarks encompass neuron damage, energy metabolic dysfunction, inflammation and oxidative stress resulting from cerebral ischemia. These processes act synergistically to induce severe brain dysfunction and neurological sequelae [5, 6]. Currently, intravenous thrombolysis (e.g., tPA) and mechanical thrombectomy serve as the primary treatments for ischemic stroke [7]. While these therapies significantly reduce disability and mortality by restoring cerebral blood flow, their efficacy is limited to narrow therapeutic windows (e.g., tPA within 3–4.5 hours post-onset) and entails risks of reperfusion injury and hemorrhagic complications [8]. Consequently, identifying novel targets and developing safer therapeutic strategies have emerged as key priorities in stroke research.
Ferroptosis, a novel form of regulated cell death, has garnered significant attention in the field of neurological diseases. Initially introduced by Dixon et al. in 2012 [9], ferroptosis differs from conventional cell death forms such as apoptosis, necrosis, and autophagy. Its central mechanism involves iron-dependent lipid peroxidation, driven by the interplay of iron metabolic disturbances and antioxidant defense systems failures. This process results in irreversible damage to neurons and glial cells. This discovery has provided a fresh perspective for understanding the mechanisms underlying ischemic stroke damage. Studies has shown that ferroptosis plays a critical role in the pathological progression of ischemic stroke [10, 11]. Following cerebral ischemia, the abnormal accumulation of iron ions catalyzes the production of excessive reactive oxygen species (ROS) via the Fenton reaction. This triggers a cascade of lipid peroxidation reactions that directly initiating ferroptosis. In return, ferroptosis accelerates disease progression by inducing neuronal programmed death, activating neuroinflammatory cascades, and compromising the integrity of the blood-brain barrier, thereby forming a vicious cycle [12, 13]. Furthermore, alterations in the expression profiles of key ferroptosis regulatory proteins have provided crucial clues for potential intervention strategies. For instance, reduced activity of Glutathione peroxidase 4 (GPX4) and increased expression of long-chain Acyl-CoA synthetase 4 (ACSL4) have been closely associated with the severity of post-ischemic neural damage. Ferroptosis regulation surpasses the limitations of traditional treatments that focus merely on blood flow pathways. It offers therapeutic benefits in both temporal and spatial dimensions, potentially extending the treatment window and effectively mitigating reperfusion injury [14]. These studies not only clarify the molecular mechanisms of ferroptosis in ischemic stroke but also provide new directions for targeted therapies. By modulating iron metabolism and activating antioxidant pathways, disease progression can be effectively intervened. This will lay the foundation for the development of novel diagnostic biomarkers and personalized treatment strategies.
Ferroptosis is a novel form of cell death distinct from apoptosis and necrosis, characterized by mitochondrial shrinkage and structural changes such as thinner outer membranes and fewer cristae [15]. Lipid peroxidation disrupts membrane integrity, increasing permeability and causing membrane rupture [9]. The nucleus retains its normal structure without apoptosis-such as chromatin condensation [16], and these morphological differences serve as key indicators for identifying ferroptosis. Biochemically, ferroptosis involves the synergistic interaction of iron metabolism imbalance and lipid peroxidation [17]. Elevated intracellular free iron drives the Fenton reaction with hydrogen peroxide, generating hydroxyl radicals and exacerbating lipid peroxidation [18, 19]. Lipid peroxidation primarily involves the conversion of polyunsaturated fatty acids (PUFAs) to lipid peroxides (LPO), damaging membrane integrity and triggering oxidative stress cascades [20, 21]. Normal cells counteract LPO via antioxidant systems such as the GPX4/GSH pathway to maintain membrane stability [22]. When glutathione is depleted or GPX4 activity declines, cells lose their defense against lipid peroxidation, triggering ferroptosis [23]. Unlike apoptosis and necrosis, ferroptosis is highly dependent on iron and oxidative stress and does not involve Caspase family activation or energy metabolism collapse [24, 25, 26, 27].
Disrupted iron metabolism and lipid peroxidation are pivotal in ischemic stroke-induced ferroptosis [28]. Disruption of the blood-brain barrier leads to free iron accumulation. Iron ions trigger lipid peroxidation via the Fenton reaction to generate hydroxyl radicals [29, 30, 31]. During ferroptosis, upregulated iron transporters (e.g., transferrin receptor TfR1) enhance iron ions’ transmembrane transport and intracellular accumulation, exacerbating oxidative stress and cellular damage [32, 33]. Ischemia also induces hepcidin expression, inhibiting Ferroportin and exacerbating brain iron overload [34, 35]. Additionally, ischemia leads to abnormal H-ferritin and L-ferritin expression in the brain, altering iron storage and release dynamics. Moreover, ischemia leads to abnormal H-ferritin and L-ferritin expression in the brain, altering iron storage and release dynamics. As the main components of cell membrane phospholipids, polyunsaturated fatty acids (PUFAs) are prone to oxidation due to their double-bond structures, producing lipid peroxides (LPO) [36]. Under the action of ACSL4 and LPCAT3, PUFAs are incorporated into cell membranes and oxidized into LPO. Simultaneously the impaired GPX4/GSH pathway reduces clearance efficiency, and enzymatic reactions such as lipoxygenase exacerbate oxidative damage [37, 38]. During ischemic stroke, the overwhelmed antioxidant system leads to peroxide accumulation. Key enzymes such as lipoxygenase accelerate phospholipid peroxidation, and abnormal mitochondrial metabolism further drives this process [39, 40, 41]. Ultimately, LPO disrupts membrane structure, leading to iron imbalance, neuronal death, and impaired brain function [42, 43]. Under normal conditions, cells resist ferroptosis via the GPX4/GSH pathway. In ischemic stroke, reduced GPX4 expression and GSH depletion impair the antioxidant system. SystemXc– malfunction also reduces cystine uptake, hindering GSH synthesis [44, 45, 46]. Furthermore, the inactivated NRF2 pathway fails to regulate oxidation and iron-metabolism-related genes, reducing cells’ oxidative-stress adaptation and blocking GPX4 recovery, thereby exacerbating neuronal injury [47, 48]. Ferroptosis is closely tied to intracellular free iron homeostasis, with excess iron causing damage via lipid peroxidation and ROS accumulation [49]. Multiple systems counteract ferroptosis. For example, ferroptosis suppressor protein 1 (FSP1) on the cell membrane scavenging free radicals with Coenzyme Q10 via myristoylation, and tetrahydrobiopterin inhibits ROS generation [50, 51, 52]. The glutathione peroxidase, AMPK, and MAPK signaling pathways also participate in ferroptosis regulation to maintain cellular redox balance [53, 54]. The core molecular mechanisms of ferroptosis are summarized in Fig. 1.
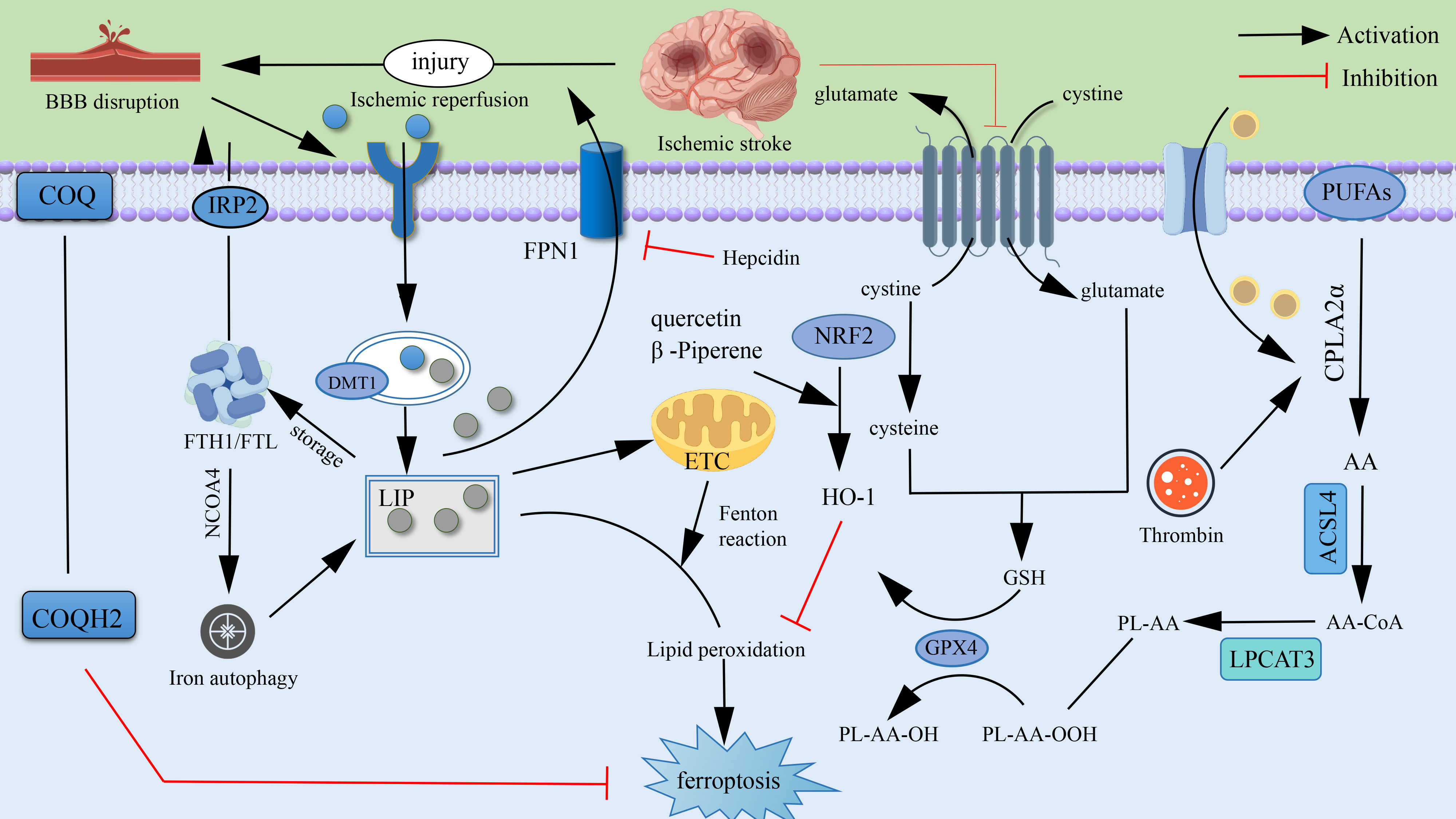 Fig. 1.
Fig. 1.
Mechanism of ferroptosis Molecules.
Ferroptosis research extends across oncology, metabolic diseases, and
immune-system disorders. In oncology, ferroptosis exhibits dual-edged regulation.
For instance, the System Xc– inhibitor erastin kills cancer cells, and GPX4
plays a key regulatory role [55, 56]; In digestive system diseases, ferroptosis
also has a double-edged effect. In mesenchymal colorectal cancer cells, the
WNT/
The molecular mechanisms of ferroptosis are implicated in several pathological
processes of ischemic stroke, including direct neuronal damage, enlargement of
the ischemic penumbra, and promotion of inflammatory and immune responses
[65, 66]. Collectively, these processes exacerbate the pathological progression of
ischemic stroke. Ferroptosis directly induces neuronal death through iron
accumulation and lipid peroxidation, causing irreversible damage to the ischemic
core [67]. In ischemic stroke, ferroptosis is closely associated with abnormally
elevated levels of intracellular free iron. Free iron reacts with hydrogen
peroxide via the Fenton reaction to generate hydroxyl radicals (•OH),
exacerbating lipid peroxidation. The accumulation of lipid peroxides not only
compromises cell membrane integrity but also triggers downstream oxidative stress
responses, thereby accelerating cell death [68]. Additionally, during
ferroptosis, depletion of glutathione (GSH) and reduced activity of GPX4
eliminate the cell’s protective capacity against lipid peroxidation, ultimately
resulting in causing irreversible neuronal damage. In the ischemic penumbra,
ferroptosis mediates sublethal neuronal states, thereby accelerating damage
expansion. The ischemic penumbra is the region between the ischemic core and
normal brain tissue, where neurons are not completely dead initially but are in a
state of functional impairment and metabolic dysfunction. Ferroptosis induces
lipid peroxidation and iron accumulation in neurons, further damaging them and
ultimately resulting in their death. This damage expansion not only exacerbates
cerebral ischemia and hypoxia but also further worsens the patient’s clinical
outcome. Ferroptosis-derived oxidative products activate inflammatory signaling
pathways in the brain, further aggravating neuroinflammation and impairing brain
tissue repair [69]. Lipid peroxides and oxidative stress products generated
during ferroptosis, such as malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE),
exhibit strong inflammation-inducing effects [70, 71]. These products activate
microglia and astrocytes in the brain, leading to the release of inflammatory
factors such as tumor necrosis factor-
Pharmacological inhibition and genetic regulation strategies show significant therapeutic potential in ferroptosis-targeted interventions. In pharmacological development, Liproxstatin-1, as a free radical-scavenging iron deposition inhibitor, reduces the volume of cerebral infarction volume by clearing lipid-derived reactive oxygen species and inhibiting the oxidative stress response in cerebral ischemia models. When evaluated using standardized scales such as the Modified Neurological Severity Scale (mNSS), the neurological function score improves by 2–3 points [78, 79]. Rosiglitazone, as an ACSL4-specific inhibitor, reduces ferroptosis marker COX2 expression by 60% through blocking the abnormal metabolism of polyunsaturated fatty acids to phosphatidylethanolamine. It also effectively ameliorates motor dysfunction in diabetic nephropathy and cerebral ischemia models. Genetic modulation via ACSL4 knockdown (using siRNA or CRISPR) reduces PUFA-PE synthesis, significantly decreases lipid peroxidation product 4-HNE levels, and achieves approximately 50% infarct volume reduction in MCAO models [80]. Adenoviral vector-mediated GPX4 overexpression increases ischemic neuron survival by 60% through reconstructing the GSH metabolic network and suppressing the generation of toxic metabolite 4-HNE generation. These interventions establish novel therapeutic pathways for cerebral ischemic injury by precisely targeting the core regulatory nodes of ferroptosis.
Ferroptosis pathogenesis is characterized by a tripartite interplay among iron
metabolism dysregulation, mitochondrial dysfunction, and neuroinflammatory
responses. Ischemic stress induces a 3-fold upregulation of the transferrin
receptor (TFR1), leading to abnormal intracellular free iron accumulation. Excess
Fe2+ generates highly reactive hydroxyl radicals via the Fenton reaction,
initiating an ACSL4-catalyzed lipid peroxidation cascade the targets membrane
polyunsaturated fatty acids (e.g., arachidonic acid) and ultimately causes
membrane disintegration [81]. Concurrently, iron overload induces mitochondrial
ultrastructural damage and functional decompensation. This is characterized by a
50% reduction in ATP synthesis efficiency and 200% increase in mitochondrial
ROS production, creating dual insults of oxidative damage and metabolic collapse
[82]. Ferroptotic neurons release damage-associated molecules (e.g., HMGB1),
which activate microglia to overproduce proinflammatory cytokines (e.g., a 300%
increase in TNF-
Molecular regulation of ferroptosis centers on three core pathways. Herbal components demonstrate significant intervention effects through multi-target synergy [85]. Regarding GPX4 regulation, herbal formulas double GPX4 expression by activating specific protective proteins. This effectively scavenges lipid peroxides, reducing cerebral infarct volume by 35% and improving neurological function in ischemia models. In GSH metabolism modulation, baicalin elevates GSH levels by 50% through enhancing cystine transporter SLC7A11 functionality. This reduces oxidative damage marker malondialdehyde by 40%, sharing mechanistic commonality with clinical oncology strategies [86]. Regarding antioxidant defense, gastrodin and related compounds activate the Nrf2 pathway, doubling HO-1 expression and decreasing cerebral iron deposition by 30%. This provides blood-brain barrier protective effects analogous to those of crocin [87, 88]. Notably, Nrf2 activation synergizes with the coenzyme Q10 (CoQ10) antioxidant system through reduced CoQ10-mediated lipid radical neutralization. This dual-pathway mechanism, validated in isoliquiritigenin and other herbal compounds, provids novel insights for multi-target therapeutic development [89].
GPX4 and NRF2 are key targets for stroke treatment, with their core mechanisms and clinical applications being crucial [90]. The viewpoint supporting targeting GPX4 suggests that it can reduce harmful lipid peroxides. Clinical observations have found that GPX4 activity is generally reduced in stroke patients, indicating its association with disease risk. However, GPX4 activity is highly dependent on sufficient glutathione and NADPH as cofactors. Their supply is often inadequate during cerebral ischemia, thereby limiting its therapeutic efficacy. In contrast, as an upstream regulatory factor, NRF2 activation can simultaneously induce the expression of multiple antioxidant proteins, including GPX4 (e.g., HO-1, NQO1, SLC7A11), thereby forming a broader cellular protective network. Importantly, Nrf2 activators may still hold therapeutic potential in the post-stroke reperfusion stage. From the perspective of drug development and clinical application, it is difficult to developing effective small-molecule GPX4 activators is challenging. In contrast, research on compounds targeting the Nrf2 pathway is relatively more advanced. Additionally, different subtypes of stroke (e.g., embolic stroke and lacunar stroke) may respond differently to these two targets. Therefore, future research should focus on exploring dual-functional therapeutic approaches capable of synergistically activating the NRF2 pathway and effectively enhancing GPX4 activity.
Recent therapeutic advances in targeting ferroptosis for ischemic stroke have been achieved through five primary approaches: iron chelators, antioxidants, genetic regulation, small-molecule compounds, and drugs targeting ferroptosis signature pathways. A systematic overview of these therapeutic targets and corresponding strategies is presented in Fig. 2 and Table 1 (Ref. [29, 30, 31, 37, 38, 44, 45, 46, 47, 48, 54, 55, 56, 57, 58, 70, 71, 80, 87, 88, 89, 91, 92, 93, 94, 95]).
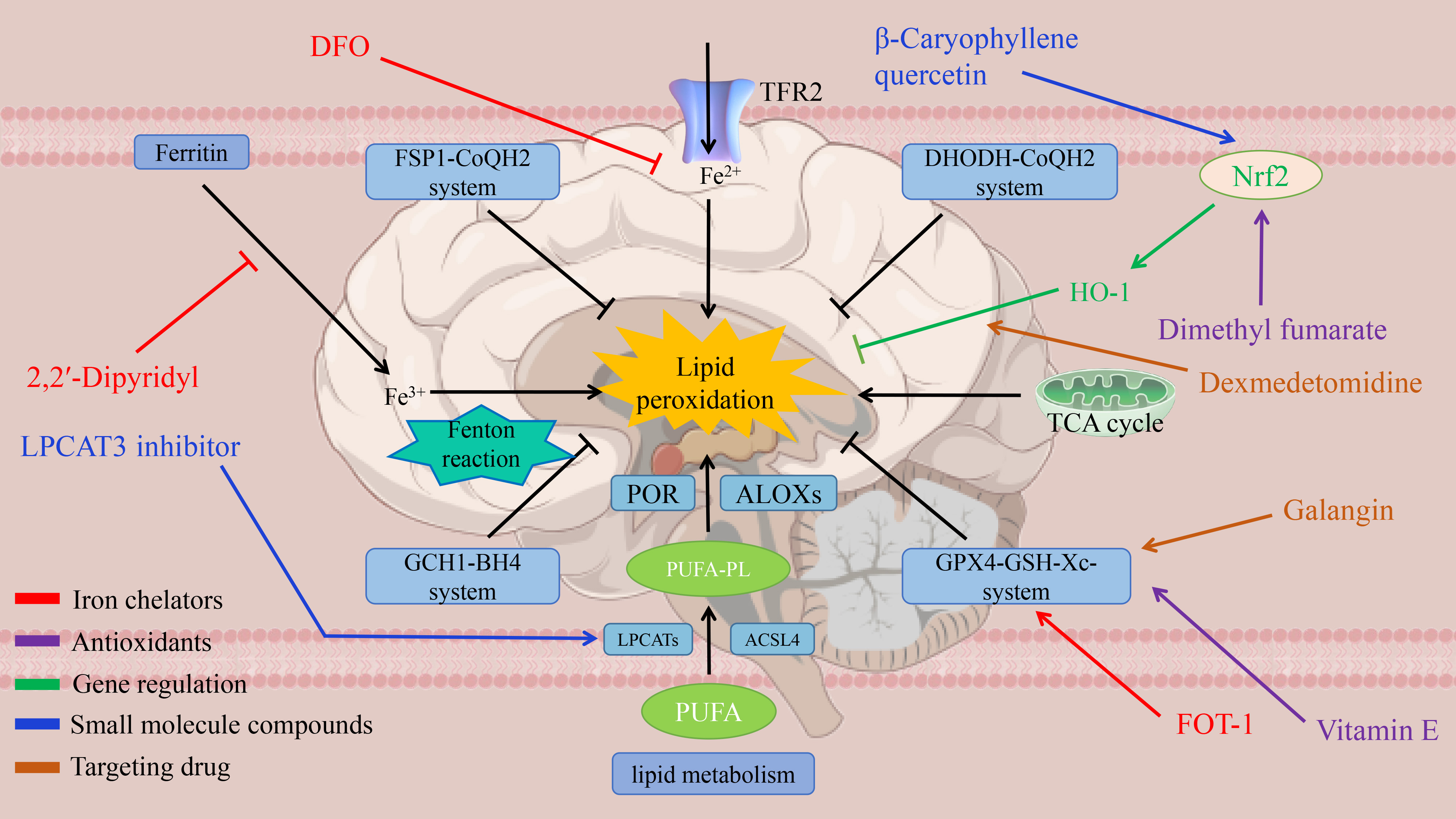 Fig. 2.
Fig. 2.
Therapeutic targets for ferroptosis.
| Categories | Core pathogenesis | Targeted intervention approaches and effects | Clinical translational significance |
| Iron metabolism disorder | Ischemia leads to abnormal free iron deposition and the Fenton reaction catalyzes an increase in ROS, inducing lipid peroxidation to damage neurons | Iron chelator Deferoxamine (DFO): reduction of infarct volume and improvement of neurological function in an animal model [29, 30, 31] | Possible prolongation of the thrombolytic time window and reduction of reperfusion injury |
| Novel chelator FerroTerminator1 (FOT1): inhibition of iron death in a liver disease model [91, 92, 93] | |||
| Lipid peroxidation | Up-regulation of (Acyl-CoA synthetase 4) ACSL4 leads to PUFA-PE synthesis and down-regulation of Glutathione peroxidase 4 (GPX4) leads to lipid peroxide scavenging and triggers cell membrane division | ACSL4 Knockout: Reduced Infarct Volume in the (Middle Cerebral Artery Occlusion) MCAO Model [37, 38] | ACSL4 is a differentiated target in lacunar versus nonlacunar stroke |
| Rosiglitazone (ACSL4 inhibitor): reduced Cyclooxygenase-2 (COX2) expression [80] | |||
| Neuro-inflammation | Ferroptosis products activate microglia and produce the inflammatory factors TNF- |
Liproxstatin-1 (free radical trapping agent): infarct volume decreased, the Increased neural scores [70, 71] | Blocking the iron death-inflammation cycle and attenuating secondary injury |
| GPX4 pathway exhaustion | SystemXc-function downregulation leads to glutathione depletion, GPX4 inactivation, and antioxidant imbalance | GPX4 overexpression: increased neuronal survival [44, 45, 46] | Combination with thrombolysis may enhance efficacy |
| Dimethyl fumarate: activation of nuclear factor E2-related factor 2 (NRF2) pathway enhances GPX4 activity [94, 95] | |||
| NRF2 pathway inhibition | NRF2 inactivation leads to abnormal Heme Oxygenase-1 (HO-1) regulation and increased brain iron deposition | Aspalathin: activation of NRF2 pathway, elevated HO-1 expression, decreased brain iron deposition [47, 48, 87, 88, 89] | Multi-target synergy of natural compounds (antioxidant and iron regulation) |
| Cross-disease modulation | Tumor therapy induces iron death to promote cell death; inhibition of ferroptosis needed to protect neurons in stroke | Stroke-targeted inhibition of ACSL4; activation of ACSL4 by cancer therapy [54, 55, 56, 57, 58] | Intervention programs need to be tailored to the disease mechanism |
GPX4, Glutathione peroxidase 4; NRF2, nuclear factor E2-related factor 2; ROS,
reactive oxygen species; ACSL4, Acyl-CoA synthetase 4; PUFA, polyunsaturated
fatty acids; TNF-
Iron chelators suppress ferroptosis and associated oxidative damage by sequestering free iron, thereby reducing hydroxyl radical (•OH) generation via the Fenton reaction [91]. For instance, Deferoxamine (DFO), as a classic iron chelator, has been shown to significantly reduces infarct volume and improves neurological function in stroke animal models [92]. The classic chelator DFO has demonstrated significant infarct volume reduction and neurological improvement in stroke animal models [93]. The novel chelator FerroTerminator1 (FOT1) demonstrates therapeutic efficacy in metabolic liver disease models by significantly decreasing hepatic lipid peroxidation and suppressing ferroptosis [96].
Antioxidants prevent ferroptosis by directly scavenging reactive oxygen species (ROS) scavenging or by enhancing of endogenous antioxidant systems [97, 98]. Vitamin E, a potent lipid-soluble antioxidant, protects cellular membrane integrity by reducing lipid peroxidation and shows significant neuroprotective effects in stroke models [99]. Dimethyl fumarate (DMF) activates the Nrf2/ARE signaling pathway to upregulate GPX4 expression, thereby effectively mitigating lipid peroxidation in ischemic brain injury [94].
Genetic modulation of ferroptosis-related genes effectively alleviates pathological processes in ischemic stroke. Downregulating TRF2 and ACSL4 expression significantly reduces lipid peroxidation and iron accumulation, thereby counteracting the ferroptosis-promoting effects of their overexpression [100]. Nrf2, a master regulator of antioxidant genes, exerts anti-ferroptotic effects by modulating antioxidant enzymes such as HO-1 and GPX4 [95]. A study has shown demonstrate that activating the Nrf2 pathway activation significantly enhances neurological functional recovery in ischemic stroke [101].
Various novel small-molecule compounds have been developed for ferroptosis
intervention.
Specific drugs show therapeutic potential by targeting signature ferroptosis
pathways [105]. Galangin restores cellular antioxidant capacity and inhibits
ferroptosis by activating the SLC7A11/GPX4 pathway [106]. Furthermore,
Dexmedetomidine attenuates ischemia-reperfusion injury by modulating the
HIF-1
Although advances have been made in elucidating ferroptosis mechanisms in ischemic stroke, substantial challenges persist concerning its multidimensional regulatory networks and clinical translation bottlenecks. Current basic research urgently requires investigation into the spatiotemporal heterogeneity of iron metabolism and the interaction mechanisms between lipid peroxidation and mitochondrial metabolism. Despite extensive research on iron homeostasis, the spatiotemporal dynamics of ferroptosis signaling across brain regions (e.g., hippocampus, cortex) and ischemic phases (acute/subacute) are not well defined. Future studies should pay particular attention to the differences in ferroptosis between lacunar and nonlacunar ischemic stroke. Lacunar stroke, a type of small-vessel disease, has unique pathological mechanisms, such as microscopic foci and BBB leakage. These may cause iron metabolism disorders and lipid peroxidation, which differ significantly from nonlacunar stroke in pathology. Therefore, it is imperative to investigate the differences in key ferroptosis pathways, neuroinflammatory responses, and therapeutic approaches across different stroke subtypes. Establishing a lacunar stroke-specific model, comparing key factor expression profiles via single-cell sequencing, and developing subtype-specific nanodelivery systems are recommended. The authors have systematically summarized the molecular mechanisms of ferroptosis and their cross-disease relevance. They have also detailed elucidation of emerging therapeutic targets and validated pharmacological agents from molecular mechanistic perspectives.
Iron metabolism regulation involves complex coordination among multiple factors, including iron transporters (TfR1, FPN), Ferritin, and Hepcidin [108]. Ischemic stroke disrupts regulatory networks, but the relationship between iron metabolism alterations and ferroptosis remains incompletely defined. Further investigation into region-specific and time-dependent variations in iron metabolism is needed. Future studies should use single-cell sequencing to clarify cell-type-specific iron distribution patterns and ferroptosis mechanisms. Therapeutic development faces two major challenges: drug delivery efficiency and coordination of treatment efficacy. Current iron chelators and antioxidants show limited clinical translation potential due to challenges in penetrating the blood-brain barrier and insufficient targeting specificity [109, 110]. Nanocarrier-based intelligent drug delivery systems enable precise ischemic region enrichment in ischemic regions through surface modification strategies. Monotherapy targeting ferroptosis shows limited efficacy and requires combination strategies with thrombolysis and other interventions. Combining ferroptosis inhibitors with GPX4 activators modulates metabolism and improves neurological function [111]. Future research should develop dual-optimized systems that integrate delivery efficiency and therapeutic potency. Developing sensitive non-invasive detection methods, such as advanced neuroimaging, will accelerate the translational applications of ferroptosis research. Engineered nanoparticles such as Pt@LF show blood-brain barrier penetration and ischemic region targeting capabilities. They enable the deep delivery of neuroprotectants while suppressing ferroptosis in stroke [112]. The multi-component synergistic effects of traditional Chinese medicine offer novel approaches for ferroptosis intervention. Tanshinone IIA inhibits lipid peroxidation via Nrf2 activation, while baicalein downregulates ACSL4 and chelates free iron. These actions suggest multi-target synergy through “antioxidant-iron regulation-anti-inflammatory” mechanisms when combined. We propose developing AI-driven platforms that integrate molecular docking and network pharmacology. These platforms can identify natural compoundssuch as celastrol and andrographolide, which have GPX4-activating or FSP1-upregulating effects. This approach can overcome single-target limitations by designing integrated Chinese-Western formulations [113]. These future strategies for treating ischemic stroke by targeting ferroptosis are conceptually integrated in Fig. 3.
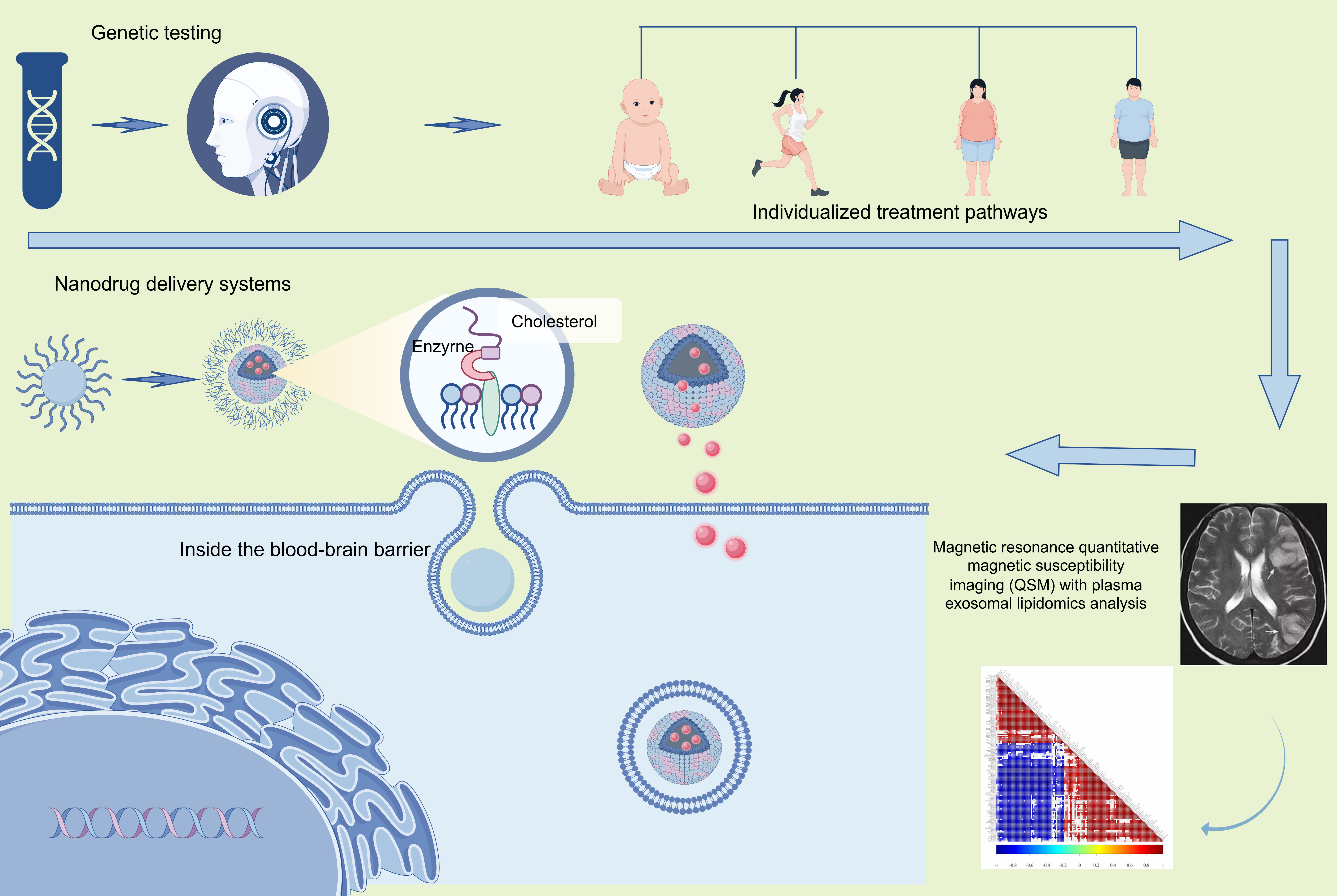 Fig. 3.
Fig. 3.
Future strategies based on ferroptosis for the treatment of ischemic stroke.
The lack of specific biomarkers reflecting ferroptosis progression currently limits the clinical implementation of related therapies. Dynamic monitoring of potential biomarkers, including serum ferritin, TFR1, and lipid peroxidation products such as MDA, may enable the assessment of ferroptosis levels. Developing non-invasive detection methodologies is a crucial step toward personalized treatment in biomarker research. Future directions suggest integrating quantitative susceptibility mapping (QSM) MRI with plasma exosome lipidomics. Machine learning can be used to construct ferroptosis signatures, such as exosomal FTH1/ACSL4 mRNA ratios and MDA-ferritin light chain dynamics. Combined with radiomics, these approaches can map the spatial patterns of iron deposition and lipid peroxidation patterns in the ischemic penumbra. CRISPR-Cas12a-driven liquid biopsy technology may enable real-time monitoring of ferroptosis-associated circRNAs, such as circGPX4, in peripheral blood. This technology offers novel tools for determining therapeutic windows determination and evaluating efficacy. These interdisciplinary innovations will advance ferroptosis research from mechanistic exploration to precision medicine, thereby establishing novel pathways for ischemic stroke prevention and treatment.
Although this review systematically summarizes the latest advances in the pathological mechanisms and intervention measures of ferroptosis in ischemic stroke, there are still several limitations and challenges remain in current research. The molecular mechanism of ferroptosis is highly complex, involving the interplay of multiple pathways, including iron metabolism, lipid metabolism, oxidative stress, and inflammation. Currently, there is insufficient research on the specific regulatory mechanisms and interactions across different stages, brain regions, and cell types of ischemic stroke; To date, research on ferroptosis has primarily relied on animal models, and the regulatory effects observed in these models have not been confirmed in clinical studies; Developing efficient brain-targeted delivery vectors is an urgent issue that needs to be addressed; The regulatory mechanisms and intervention effects of ferroptosis may vary at different time points, making it necessary to consider the treatment time window; The synergistic effects of ferroptosis inhibitors with existing therapies (thrombolysis, thrombectomy, anti-inflammatory treatments, etc.) are still in the early stages of research. It is necessary to optimize the combination methods to avoid potential drug interactions and adverse reactions. Recognizing the limitations mentioned above is crucial for objectively evaluating the current research status, planning future research directions, and conducting in-depth research on ferroptosis.
PUFAs, Polyunsaturated Fatty Acids; LPO, lipid peroxides; GSH, glutathione; GPX4, Glutathione peroxidase 4; ACSL4, HO-1, Heme Oxygenase-1; MCAO, Middle Cerebral Artery Occlusion; COX2, Cyclooxygenase-2; IL-6, interleukin-6; Acyl-CoA synthetase 4; NRF2, nuclear factor E2 related factor 2; LPO, lipid peroxides; ROS, reactive oxygen species; 4-HNE, 4-hydroxynonenal; MDA, malondialdehyde; TFR1, transferrin receptor; DFO, Deferoxamine; IFN-
TW: Methodology, Writing—review & editing. JWZ: Conceptualization, Writing—original draft. JHZ: Creating charts, Software and Drafting. XXS: Search for references and Graphic production. CJ: Methodology, Conceptualization, Writing—review and editing. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work. All authors contributed to editorial changes in the manuscript.
Not applicable.
Not applicable.
The Project of Shaanxi Key Laboratory of BrainDisorders (No.20NBZD02); the Major scientific research project of the “Buchang Cup” Brain-Heart Collaborative Research Fund in 2022 (No.NXTZ20221101), Xi’an Science and Technology Support Project (No.22YXYJ0096), National Science Foundation Incubation Program of the Second Afffliated Hospital of Xi’an Medical University (No.23KY0101); Wu Jieping Medical Foundation Scientific Research Special Grant Program (No.320.6750.2024-01-14); Shaanxi College Students’ Innovation and Entrepreneurship Training Program (No.S202411840081).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.